
Nuclear Small Dystrophin Isoforms during Muscle Differentiation
 , and
, and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tissue Culture
2.2. Immunofluorescence Staining
2.3. Western Blot
2.4. RNA-Sequencing
2.5. Statistical Analysis
3. Results
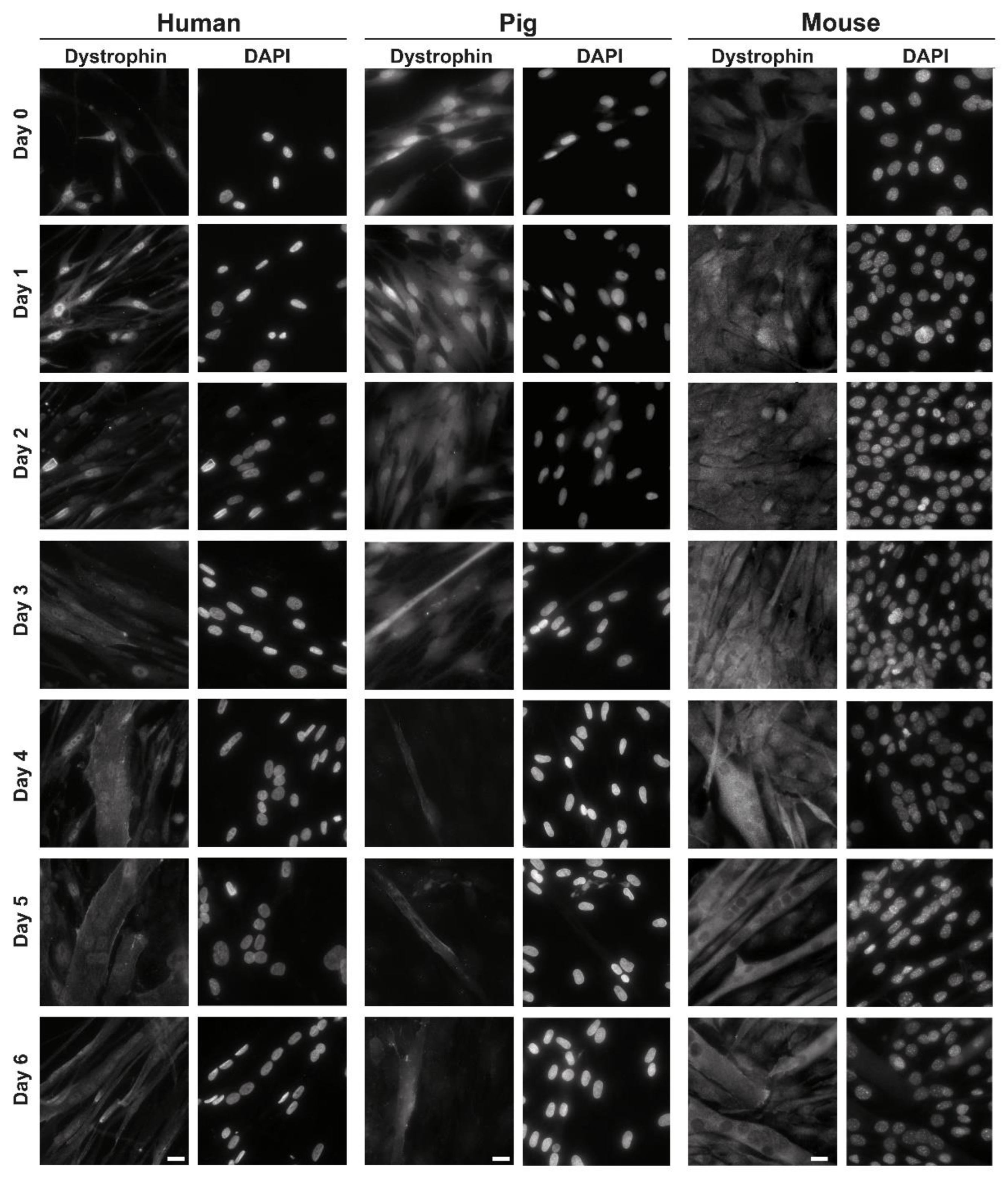
3.1. Localization of C-Terminal Dys during Muscle Differentiation
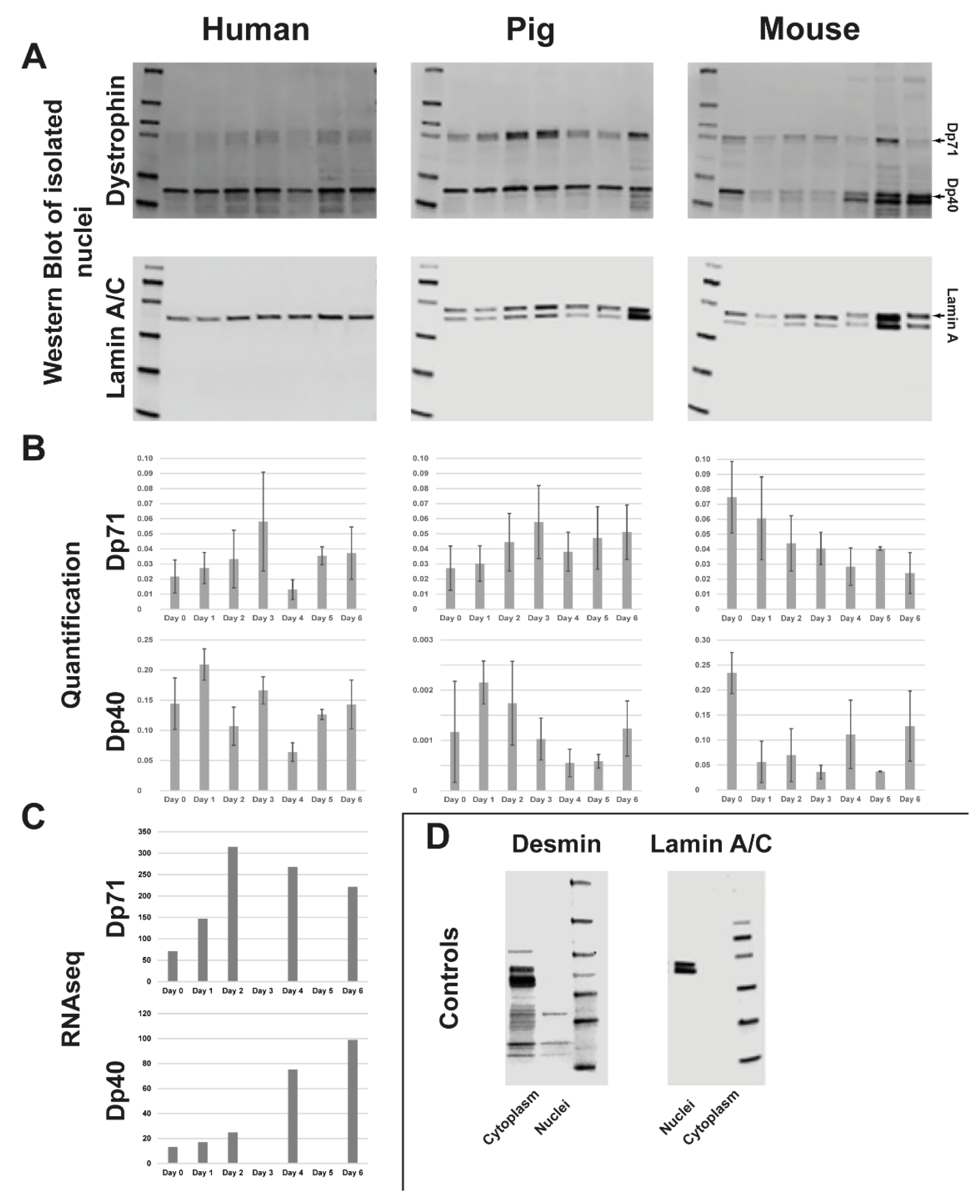
3.2. Quantification of C-Terminal Dystrophin during Muscle Differentiation and RNAseq of Human Myoblast Differentiation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.M.; Blake, D.J.; Davies, K.E. Davies, Apo-dystrophin-3: A 2.2kb transcript from the DMD locus encoding the dystrophin glycoprotein binding site. Hum. Mol. Genet. 1993, 2, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Feener, C.A.; Koenig, M.; Kunkel, L.M. Alternative splicing of human dystrophin mRNA generates isoforms at the carboxy terminus. Nature 1989, 338, 509–511. [Google Scholar] [CrossRef] [PubMed]
- García-Cruz, C.J.; Lourdel, S.; Annan, A.; Roger, J.E.; Montanez, C.; Vaillend, C. Tissue- and cell-specific whole-transcriptome meta-analysis from brain and retina reveals differential expression of dystrophin complexes and new dystrophin spliced isoforms. Hum. Mol. Genet. 2023, 32, 659–676. [Google Scholar] [CrossRef]
- Deconinck, N.; Dan, B. Pathophysiology of Duchenne muscular dystrophy: Current hypotheses. Pediatr. Neurol. 2007, 36, 1–7. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Koenig, M.; Beggs, A.H.; Moyer, M.; Scherpf, S.; Heindrich, K.; Bettecken, T.; Meng, G.; Müller, C.R.; Lindlöf, M.; Kaariainen, H.; et al. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am. J. Hum. Genet. 1989, 45, 498–506. [Google Scholar]
- Chesshyre, M.; Ridout, D.; Hashimoto, Y.; Ookubo, Y.; Torelli, S.; Maresh, K.; Ricotti, V.; Abbott, L.; Gupta, V.A.; Main, M.; et al. Investigating the role of dystrophin isoform deficiency in motor function in Duchenne muscular dystrophy. J. Cachex- Sarcopenia Muscle 2022, 13, 1360–1372. [Google Scholar] [CrossRef]
- Iskandar, K.; Triono, A.; Sunartini; Dwianingsih, E.K.; Indraswari, B.W.; Kirana, I.R.; Ivana, G.; Sutomo, R.; Patria, S.Y.; Herini, E.S.; et al. Dp71 and intellectual disability in Indonesian patients with Duchenne muscular dystrophy. PLoS ONE 2022, 17, e0276640. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Shah, N.A.; Woo, S.; Wilton-Clark, H.; Zhabyeyev, P.; Wang, F.; Maruyama, R.; Oudit, G.Y.; Yokota, T. Natural History of a Mouse Model Overexpressing the Dp71 Dystrophin Isoform. Int. J. Mol. Sci. 2021, 22, 12617. [Google Scholar] [CrossRef]
- Schorling, D.; Müller, C.; Pechmann, A.; Borell, S.; Rosenfelder, S.; Kölbel, H.; Schara, U.; Zieger, B.; Kirschner, J. Impaired secretion of platelet granules in patients with Duchenne muscular dystrophy—Results of a prospective diagnostic study. Neuromuscul. Disord. 2020, 31, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Awano, H.; Lee, T.; Takeshima, Y.; Matsuo, M.; Iijima, K. Patients with Duchenne muscular dystrophy are significantly shorter than those with Becker muscular dystrophy, with the higher incidence of short stature in Dp71 mutated subgroup. Neuromuscul. Disord. 2017, 27, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Farea, M.; Maeta, K.; Nishio, H.; Matsuo, M. Human Dystrophin Dp71ab Enhances the Proliferation of Myoblasts Across Species But Not Human Nonmyoblast Cells. Front. Cell Dev. Biol. 2022, 10, 877612. [Google Scholar] [CrossRef]
- Paúl-González, S.; Aragón, J.; Rodríguez-Martínez, G.; Romo-Yáñez, J.; Montanez, C. Differential expression of Dp71 and Dp40 isoforms in proliferating and differentiated neural stem cells: Identification of Dp40 splicing variants. Biochem. Biophys. Res. Commun. 2021, 560, 152–158. [Google Scholar] [CrossRef]
- Villarreal-Silva, M.; Suárez-Sánchez, R.; Rodríguez-Muñoz, R.; Mornet, D.; Cisneros, B. Dystrophin Dp71 is critical for stability of the DAPs in the nucleus of PC12 cells. Neurochem. Res. 2010, 35, 366–373. [Google Scholar] [CrossRef]
- Fuentes-Mera, L.; Rodríguez-Muñoz, R.; González-Ramírez, R.; García-Sierra, F.; González, E.; Mornet, D.; Cisneros, B. Characterization of a novel Dp71 dystrophin-associated protein complex (DAPC) present in the nucleus of HeLa cells: Members of the nuclear DAPC associate with the nuclear matrix. Exp. Cell Res. 2006, 312, 3023–3035. [Google Scholar] [CrossRef] [Green Version]
- González-Ramírez, R.; Morales-Lázaro, S.; Tapia-Ramírez, V.; Mornet, D.; Cisneros, B. Nuclear and nuclear envelope localization of dystrophin Dp71 and dystrophin-associated proteins (DAPs) in the C2C12 muscle cells: DAPs nuclear localization is modulated during myogenesis. J. Cell. Biochem. 2008, 105, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Sanchez, R.; Aguilar, A.; Wagstaff, K.M.; Velez, G.; Azuara-Medina, P.M.; Gomez, P.; Vasquez-Limeta, A.; Hernandez-Hernandez, O.; Lieu, K.G.; Jans, D.A.; et al. Nucleocytoplasmic shuttling of the Duchenne muscular dystrophy gene product dystrophin Dp71d is dependent on the importin alpha/beta and CRM1 nuclear transporters and microtubule motor dynein. Biochim. Biophys. Acta 2014, 1843, 985–1001. [Google Scholar] [CrossRef] [Green Version]
- Nishida, A.; Yasuno, S.; Takeuchi, A.; Awano, H.; Lee, T.; Niba, E.T.E.; Fujimoto, T.; Itoh, K.; Takeshima, Y.; Nishio, H.; et al. HEK293 cells express dystrophin Dp71 with nucleus-specific localization of Dp71ab. Histochem. Cell. Biol. 2016, 146, 301–309. [Google Scholar] [CrossRef]
- Niba, E.T.E.; Awano, H.; Lee, T.; Takeshima, Y.; Shinohara, M.; Nishio, H.; Matsuo, M. Dystrophin Dp71 Subisoforms Localize to the Mitochondria of Human Cells. Life 2021, 11, 978. [Google Scholar] [CrossRef]
- Aragón, J.; Martínez-Herrera, A.; Bermúdez-Cruz, R.M.; Bazán, M.L.; Soid-Raggi, G.; Ceja, V.; Coy-Arechavaleta, A.S.; Alemán, V.; Depardón, F.; Montañez, C. EF-hand domains are involved in the differential cellular distribution of dystrophin Dp40. Neurosci. Lett. 2015, 600, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, A.; Aragón, J.; Ceja, V.; Rendon, A.; Montanez, C. Nuclear transport and subcellular localization of the dystrophin Dp71 and Dp40 isoforms in the PC12 cell line. Biochem. Biophys. Res. Commun. 2022, 630, 125–132. [Google Scholar] [CrossRef] [PubMed]
- García-Cruz, C.; Merino-Jiménez, C.; Aragón, J.; Ceja, V.; González-Assad, B.; Reyes-Grajeda, J.P.; Montanez, C. Overexpression of the dystrophins Dp40 and Dp40(L170P) modifies neurite outgrowth and the protein expression profile of PC12 cells. Sci. Rep. 2022, 12, 1410. [Google Scholar] [CrossRef] [PubMed]
- Tozawa, T.; Itoh, K.; Yaoi, T.; Tando, S.; Umekage, M.; Dai, H.; Hosoi, H.; Fushiki, S. The shortest isoform of dystrophin (Dp40) interacts with a group of presynaptic proteins to form a presumptive novel complex in the mouse brain. Mol. Neurobiol. 2012, 45, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donandt, T.; Hintze, S.; Krause, S.; Wolf, E.; Schoser, B.; Walter, M.C.; Meinke, P. Isolation and Characterization of Primary DMD Pig Muscle Cells as an In Vitro Model for Preclinical Research on Duchenne Muscular Dystrophy. Life 2022, 12, 1668. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Colvin, M.K.; Cripe, L.; Herron, A.R.; Kennedy, A.; Kinnett, K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: Primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 2018, 17, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Muntoni, F.; Cau, M.; Ganau, A.; Congiu, R.; Arvedi, G.; Mateddu, A.; Marrosu, M.G.; Cianchetti, C.; Realdi, G.; Cao, A.; et al. Brief report: Deletion of the dystrophin muscle-promoter region associated with X-linked dilated cardiomyopathy. N. Engl. J. Med. 1993, 329, 921–925. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Van Deutekom, J.C.T.; Fokkema, I.F.; Van Ommen, G.J.B.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef]
- Naidoo, M.; Anthony, K. Dystrophin Dp71 and the Neuropathophysiology of Duchenne Muscular Dystrophy. Mol. Neurobiol. 2019, 57, 1748–1767. [Google Scholar] [CrossRef] [Green Version]
- Tadayoni, R.; Rendon, A.; Soria-Jasso, L.E.; Cisneros, B. Dystrophin Dp71: The smallest but multifunctional product of the Duchenne muscular dystrophy gene. Mol. Neurobiol. 2011, 45, 43–60. [Google Scholar] [CrossRef]
- De Brouwer, A.P.; Nabuurs, S.B.; Verhaart, I.E.; Oudakker, A.R.; Hordijk, R.; Yntema, H.G.; Hordijk-Hos, J.M.; Voesenek, K.; De Vries, B.B.; Van Essen, T.; et al. A 3-base pair deletion, c.9711_9713del, in DMD results in intellectual disability without muscular dystrophy. Eur. J. Hum. Genet. 2013, 22, 480–485. [Google Scholar] [CrossRef] [Green Version]
- Redin, C.; Gerard, B.; Lauer, J.; Herenger, Y.; Muller, J.; Quartier, A.; Masurel-Paulet, A.; Willems, M.; Lesca, G.; El-Chehadeh, S.; et al. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J. Med. Genet. 2014, 51, 724–736. [Google Scholar] [CrossRef] [Green Version]
- Tunnah, D.; Sewry, C.A.; Vaux, D.; Schirmer, E.C.; Morris, G.E. The apparent absence of lamin B1 and emerin in many tissue nuclei is due to epitope masking. J. Mol. Histol. 2005, 36, 337–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteves de Lima, J.; Relaix, F. Master regulators of skeletal muscle lineage development and pluripotent stem cells differentiation. Cell. Regen. 2021, 10, 31. [Google Scholar] [CrossRef]
- Stirm, M.; Fonteyne, L.M.; Shashikadze, B.; Lindner, M.; Chirivi, M.; Lange, A.; Kaufhold, C.; Mayer, C.; Medugorac, I.; Kessler, B.; et al. A scalable, clinically severe pig model for Duchenne muscular dystrophy. Dis. Model. Mech. 2021, 14, dmm049285. [Google Scholar] [CrossRef] [PubMed]
- Zaynitdinova, M.I.; Lavrov, A.V.; Smirnikhina, S.A. Animal models for researching approaches to therapy of Duchenne muscular dystrophy. Transgenic Res. 2021, 30, 709–725. [Google Scholar] [CrossRef] [PubMed]
- Stirm, M.; Fonteyne, L.M.; Shashikadze, B.; Stöckl, J.B.; Kurome, M.; Keßler, B.; Zakhartchenko, V.; Kemter, E.; Blum, H.; Arnold, G.J.; et al. Pig models for Duchenne muscular dystrophy—from disease mechanisms to validation of new diagnostic and therapeutic concepts. Neuromuscul. Disord. 2022, 32, 543–556. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donandt, T.; Todorow, V.; Hintze, S.; Graupner, A.; Schoser, B.; Walter, M.C.; Meinke, P. Nuclear Small Dystrophin Isoforms during Muscle Differentiation. Life 2023, 13, 1367. https://doi.org/10.3390/life13061367
Donandt T, Todorow V, Hintze S, Graupner A, Schoser B, Walter MC, Meinke P. Nuclear Small Dystrophin Isoforms during Muscle Differentiation. Life. 2023; 13(6):1367. https://doi.org/10.3390/life13061367
Chicago/Turabian StyleDonandt, Tina, Vanessa Todorow, Stefan Hintze, Alexandra Graupner, Benedikt Schoser, Maggie C. Walter, and Peter Meinke. 2023. "Nuclear Small Dystrophin Isoforms during Muscle Differentiation" Life 13, no. 6: 1367. https://doi.org/10.3390/life13061367