
The Roles of Coenzyme A Binding Pocket Residues in Short and Medium Chain Acyl-CoA Synthetases


Abstract
:1. Introduction
2. Materials and Methods
2.1. Site-Directed Mutagenesis
2.2. Purification of MacsMa and AcsMt Enzymes
2.3. Assay for Acyl-CoA and Acyl-Adenylate Production
2.4. Assay for Inorganic Pyrophosphate Production
2.5. Assay for Acyl-CoA Thioester Bond Formation
3. Results
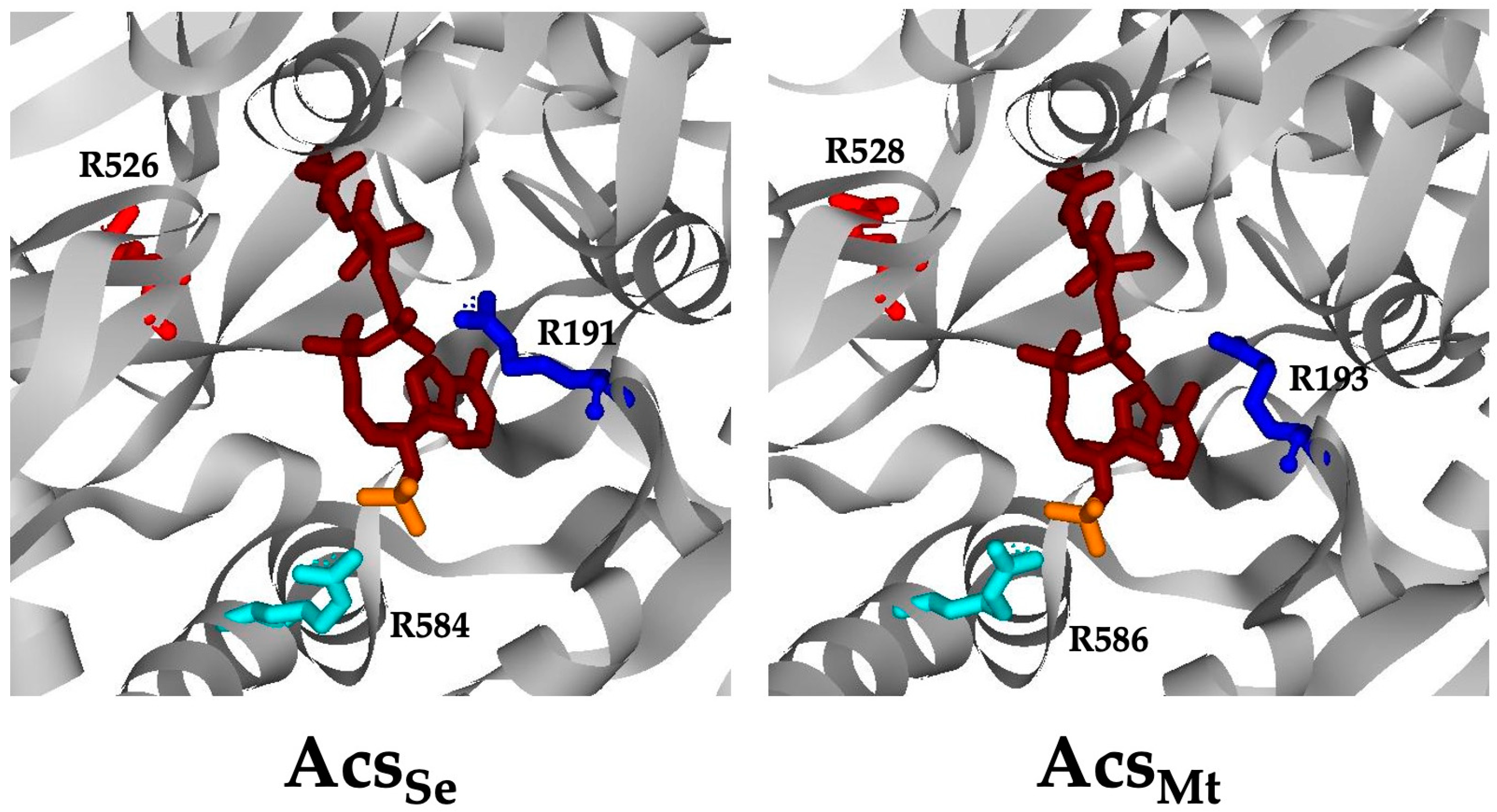
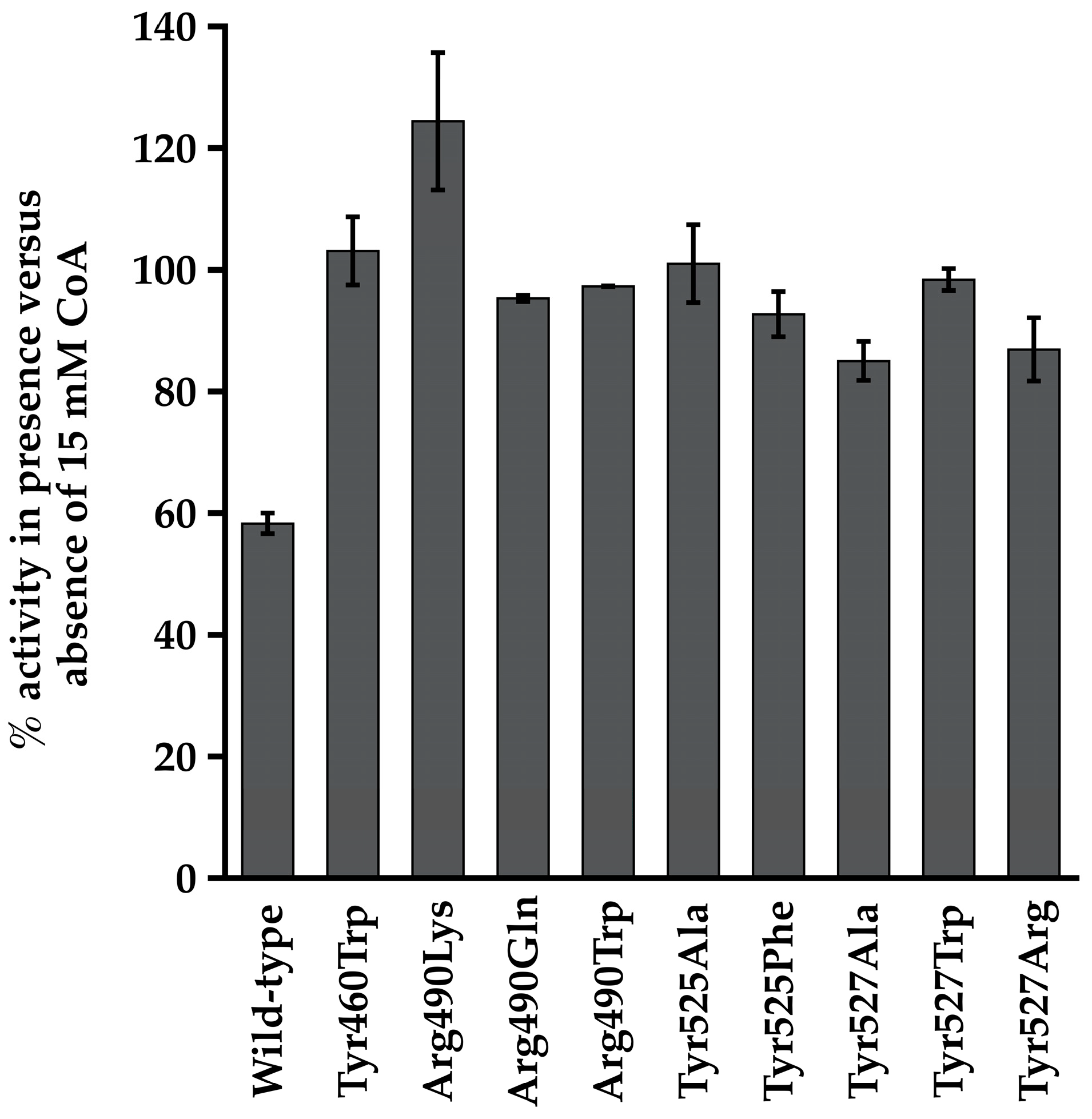
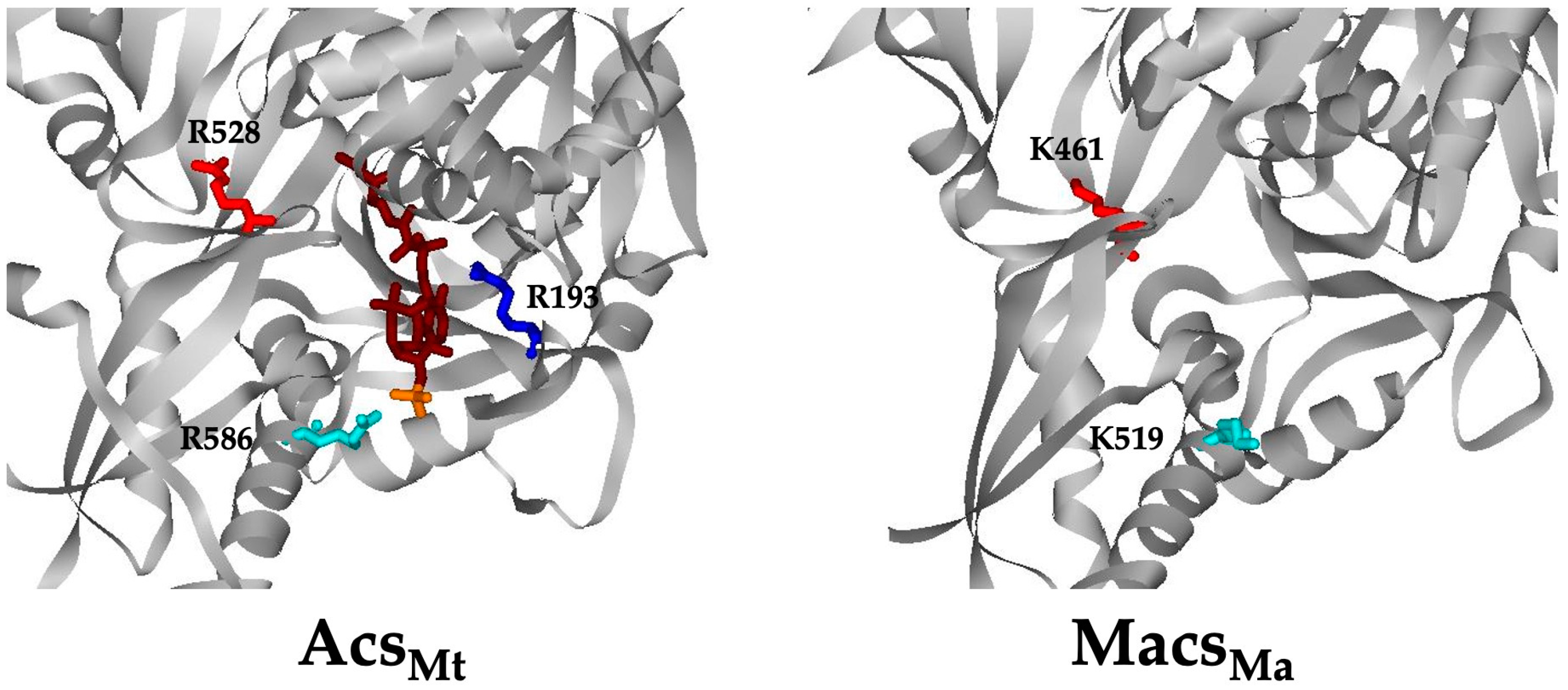
3.1. Conserved Arg Residues in Acs Interact with CoA
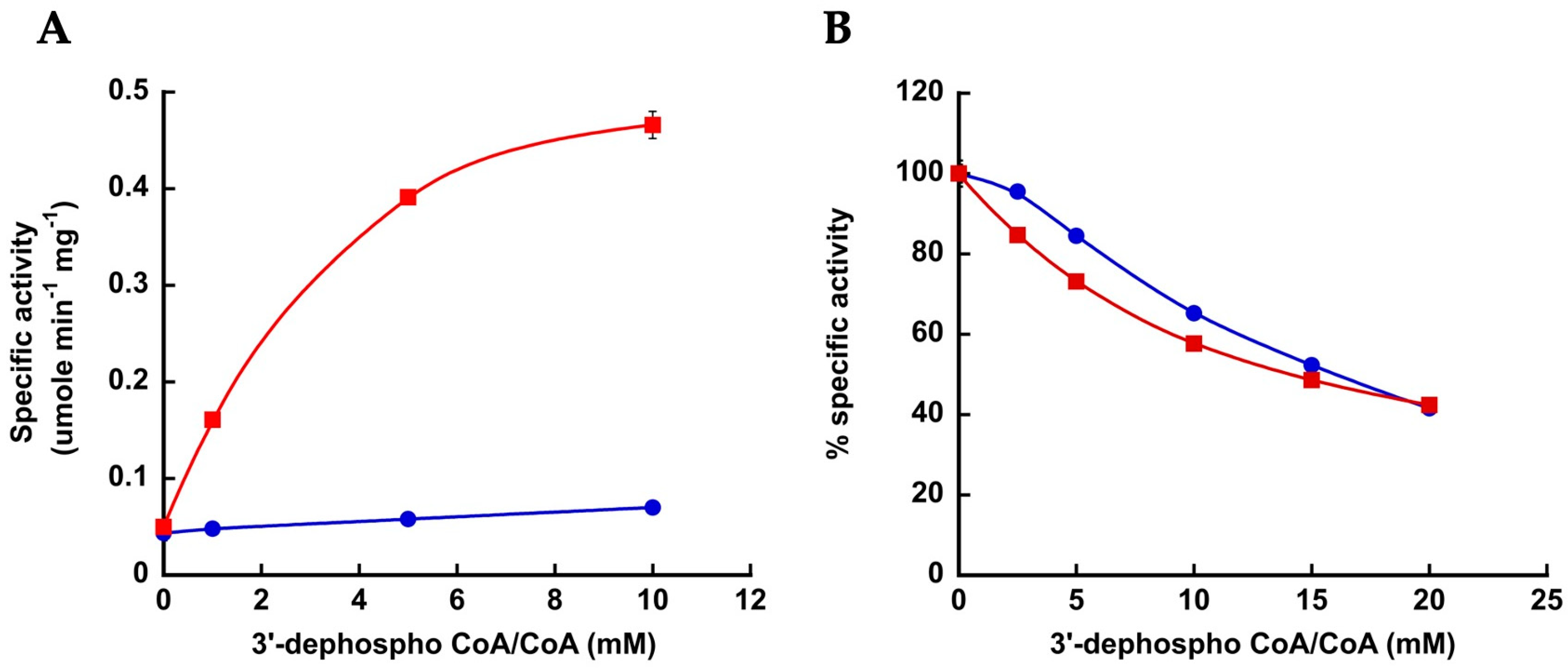
3.2. Interaction between Arg586 and the 3′-Phosphate Group of CoA Is Important for Substrate Binding and Catalysis
3.3. Electrostatic Interaction between MacsMa and the 3′-Phosphate Group of CoA Is Important
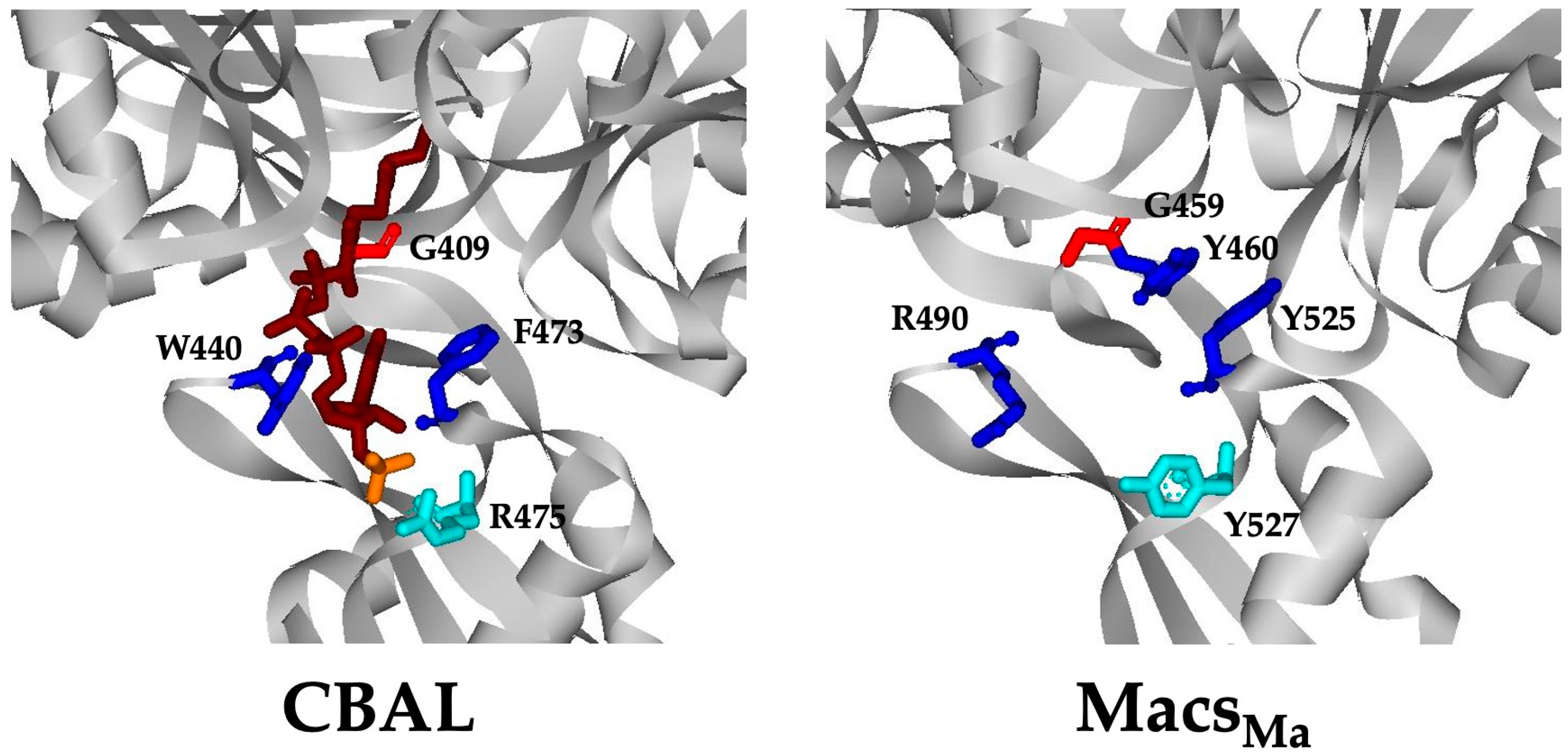
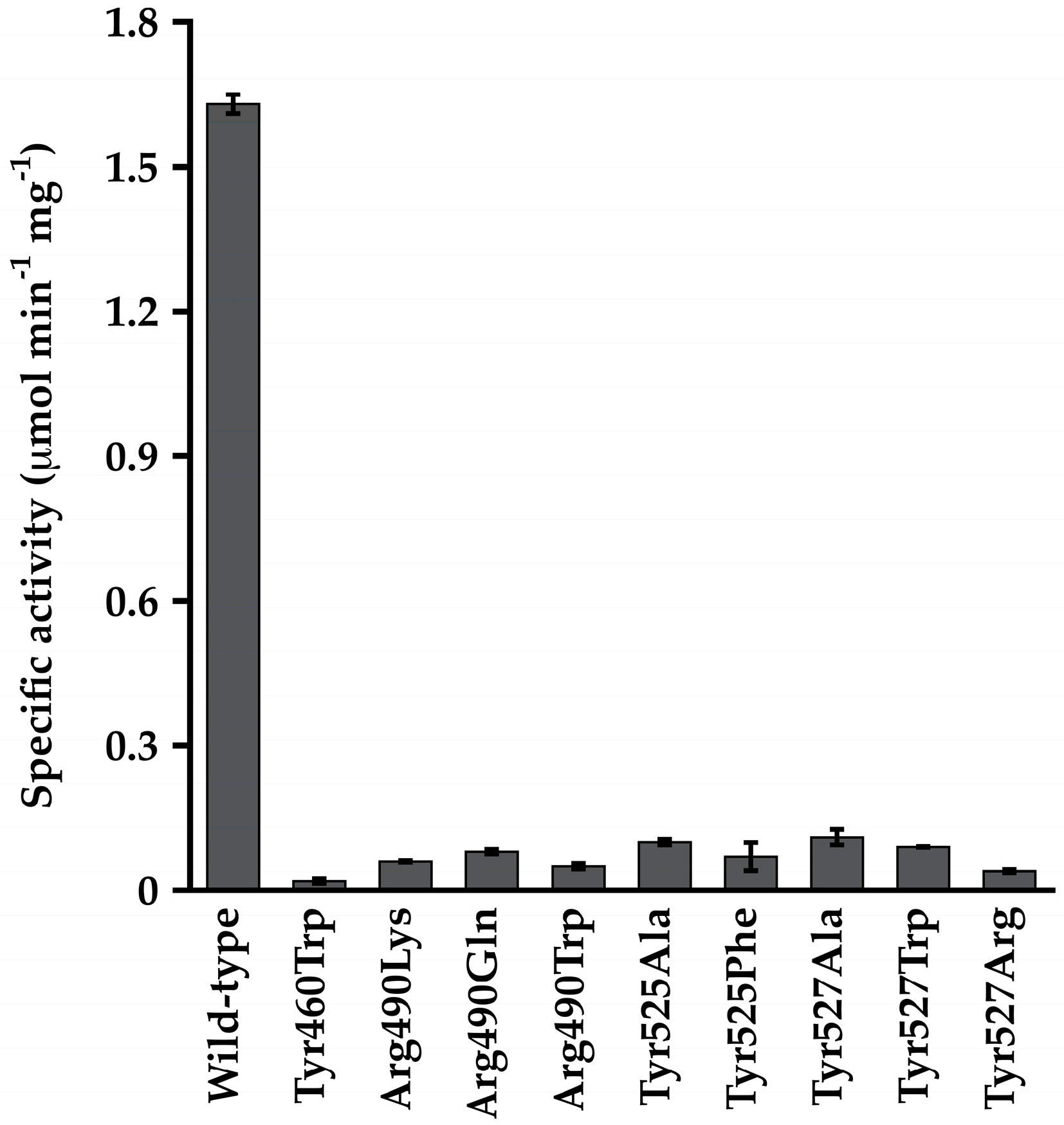
3.4. The CoA Binding Pocket in MacsMa Resembles That in CBAL
3.5. The Corresponding Lys Residues in MacsMa Do Not Play a Major Role in CoA Binding
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moffett, J.R.; Puthillathu, N.; Vengilote, R.; Jaworski, D.M.; Namboodiri, A.M. Acetate revisited: A key biomolecule at the nexus of metabolism, epigenetics, and oncogenesis—Part 2: Acetate and ACSS2 in health and disease. Front. Physiol. 2020, 11, 580171. [Google Scholar] [CrossRef]
- Moffett, J.R.; Puthillathu, N.; Vengilote, R.; Jaworski, D.M.; Namboodiri, A.M. Acetate revisited: A key biomolecule at the nexus of metabolism, epigenetics and oncogenesis—Part 1: Acetyl-CoA, acetogenesis and acyl-CoA short-chain synthetases. Front. Physiol. 2020, 11, 580167. [Google Scholar] [CrossRef] [PubMed]
- VanDrisse, C.M.; Escalante-Semerena, J.C. Protein acetylation in bacteria. Annu. Rev. Microbiol. 2019, 73, 111–132. [Google Scholar] [CrossRef]
- Jogl, G.; Tong, L. Crystal structure of yeast acetyl-coenzyme A synthetase in complex with AMP. Biochemistry 2004, 43, 1425–1431. [Google Scholar] [CrossRef]
- Gulick, A.M.; Starai, V.J.; Horswill, A.R.; Homick, K.M.; Escalante-Semerena, J.C. The 1.75 Å crystal structure of acetyl-CoA synthetase bound to adenosine-5′-propylphosphate and coenzyme A. Biochemistry 2003, 42, 2866–2873. [Google Scholar] [CrossRef] [PubMed]
- Reger, A.S.; Carney, J.M.; Gulick, A.M. Biochemical and crystallographic analysis of substrate binding and conformational changes in acetyl-CoA synthetase. Biochemistry 2007, 46, 6536–6546. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.B.; Ingram-Smith, C.; Cooper, L.L.; Qu, J.; Meng, Y.; Smith, K.S.; Gulick, A.M. The 2.1 Å crystal structure of an acyl-CoA synthetase from Methanosarcina acetivorans reveals an alternate acyl-binding pocket for small branched acyl substrates. Proteins 2009, 77, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, M.; Barad, S.; Peleg, Y.; Albeck, S.; Dym, O.; Brandis, A.; Mehlman, T.; Reich, Z. The identification and characterization of an oxalyl-CoA synthetase from grass pea (Lathyrus sativus L.). RSC Chem. Biol. 2022, 3, 320–333. [Google Scholar] [CrossRef]
- Ingram-Smith, C.; Smith, K.S. AMP-forming acetyl-CoA synthetases in Archaea show unexpected diversity in substrate utilization. Archaea 2007, 2, 95–107. [Google Scholar] [CrossRef] [Green Version]
- Brasen, C.; Urbanke, C.; Schonheit, P. A novel octameric AMP-forming acetyl-CoA synthetase from the hyperthermophilic crenarchaeon Pyrobaculum aerophilum. FEBS Lett. 2005, 579, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Ingram-Smith, C.; Cooper, L.L.; Smith, K.S. Characterization of an archaeal medium-chain acyl coenzyme A synthetase from Methanosarcina acetivorans. J. Bacteriol. 2010, 192, 5982–5990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingram-Smith, C.; Woods, B.I.; Smith, K.S. Characterization of the acyl substrate binding pocket of acetyl-CoA synthetase. Biochemistry 2006, 45, 11482–11490. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, H.K.; Derbyshire, E.R. Investigating the role of class I adenylate-forming enzymes in natural product biosynthesis. ACS Chem. Biol. 2020, 15, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Mouterde, L.M.M.; Stewart, J.D. Application of acetyl-CoA synthetase from Methanothermobacter thermautotrophicus to non-native substrates. Enzym. Microb. Technol. 2019, 128, 67–71. [Google Scholar] [CrossRef]
- Sofeo, N.; Hart, J.H.; Butler, B.; Oliver, D.J.; Yandeau-Nelson, M.D.; Nikolau, B.J. Altering the substrate specificity of acetyl-CoA synthetase by rational mutagenesis of the carboxylate binding pocket. ACS Synth. Biol. 2019, 8, 1325–1336. [Google Scholar] [CrossRef] [Green Version]
- Ingram-Smith, C.; Thurman, J.L., Jr.; Zimowski, K.; Smith, K.S. Role of motif III in catalysis by acetyl-CoA synthetase. Archaea 2012, 2012, 509579. [Google Scholar] [CrossRef] [Green Version]
- Gallego-Jara, J.; Terol, G.L.; Ecija Conesa, A.; Zambelli, B.; Canovas Diaz, M.; de Diego Puente, T. Characterization of acetyl-CoA synthetase kinetics and ATP-binding. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1040–1049. [Google Scholar] [CrossRef]
- Chang, K.H.; Xiang, H.; Dunaway-Mariano, D. Acyl-adenylate motif of the acyl-adenylate/thioester-forming enzyme superfamily: A site-directed mutagenesis study with the Pseudomonas sp. strain CBS3 4-chlorobenzoate:coenzyme A ligase. Biochemistry 1997, 36, 15650–15659. [Google Scholar] [CrossRef]
- Chang, K.H.; Liang, P.H.; Beck, W.; Scholten, J.D.; Dunaway-Mariano, D. Isolation and characterization of the three polypeptide components of 4-chlorobenzoate dehalogenase from Pseudomonas sp. strain CBS-3. Biochemistry 1992, 31, 5605–5610. [Google Scholar] [CrossRef]
- Gulick, A.M.; Lu, X.; Dunaway-Mariano, D. Crystal structure of 4-chlorobenzoate:CoA ligase/synthetase in the unliganded and aryl substrate-bound states. Biochemistry 2004, 43, 8670–8679. [Google Scholar] [CrossRef]
- Reger, A.S.; Wu, R.; Dunaway-Mariano, D.; Gulick, A.M. Structural characterization of a 140 degrees domain movement in the two-step reaction catalyzed by 4-chlorobenzoate:CoA ligase. Biochemistry 2008, 47, 8016–8025. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Cao, J.; Lu, X.; Reger, A.S.; Gulick, A.M.; Dunaway-Mariano, D. Mechanism of 4-chlorobenzoate:coenzyme a ligase catalysis. Biochemistry 2008, 47, 8026–8039. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Reger, A.S.; Lu, X.; Gulick, A.M.; Dunaway-Mariano, D. The mechanism of domain alternation in the acyl-adenylate forming ligase superfamily member 4-chlorobenzoate: Coenzyme A ligase. Biochemistry 2009, 48, 4115–4125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Lipmann, F.; Tuttle, L.C. A specific micromethod for determination of acyl phosphates. J. Biol. Chem. 1945, 159, 21–28. [Google Scholar] [CrossRef]
- Rose, I.A.; Grunberg-Manago, M.; Korey, S.F.; Ochoa, S. Enzymatic phosphorylation of acetate. J. Biol. Chem. 1954, 211, 737–756. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Na, K.B.; Koo, H.M.; Kim, Y.S. Identification of active site residues in Bradyrhizobium japonicum acetyl-CoA synthetase. J. Biochem. 2001, 130, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Zahn, M.; Kurteva-Yaneva, N.; Schuster, J.; Krug, U.; Georgi, T.; Muller, R.H.; Rohwerder, T.; Strater, N. Structures of 2-hydroxyisobutyric acid-CoA ligase reveal determinants of substrate specificity and describe a multi-conformational catalytic cycle. J. Mol. Biol. 2019, 431, 2747–2761. [Google Scholar] [CrossRef]
- Du, J.; Wang, X.; Nie, Q.; Yang, J.; Yao, X. Computational study of the binding mechanism of medium chain acyl-CoA synthetase with substrate in Methanosarcina acetivorans. J. Biotechnol. 2017, 259, 160–167. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Km CoA (mM) | kcat (sec−1) | kcat/Km (sec−1 mM−1) |
|---|---|---|---|
| Wild-type a | 0.19 ± 0.003 | 81.6 ± 0.7 | 423.7 ± 4.7 |
| Arg193Ala | Unsaturable b | ||
| Arg193Lys | 3.6 ± 0.37 | 0.51 ± 0.01 | 0.14 ± 0.01 |
| Arg193Gln | 7.8 ± 0.40 | 0.28 ± 0.004 | 0.036 ± 0.002 |
| Arg528Ala | Unsaturable b | ||
| Arg528Lys | 0.45 ± 0.03 | 0.25 ± 0.01 | 0.54 ± 0.02 |
| Arg528Gln | 1.67 ± 0.05 | 0.63 ± 0.04 | 0.38 ± 0.06 |
| Arg586Ala | 1.19 ± 0.08 | 2.13 ± 0.09 | 1.80 ± 0.06 |
| Arg586Lys | 0.33 ± 0.013 | 2.39 ± 0.016 | 7.34 ± 0.24 |
| Arg586Gln | 0.28 ± 0.009 | 0.12 ± 0.003 | 0.45 ± 0.028 |
| Enzyme | Substrate | Km (mM) | kcat (sec−1) | kcat/Km (sec−1 mM−1) | (kcat/Km CoA)/ (kcat/Km deCoA) |
|---|---|---|---|---|---|
| Wild-type | CoA | 0.19 ± 0.003 | 81.6 ± 0.7 | 423.7 ± 4.7 | 26.5 |
| deCoA | 2.16 ± 0.50 | 34.5 ± 2.9 | 16.0 ± 2.5 | ||
| Arg586Ala | CoA | 1.19 ± 0.08 | 2.13 ± 0.09 | 1.80 ± 0.06 | 2.2 |
| deCoA | 2.05 ± 0.40 | 1.66 ± 0.11 | 0.81 ± 0.11 | ||
| Arg586Lys | CoA | 0.33 ± 0.01 | 2.39 ± 0.02 | 7.34 ± 0.20 | 3.7 |
| deCoA | 0.86 ± 0.04 | 1.72 ± 0.01 | 2.01 ± 0.09 |
| Enzyme | kcat (sec−1) | Km CoA (mM) |
|---|---|---|
| Wild-type | 2.15 ± 0.10 | 4.09 ± 0.45 |
| Gly459Ala | 0.41 ± 0.01 | 2.09 ± 0.06 |
| Lys461Ala | 0.55 ± 0.03 | 2.19 ± 0.18 |
| Lys461Arg | 0.46 ± 0.10 | 1.38 ± 0.14 |
| Lys519Ala | * | * |
| Lys519Arg | 1.07 ± 0.02 | 7.18 ± 0.29 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, Y.; Ingram-Smith, C.; Ahmed, O.; Smith, K. The Roles of Coenzyme A Binding Pocket Residues in Short and Medium Chain Acyl-CoA Synthetases. Life 2023, 13, 1643. https://doi.org/10.3390/life13081643
Meng Y, Ingram-Smith C, Ahmed O, Smith K. The Roles of Coenzyme A Binding Pocket Residues in Short and Medium Chain Acyl-CoA Synthetases. Life. 2023; 13(8):1643. https://doi.org/10.3390/life13081643
Chicago/Turabian StyleMeng, Yu, Cheryl Ingram-Smith, Oly Ahmed, and Kerry Smith. 2023. "The Roles of Coenzyme A Binding Pocket Residues in Short and Medium Chain Acyl-CoA Synthetases" Life 13, no. 8: 1643. https://doi.org/10.3390/life13081643
APA StyleMeng, Y., Ingram-Smith, C., Ahmed, O., & Smith, K. (2023). The Roles of Coenzyme A Binding Pocket Residues in Short and Medium Chain Acyl-CoA Synthetases. Life, 13(8), 1643. https://doi.org/10.3390/life13081643