
The Effects of Artificial Sweeteners on Intestinal Nutrient-Sensing Receptors: Dr. Jekyll or Mr. Hyde?

 , ,
, ,
Abstract
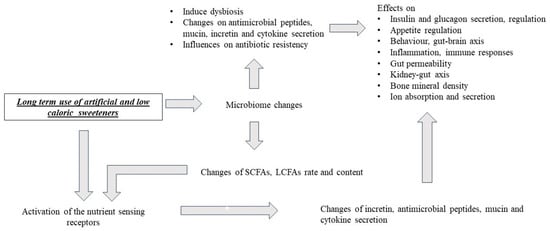
:
1. Introduction
2. Materials and Methods
3. The Role of Nutrient-Sensing Receptors outside and inside the Gastrointestinal System
3.1. Sweet and Bitter Taste Receptors (T1Rs and T2Rs)
3.2. Amino Acid-Sensing Receptors: Calcium-Sensing Receptor (CaSR), GPRC6A and GPR92
3.3. Lipid-Sensing Receptors: Free Fatty Acid Receptors 1–4 (FFAR1–4)
4. Effects of Artificial and Low-Calorie Sweeteners on Nutrient-Sensing Receptors, Gut Microbiota and Metabolism
4.1. Saccharin
4.2. Sucralose
4.3. Acesulfame Potassium
4.4. Aspartame
4.5. Other Studies with Artificial Sweeteners (Cohort, Clinical and Animal)
4.6. Steviol Glycosides and Sugar Alcohols
5. Discussion
6. Future Directions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Scheithauer, T.P.M.; Rampanelli, E.; Nieuwdorp, M.; Vallance, B.A.; Verchere, C.B.; van Raalte, D.H.; Herrema, H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front. Immunol. 2020, 11, 571731. [Google Scholar] [CrossRef]
- Shon, W.J.; Jung, M.H.; Kim, Y.; Kang, G.H.; Choi, E.Y.; Shin, D.M. Sugar-sweetened beverages exacerbate high-fat diet-induced inflammatory bowel disease by altering the gut microbiome. J. Nutr. Biochem. 2023, 113, 109254. [Google Scholar] [CrossRef] [PubMed]
- Sylvetsky, A.C.; Welsh, J.A.; Brown, R.J.; Vos, M.B. Low-calorie sweetener consumption is increasing in the United States. Am. J. Clin. Nutr. 2012, 96, 640–646. [Google Scholar] [CrossRef]
- Sylvetsky, A.C. Trends in the consumption of low-calorie sweeteners. Physiol. Behav. 2016, 164, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ojeda, F.J.; Plaza-Diaz, J.; Saez-Lara, M.J.; Gil, A. Effects of Sweeteners on the Gut Microbiota: A Review of Experimental Studies and Clinical Trials. Adv. Nutr. 2019, 10, S31–S48. [Google Scholar] [CrossRef]
- Qin, X. Etiology of inflammatory bowel disease: A unified hypothesis. World J. Gastroenterol. 2012, 18, 1708–1722. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.; Veysey, M.; Keely, S.; Scarlett, C.J.; Lucock, M.; Beckett, E.L. Intense Sweeteners, Taste Receptors and the Gut Microbiome: A Metabolic Health Perspective. Int. J. Environ. Res. Public Health 2020, 17, 4094. [Google Scholar] [CrossRef] [PubMed]
- Li, X. T1R receptors mediate mammalian sweet and umami taste. Am. J. Clin. Nutr. 2009, 90, 733S–737S. [Google Scholar] [CrossRef]
- Hoon, M.A.; Adler, E.; Lindemeier, J.; Battey, J.F.; Ryba, N.J.; Zuker, C.S. Putative mammalian taste receptors: A class of taste-specific GPCRs with distinct topographic selectivity. Cell 1999, 96, 541–551. [Google Scholar] [CrossRef]
- Rozengurt, E. Taste receptors in the gastrointestinal tract. I. Bitter taste receptors and alpha-gustducin in the mammalian gut. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G171–G177. [Google Scholar] [CrossRef]
- Crowe, M.S.; Wang, H.; Blakeney, B.A.; Mahavadi, S.; Singh, K.; Murthy, K.S.; Grider, J.R. Expression and function of umami receptors T1R1/T1R3 in gastric smooth muscle. Neurogastroenterol. Motil. 2020, 32, e13737. [Google Scholar] [CrossRef]
- Dias, A.G.; Eny, K.M.; Cockburn, M.; Chiu, W.; Nielsen, D.E.; Duizer, L.; El-Sohemy, A. Variation in the TAS1R2 Gene, Sweet Taste Perception and Intake of Sugars. J. Nutr. Nutr. 2015, 8, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Dinehart, M.E.; Hayes, J.E.; Bartoshuk, L.M.; Lanier, S.L.; Duffy, V.B. Bitter taste markers explain variability in vegetable sweetness, bitterness, and intake. Physiol. Behav. 2006, 87, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Behrens, M.; Lang, T. Extra-Oral Taste Receptors-Function, Disease, and Perspectives. Front. Nutr. 2022, 9, 881177. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Kim, H.K.; Seo, D.W.; Ki, S.Y.; Park, S.; Choi, S.H.; Kim, D.H.; Moon, S.J.; Jeong, Y.T. Whole-Brain Mapping of the Expression Pattern of T1R2, a Subunit Specific to the Sweet Taste Receptor. Front. Neuroanat. 2021, 15, 751839. [Google Scholar] [CrossRef] [PubMed]
- Kohno, D.; Koike, M.; Ninomiya, Y.; Kojima, I.; Kitamura, T.; Yada, T. Sweet Taste Receptor Serves to Activate Glucose- and Leptin-Responsive Neurons in the Hypothalamic Arcuate Nucleus and Participates in Glucose Responsiveness. Front. Neurosci. 2016, 10, 502. [Google Scholar] [CrossRef] [PubMed]
- Wolfle, U.; Elsholz, F.A.; Kersten, A.; Haarhaus, B.; Schumacher, U.; Schempp, C.M. Expression and Functional Activity of the Human Bitter Taste Receptor TAS2R38 in Human Placental Tissues and JEG-3 Cells. Molecules 2016, 21, 306. [Google Scholar] [CrossRef] [PubMed]
- Taher, S.; Borja, Y.; Cabanela, L.; Costers, V.J.; Carson-Marino, M.; Bailes, J.C.; Dhar, B.; Beckworth, M.T.; Rabaglino, M.B.; Post Uiterweer, E.D.; et al. Cholecystokinin, gastrin, cholecystokinin/gastrin receptors, and bitter taste receptor TAS2R14, trophoblast expression and signaling. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 316, R628–R639. [Google Scholar] [CrossRef]
- Chisini, L.A.; Cademartori, M.G.; Conde, M.C.M.; Costa, F.D.S.; Salvi, L.C.; Tovo-Rodrigues, L.; Correa, M.B. Single nucleotide polymorphisms of taste genes and caries: A systematic review and meta-analysis. Acta Odontol. Scand. 2021, 79, 147–155. [Google Scholar] [CrossRef]
- De Jesus, V.C. Association of Bitter Taste Receptor T2R38 Polymorphisms, Oral Microbiota, and Rheumatoid Arthritis. Curr. Issues Mol. Biol. 2021, 43, 1460–1472. [Google Scholar] [CrossRef]
- Schneider, C. Tuft cell integration of luminal states and interaction modules in tissues. Pflug. Arch. 2021, 473, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.J.; Kofonow, J.M.; Rosen, P.L.; Siebert, A.P.; Chen, B.; Doghramji, L.; Xiong, G.; Adappa, N.D.; Palmer, J.N.; Kennedy, D.W.; et al. Bitter and sweet taste receptors regulate human upper respiratory innate immunity. J. Clin. Investig. 2014, 124, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.J.; Cohen, N.A. Taste receptors in innate immunity. Cell. Mol. Life Sci. 2015, 72, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, M. Bitter taste receptor T2R38 is expressed on skin-infiltrating lymphocytes and regulates lymphocyte migration. Sci. Rep. 2022, 12, 11790. [Google Scholar] [CrossRef] [PubMed]
- Malki, A.; Fiedler, J.; Fricke, K.; Ballweg, I.; Pfaffl, M.W.; Krautwurst, D. Class I odorant receptors, TAS1R and TAS2R taste receptors, are markers for subpopulations of circulating leukocytes. J. Leukoc. Biol. 2015, 97, 533–545. [Google Scholar] [CrossRef]
- Gaida, M.M.; Dapunt, U.; Hansch, G.M. Sensing developing biofilms: The bitter receptor T2R38 on myeloid cells. Pathog. Dis. 2016, 74, ftw004. [Google Scholar] [CrossRef] [PubMed]
- Gopallawa, I.; Freund, J.R.; Lee, R.J. Bitter taste receptors stimulate phagocytosis in human macrophages through calcium, nitric oxide, and cyclic-GMP signaling. Cell. Mol. Life Sci. 2021, 78, 271–286. [Google Scholar] [CrossRef]
- Sharma, P.; Yi, R.; Nayak, A.P.; Wang, N.; Tang, F.; Knight, M.J.; Pan, S.; Oliver, B.; Deshpande, D.A. Bitter Taste Receptor Agonists Mitigate Features of Allergic Asthma in Mice. Sci. Rep. 2017, 7, 46166. [Google Scholar] [CrossRef]
- Ekoff, M.; Choi, J.H.; James, A.; Dahlen, B.; Nilsson, G.; Dahlen, S.E. Bitter taste receptor (TAS2R) agonists inhibit IgE-dependent mast cell activation. J. Allergy Clin. Immunol. 2014, 134, 475–478. [Google Scholar] [CrossRef]
- Zhou, L.; Huang, W.; Xu, Y.; Gao, C.; Zhang, T.; Guo, M.; Liu, Y.; Ding, J.; Qin, L.; Xu, Z.; et al. Sweet Taste Receptors Mediated ROS-NLRP3 Inflammasome Signaling Activation: Implications for Diabetic Nephropathy. J. Diabetes Res. 2018, 2018, 7078214. [Google Scholar] [CrossRef]
- Depoortere, I. Taste receptors of the gut: Emerging roles in health and disease. Gut 2014, 63, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Dotson, C.D.; Zhang, L.; Xu, H.; Shin, Y.K.; Vigues, S.; Ott, S.H.; Elson, A.E.; Choi, H.J.; Shaw, H.; Egan, J.M.; et al. Bitter taste receptors influence glucose homeostasis. PLoS ONE 2008, 3, e3974. [Google Scholar] [CrossRef]
- Young, R.L.; Chia, B.; Isaacs, N.J.; Ma, J.; Khoo, J.; Wu, T.; Horowitz, M.; Rayner, C.K. Disordered control of intestinal sweet taste receptor expression and glucose absorption in type 2 diabetes. Diabetes 2013, 62, 3532–3541. [Google Scholar] [CrossRef] [PubMed]
- Worthington, J.J.; Reimann, F.; Gribble, F.M. Enteroendocrine cells-sensory sentinels of the intestinal environment and orchestrators of mucosal immunity. Mucosal Immunol. 2018, 11, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Cekic, C.; Arabul, M.; Alper, E.; Pakoz, Z.B.; Saritas, E.; Yuksel Unsal, B. Evaluation of the relationship between serum ghrelin, C-reactive protein and interleukin-6 levels, and disease activity in inflammatory bowel diseases. Hepatogastroenterology 2014, 61, 1196–1200. [Google Scholar] [PubMed]
- Bendet, N.; Scapa, E.; Cohen, O.; Bloch, O.; Aharoni, D.; Ramot, Y.; Weiss, M.; Halevi, A.; Rapoport, M.J. Enhanced glucose-dependent glucagon-like peptide-1 and insulin secretion in Crohn patients with terminal ileum disease is unrelated to disease activity or ileal resection. Scand. J. Gastroenterol. 2004, 39, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Ates, Y.; Degertekin, B.; Erdil, A.; Yaman, H.; Dagalp, K. Serum ghrelin levels in inflammatory bowel disease with relation to disease activity and nutritional status. Dig. Dis. Sci. 2008, 53, 2215–2221. [Google Scholar] [CrossRef]
- Kreuch, D.; Keating, D.J.; Wu, T.; Horowitz, M.; Rayner, C.K.; Young, R.L. Gut Mechanisms Linking Intestinal Sweet Sensing to Glycemic Control. Front. Endocrinol. 2018, 9, 741. [Google Scholar] [CrossRef]
- Duan, L.; Rao, X.; Braunstein, Z.; Toomey, A.C.; Zhong, J. Role of Incretin Axis in Inflammatory Bowel Disease. Front. Immunol. 2017, 8, 1734. [Google Scholar] [CrossRef]
- Howitt, M.R.; Lavoie, S.; Michaud, M.; Blum, A.M.; Tran, S.V.; Weinstock, J.V.; Gallini, C.A.; Redding, K.; Margolskee, R.F.; Osborne, L.C.; et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 2016, 351, 1329–1333. [Google Scholar] [CrossRef]
- Howitt, M.R.; Cao, Y.G.; Gologorsky, M.B.; Li, J.A.; Haber, A.L.; Biton, M.; Lang, J.; Michaud, M.; Regev, A.; Garrett, W.S. The Taste Receptor TAS1R3 Regulates Small Intestinal Tuft Cell Homeostasis. Immunohorizons 2020, 4, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Strine, M.S.; Wilen, C.B. Tuft cells are key mediators of interkingdom interactions at mucosal barrier surfaces. PLoS Pathog. 2022, 18, e1010318. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Herring, C.A.; Chen, B.; Kim, H.; Simmons, A.J.; Southard-Smith, A.N.; Allaman, M.M.; White, J.R.; Macedonia, M.C.; McKinley, E.T.; et al. Succinate Produced by Intestinal Microbes Promotes Specification of Tuft Cells to Suppress Ileal Inflammation. Gastroenterology 2020, 159, 2101–2115.e2105. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, S.; Jensen, T.S.R.; Feddersen, U.R.; Bindslev, N.; Grunddal, K.V.; Poulsen, S.S.; Rasmussen, H.B.; Budtz-Jorgensen, E.; Berner-Hansen, M. Decreased number of colonic tuft cells in quiescent ulcerative colitis patients. Eur. J. Gastroenterol. Hepatol. 2021, 33, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Wehkamp, J.; Stange, E.F. An Update Review on the Paneth Cell as Key to Ileal Crohn’s Disease. Front. Immunol. 2020, 11, 646. [Google Scholar] [CrossRef]
- Liszt, K.I.; Wang, Q.; Farhadipour, M.; Segers, A.; Thijs, T.; Nys, L.; Deleus, E.; Van der Schueren, B.; Gerner, C.; Neuditschko, B.; et al. Human intestinal bitter taste receptors regulate innate immune responses and metabolic regulators in obesity. J. Clin. Investig. 2022, 132, e144828. [Google Scholar] [CrossRef]
- Gersemann, M.; Becker, S.; Kubler, I.; Koslowski, M.; Wang, G.; Herrlinger, K.R.; Griger, J.; Fritz, P.; Fellermann, K.; Schwab, M.; et al. Differences in goblet cell differentiation between Crohn’s disease and ulcerative colitis. Differentiation 2009, 77, 84–94. [Google Scholar] [CrossRef]
- Singh, V.; Johnson, K.; Yin, J.; Lee, S.; Lin, R.; Yu, H.; In, J.; Foulke-Abel, J.; Zachos, N.C.; Donowitz, M.; et al. Chronic Inflammation in Ulcerative Colitis Causes Long-Term Changes in Goblet Cell Function. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 219–232. [Google Scholar] [CrossRef]
- Park, J.H.; Song, D.K. Sweet taste receptors as a tool for an amplifying pathway of glucose-stimulated insulin secretion in pancreatic beta cells. Pflug. Arch. 2019, 471, 655–657. [Google Scholar] [CrossRef]
- Sanchez-Andres, J.V.; Malaisse, W.J.; Kojima, I. Electrophysiology of the pancreatic islet beta-cell sweet taste receptor TIR3. Pflug. Arch. 2019, 471, 647–654. [Google Scholar] [CrossRef]
- Murovets, V.O.; Bachmanov, A.A.; Zolotarev, V.A. Impaired Glucose Metabolism in Mice Lacking the Tas1r3 Taste Receptor Gene. PLoS ONE 2015, 10, e0130997. [Google Scholar] [CrossRef] [PubMed]
- Murovets, V.O.; Sozontov, E.A.; Zachepilo, T.G. The Effect of the Taste Receptor Protein T1R3 on the Development of Islet Tissue of the Murine Pancreas. Dokl. Biol. Sci. 2019, 484, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Eaton, M.S.; Weinstein, N.; Newby, J.B.; Plattes, M.M.; Foster, H.E.; Arthur, J.W.; Ward, T.D.; Shively, S.R.; Shor, R.; Nathan, J.; et al. Loss of the nutrient sensor TAS1R3 leads to reduced bone resorption. J. Physiol. Biochem. 2018, 74, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Chanpaisaeng, K.; Teerapornpuntakit, J.; Wongdee, K.; Charoenphandhu, N. Emerging roles of calcium-sensing receptor in the local regulation of intestinal transport of ions and calcium. Am. J. Physiol. Cell Physiol. 2021, 320, C270–C278. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Elajnaf, T.; Iamartino, L.; Mesteri, I.; Muller, C.; Bassetto, M.; Manhardt, T.; Baumgartner-Parzer, S.; Kallay, E.; Schepelmann, M. Nutritional and Pharmacological Targeting of the Calcium-Sensing Receptor Influences Chemically Induced Colitis in Mice. Nutrients 2019, 11, 3072. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kovacs-Nolan, J.; Kodera, T.; Eto, Y.; Mine, Y. Gamma-Glutamyl cysteine and gamma-glutamyl valine inhibit TNF-alpha signaling in intestinal epithelial cells and reduce inflammation in a mouse model of colitis via allosteric activation of the calcium-sensing receptor. Biochim. Biophys. Acta 2015, 1852, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Iamartino, L.; Elajnaf, T.; Kallay, E.; Schepelmann, M. Calcium-sensing receptor in colorectal inflammation and cancer: Current insights and future perspectives. World J. Gastroenterol. 2018, 24, 4119–4131. [Google Scholar] [CrossRef]
- Clemmensen, C.; Smajilovic, S.; Madsen, A.N.; Klein, A.B.; Holst, B.; Brauner-Osborne, H. Increased susceptibility to diet-induced obesity in GPRC6A receptor knockout mice. J. Endocrinol. 2013, 217, 151–160. [Google Scholar] [CrossRef]
- Paccou, J.; Boudot, C.; Renard, C.; Liabeuf, S.; Kamel, S.; Fardellone, P.; Massy, Z.; Brazier, M.; Mentaverri, R. Total calcium-sensing receptor expression in circulating monocytes is increased in rheumatoid arthritis patients with severe coronary artery calcification. Arthritis Res. Ther. 2014, 16, 412. [Google Scholar] [CrossRef]
- Hou, Q.; Huang, J.; Xiong, X.; Guo, Y.; Zhang, B. Role of Nutrient-sensing Receptor GPRC6A in Regulating Colonic Group 3 Innate Lymphoid Cells and Inflamed Mucosal Healing. J. Crohns Colitis 2022, 16, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Rettenberger, A.T.; Schulze, W.; Breer, H.; Haid, D. Analysis of the protein related receptor GPR92 in G-cells. Front. Physiol. 2015, 6, 261. [Google Scholar] [CrossRef] [PubMed]
- Schlatterer, K.; Peschel, A.; Kretschmer, D. Short-Chain Fatty Acid and FFAR2 Activation—A New Option for Treating Infections? Front. Cell. Infect. Microbiol. 2021, 11, 785833. [Google Scholar] [CrossRef] [PubMed]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef]
- Takakuwa, A.; Nakamura, K.; Kikuchi, M.; Sugimoto, R.; Ohira, S.; Yokoi, Y.; Ayabe, T. Butyric Acid and Leucine Induce alpha-Defensin Secretion from Small Intestinal Paneth Cells. Nutrients 2019, 11, 2817. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, M.; Binienda, A.; Fichna, J. The role of fatty acids in Crohn’s disease pathophysiology—An overview. Mol. Cell. Endocrinol. 2021, 538, 111448. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, A.; Tsumaya, K.; Awaji, T.; Katsuma, S.; Adachi, T.; Yamada, M.; Sugimoto, Y.; Miyazaki, S.; Tsujimoto, G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 2005, 11, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, L.; Myhre, S.; Sundqvist, M.; Ahnmark, A.; McCoull, W.; Raubo, P.; Groombridge, S.D.; Polla, M.; Nystrom, A.C.; Kristensson, L.; et al. The acute glucose lowering effect of specific GPR120 activation in mice is mainly driven by glucagon-like peptide 1. PLoS ONE 2017, 12, e0189060. [Google Scholar] [CrossRef]
- Paulsen, S.J.; Larsen, L.K.; Hansen, G.; Chelur, S.; Larsen, P.J.; Vrang, N. Expression of the fatty acid receptor GPR120 in the gut of diet-induced-obese rats and its role in GLP-1 secretion. PLoS ONE 2014, 9, e88227. [Google Scholar] [CrossRef]
- Paschoal, V.A.; Walenta, E.; Talukdar, S.; Pessentheiner, A.R.; Osborn, O.; Hah, N.; Chi, T.J.; Tye, G.L.; Armando, A.M.; Evans, R.M.; et al. Positive Reinforcing Mechanisms between GPR120 and PPARgamma Modulate Insulin Sensitivity. Cell Metab. 2020, 31, 1173–1188.e1175. [Google Scholar] [CrossRef]
- Paschoal, V.A.; Oh, D.Y. Revisiting PPARgamma as a new friend of GPR120 in the treatment of metabolic disorders. Adipocyte 2020, 9, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhou, J.; Meng, Y.; Shi, N.; Wang, X.; Zhou, M.; Li, G.; Yang, Y. DHA Sensor GPR120 in Host Defense Exhibits the Dual Characteristics of Regulating Dendritic Cell Function and Skewing the Balance of Th17/Tregs. Int. J. Biol. Sci. 2020, 16, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Rubbino, F.; Garlatti, V.; Garzarelli, V.; Massimino, L.; Spano, S.; Iadarola, P.; Cagnone, M.; Giera, M.; Heijink, M.; Guglielmetti, S.; et al. GPR120 prevents colorectal adenocarcinoma progression by sustaining the mucosal barrier integrity. Sci. Rep. 2022, 12, 381. [Google Scholar] [CrossRef] [PubMed]
- Magnuson, B.A.; Carakostas, M.C.; Moore, N.H.; Poulos, S.P.; Renwick, A.G. Biological fate of low-calorie sweeteners. Nutr. Rev. 2016, 74, 670–689. [Google Scholar] [CrossRef] [PubMed]
- Basson, A.R.; Rodriguez-Palacios, A.; Cominelli, F. Artificial Sweeteners: History and New Concepts on Inflammation. Front. Nutr. 2021, 8, 746247. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, C.; Bufe, B.; Winnig, M.; Hofmann, T.; Frank, O.; Behrens, M.; Lewtschenko, T.; Slack, J.P.; Ward, C.D.; Meyerhof, W. Bitter taste receptors for saccharin and acesulfame K. J. Neurosci. 2004, 24, 10260–10265. [Google Scholar] [CrossRef] [PubMed]
- Serrano, J.; Meshram, N.N.; Soundarapandian, M.M.; Smith, K.R.; Mason, C.; Brown, I.S.; Tyrberg, B.; Kyriazis, G.A. Saccharin Stimulates Insulin Secretion Dependent on Sweet Taste Receptor-Induced Activation of PLC Signaling Axis. Biomedicines 2022, 10, 120. [Google Scholar] [CrossRef] [PubMed]
- Sunderhauf, A.; Pagel, R.; Kunstner, A.; Wagner, A.E.; Rupp, J.; Ibrahim, S.M.; Derer, S.; Sina, C. Saccharin Supplementation Inhibits Bacterial Growth and Reduces Experimental Colitis in Mice. Nutrients 2020, 12, 1122. [Google Scholar] [CrossRef]
- Suez, J.; Korem, T.; Zeevi, D.; Zilberman-Schapira, G.; Thaiss, C.A.; Maza, O.; Israeli, D.; Zmora, N.; Gilad, S.; Weinberger, A.; et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 2014, 514, 181–186. [Google Scholar] [CrossRef]
- Serrano, J.; Smith, K.R.; Crouch, A.L.; Sharma, V.; Yi, F.; Vargova, V.; LaMoia, T.E.; Dupont, L.M.; Serna, V.; Tang, F.; et al. High-dose saccharin supplementation does not induce gut microbiota changes or glucose intolerance in healthy humans and mice. Microbiome 2021, 9, 11. [Google Scholar] [CrossRef]
- Schiffman, S.S.; Scholl, E.H.; Furey, T.S.; Nagle, H.T. Toxicological and pharmacokinetic properties of sucralose-6-acetate and its parent sucralose: In vitro screening assays. J. Toxicol. Environ. Health B Crit. Rev. 2023, 6, 307–341. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Palacios, A.; Harding, A.; Menghini, P.; Himmelman, C.; Retuerto, M.; Nickerson, K.P.; Lam, M.; Croniger, C.M.; McLean, M.H.; Durum, S.K.; et al. The Artificial Sweetener Splenda Promotes Gut Proteobacteria, Dysbiosis, and Myeloperoxidase Reactivity in Crohn’s Disease-Like Ileitis. Inflamm. Bowel Dis. 2018, 24, 1005–1020. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Wang, Y.; Li, X.; Liu, X.; Guo, M.; Tan, Y.; Qin, X.; Wang, X.; Jiang, M. Sucralose Promotes Colitis-Associated Colorectal Cancer Risk in a Murine Model Along With Changes in Microbiota. Front. Oncol. 2020, 10, 710. [Google Scholar] [CrossRef] [PubMed]
- Pino-Seguel, P.; Moya, O.; Borquez, J.C.; Pino-de la Fuente, F.; Diaz-Castro, F.; Donoso-Barraza, C.; Llanos, M.; Troncoso, R.; Bravo-Sagua, R. Sucralose consumption ameliorates high-fat diet-induced glucose intolerance and liver weight gain in mice. Front. Nutr. 2022, 9, 979624. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.T.; Lin, C.H.; Pai, H.L.; Chen, Y.C.; Cheng, K.P.; Kuo, H.Y.; Li, C.H.; Ou, H.Y. Sucralose, a Non-nutritive Artificial Sweetener Exacerbates High Fat Diet-Induced Hepatic Steatosis Through Taste Receptor Type 1 Member 3. Front. Nutr. 2022, 9, 823723. [Google Scholar] [CrossRef] [PubMed]
- Zani, F.; Blagih, J.; Gruber, T.; Buck, M.D.; Jones, N.; Hennequart, M.; Newell, C.L.; Pilley, S.E.; Soro-Barrio, P.; Kelly, G.; et al. The dietary sweetener sucralose is a negative modulator of T cell-mediated responses. Nature 2023, 615, 705–711. [Google Scholar] [CrossRef]
- Lizunkova, P.; Enuwosa, E.; Chichger, H. Activation of the sweet taste receptor T1R3 by sucralose attenuates VEGF-induced vasculogenesis in a cell model of the retinal microvascular endothelium. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 71–81. [Google Scholar] [CrossRef]
- Lertrit, A.; Srimachai, S.; Saetung, S.; Chanprasertyothin, S.; Chailurkit, L.O.; Areevut, C.; Katekao, P.; Ongphiphadhanakul, B.; Sriphrapradang, C. Effects of sucralose on insulin and glucagon-like peptide-1 secretion in healthy subjects: A randomized, double-blind, placebo-controlled trial. Nutrition 2018, 55–56, 125–130. [Google Scholar] [CrossRef]
- Mendez-Garcia, L.A.; Bueno-Hernandez, N.; Cid-Soto, M.A.; De Leon, K.L.; Mendoza-Martinez, V.M.; Espinosa-Flores, A.J.; Carrero-Aguirre, M.; Esquivel-Velazquez, M.; Leon-Hernandez, M.; Viurcos-Sanabria, R.; et al. Ten-Week Sucralose Consumption Induces Gut Dysbiosis and Altered Glucose and Insulin Levels in Healthy Young Adults. Microorganisms 2022, 10, 434. [Google Scholar] [CrossRef]
- Hanawa, Y.; Higashiyama, M.; Kurihara, C.; Tanemoto, R.; Ito, S.; Mizoguchi, A.; Nishii, S.; Wada, A.; Inaba, K.; Sugihara, N.; et al. Acesulfame potassium induces dysbiosis and intestinal injury with enhanced lymphocyte migration to intestinal mucosa. J. Gastroenterol. Hepatol. 2021, 36, 3140–3148. [Google Scholar] [CrossRef]
- Bian, X. The artificial sweetener acesulfame potassium affects the gut microbiome and body weight gain in CD-1 mice. PLoS ONE 2017, 12, e0178426. [Google Scholar] [CrossRef] [PubMed]
- Palmnas, M.S.; Cowan, T.E.; Bomhof, M.R.; Su, J.; Reimer, R.A.; Vogel, H.J.; Hittel, D.S.; Shearer, J. Low-dose aspartame consumption differentially affects gut microbiota-host metabolic interactions in the diet-induced obese rat. PLoS ONE 2014, 9, e109841. [Google Scholar] [CrossRef] [PubMed]
- Higgins, K.A.; Considine, R.V.; Mattes, R.D. Aspartame consumption for 12 weeks does not affect glycemia, appetite, or body weight of healthy, lean adults in a randomized controlled trial. J. Nutr. 2018, 148, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, K.; Pilarz, A.; Rogut, A.; Maj, P.; Szymanska, J.; Olejnik, L.; Szymanski, P. Aspartame-True or False? Narrative Review of Safety Analysis of General Use in Products. Nutrients 2021, 13, 1957. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.; Hornik, C.D.; Bilbo, S.; Holzknecht, Z.E.; Gentry, L.; Rao, R.; Lin, S.S.; Herbert, M.R.; Nevison, C.D. The role of oxidative stress, inflammation and acetaminophen exposure from birth to early childhood in the induction of autism. J. Int. Med. Res. 2017, 45, 407–438. [Google Scholar] [CrossRef] [PubMed]
- Landrigan, P.J.; Straif, K. Aspartame and cancer—new evidence for causation. Environ. Health 2021, 20, 42. [Google Scholar] [CrossRef] [PubMed]
- McCullough, M.L.; Teras, L.R.; Shah, R.; Diver, W.R.; Gaudet, M.M.; Gapstur, S.M. Artificially and sugar-sweetened carbonated beverage consumption is not associated with risk of lymphoid neoplasms in older men and women. J. Nutr. 2014, 144, 2041–2049. [Google Scholar] [CrossRef] [PubMed]
- Chia, C.W.; Shardell, M.; Gravenstein, K.S.; Carlson, O.D.; Simonsick, E.M.; Ferrucci, L.; Egan, J.M. Regular low-calorie sweetener consumption is associated with increased secretion of glucose-dependent insulinotropic polypeptide. Diabetes Obes. Metab. 2018, 20, 2282–2285. [Google Scholar] [CrossRef]
- Shil, A.; Olusanya, O.; Ghufoor, Z.; Forson, B.; Marks, J.; Chichger, H. Artificial Sweeteners Disrupt Tight Junctions and Barrier Function in the Intestinal Epithelium through Activation of the Sweet Taste Receptor, T1R3. Nutrients 2020, 12, 1862. [Google Scholar] [CrossRef]
- Shil, A.; Chichger, H. Artificial Sweeteners Negatively Regulate Pathogenic Characteristics of Two Model Gut Bacteria, E. coli and E. faecalis. Int. J. Mol. Sci. 2021, 22, 5228. [Google Scholar] [CrossRef]
- Yu, Z.; Wang, Y.; Lu, J.; Bond, P.L.; Guo, J. Nonnutritive sweeteners can promote the dissemination of antibiotic resistance through conjugative gene transfer. ISME J. 2021, 15, 2117–2130. [Google Scholar] [CrossRef] [PubMed]
- De Dios, R.; Proctor, C.R.; Maslova, E.; Dzalbe, S.; Rudolph, C.J.; McCarthy, R.R. Artificial sweeteners inhibit multidrug-resistant pathogen growth and potentiate antibiotic activity. EMBO Mol. Med. 2023, 15, e16397. [Google Scholar] [CrossRef] [PubMed]
- Tsan, L.; Chometton, S.; Hayes, A.M.; Klug, M.E.; Zuo, Y.; Sun, S.; Bridi, L.; Lan, R.; Fodor, A.A.; Noble, E.E.; et al. Early-life low-calorie sweetener consumption disrupts glucose regulation, sugar-motivated behavior, and memory function in rats. JCI Insight 2022, 7, e157714. [Google Scholar] [CrossRef] [PubMed]
- Debras, C.; Chazelas, E.; Srour, B.; Druesne-Pecollo, N.; Esseddik, Y.; Szabo de Edelenyi, F.; Agaesse, C.; De Sa, A.; Lutchia, R.; Gigandet, S.; et al. Artificial sweeteners and cancer risk: Results from the NutriNet-Sante population-based cohort study. PLoS Med. 2022, 19, e1003950. [Google Scholar] [CrossRef] [PubMed]
- Debras, C.; Deschasaux-Tanguy, M.; Chazelas, E.; Sellem, L.; Druesne-Pecollo, N.; Esseddik, Y.; Szabo de Edelenyi, F.; Agaesse, C.; De Sa, A.; Lutchia, R.; et al. Artificial Sweeteners and Risk of Type 2 Diabetes in the Prospective NutriNet-Sante Cohort. Diabetes Care 2023, 46, 1681–1690. [Google Scholar] [CrossRef]
- Debras, C.; Chazelas, E.; Sellem, L.; Porcher, R.; Druesne-Pecollo, N.; Esseddik, Y.; de Edelenyi, F.S.; Agaesse, C.; De Sa, A.; Lutchia, R.; et al. Artificial sweeteners and risk of cardiovascular diseases: Results from the prospective NutriNet-Sante cohort. BMJ 2022, 378, e071204. [Google Scholar] [CrossRef] [PubMed]
- Mossavar-Rahmani, Y.; Kamensky, V.; Manson, J.E.; Silver, B.; Rapp, S.R.; Haring, B.; Beresford, S.A.A.; Snetselaar, L.; Wassertheil-Smoller, S. Artificially Sweetened Beverages and Stroke, Coronary Heart Disease, and All-Cause Mortality in the Women’s Health Initiative. Stroke 2019, 50, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Del Pozo, S.; Gomez-Martinez, S.; Diaz, L.E.; Nova, E.; Urrialde, R.; Marcos, A. Potential Effects of Sucralose and Saccharin on Gut Microbiota: A Review. Nutrients 2022, 14, 1682. [Google Scholar] [CrossRef]
- Basson, A.R.; Katz, J.; Singh, S.; Celio, F.; Cominelli, F.; Rodriguez-Palacios, A. Sweets and Inflammatory Bowel Disease: Patients Favor Artificial Sweeteners and Diet Foods/Drinks Over Table Sugar and Consume Less Fruits/Vegetables. Inflamm. Bowel. Dis. 2023, 29, 1751–1759. [Google Scholar] [CrossRef]
- Ajami, M.; Seyfi, M.; Abdollah Pouri Hosseini, F.; Naseri, P.; Velayati, A.; Mahmoudnia, F.; Zahedirad, M.; Hajifaraji, M. Effects of stevia on glycemic and lipid profile of type 2 diabetic patients: A randomized controlled trial. Avicenna J. Phytomed. 2020, 10, 118–127. [Google Scholar]
- Anton, S.D.; Martin, C.K.; Han, H.; Coulon, S.; Cefalu, W.T.; Geiselman, P.; Williamson, D.A. Effects of stevia, aspartame, and sucrose on food intake, satiety, and postprandial glucose and insulin levels. Appetite 2010, 55, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Philippaert, K.; Pironet, A.; Mesuere, M.; Sones, W.; Vermeiren, L.; Kerselaers, S.; Pinto, S.; Segal, A.; Antoine, N.; Gysemans, C.; et al. Steviol glycosides enhance pancreatic beta-cell function and taste sensation by potentiation of TRPM5 channel activity. Nat. Commun. 2017, 8, 14733. [Google Scholar] [CrossRef] [PubMed]
- Samuel, P.; Ayoob, K.T.; Magnuson, B.A.; Wölwer-Rieck, U.; Jeppesen, P.B.; Rogers, P.J.; Rowland, I.; Mathew, R.S. Stevia leaf to stevia sweetener: Exploring its science, benefits, and future potential. J. Nutr. 2018, 147, 1186S–1205S. [Google Scholar] [CrossRef] [PubMed]
- Van der Wielen, N.; Ten Klooster, J.P.; Muckenschnabl, S.; Pieters, R.; Hendriks, H.F.; Witkamp, R.F.; Meijerink, J. The Noncaloric Sweetener Rebaudioside A Stimulates Glucagon-Like Peptide 1 Release and Increases Enteroendocrine Cell Numbers in 2-Dimensional Mouse Organoids Derived from Different Locations of the Intestine. J. Nutr. 2016, 146, 2429–2435. [Google Scholar] [CrossRef] [PubMed]
- Kasti, A.N.; Nikolaki, M.D.; Synodinou, K.D.; Katsas, K.N.; Petsis, K.; Lambrinou, S.; Pyrousis, I.A.; Triantafyllou, K. The Effects of Stevia Consumption on Gut Bacteria: Friend or Foe? Microorganisms 2022, 10, 744. [Google Scholar] [CrossRef] [PubMed]
- Wölnerhanssen, B.K.; Cajacob, L.; Keller, N.; Doody, A.; Rehfeld, J.F.; Drewe, J.; Peterli, R.; Beglinger, C.; Meyer-Gerspach, A.C. Gut hormone secretion, gastric emptying, and glycemic responses to erythritol and xylitol in lean and obese subjects. Am. J. Physiol.-Endocrinol. Metab. 2016, 310, E1053–E1061. [Google Scholar] [CrossRef]
- Teysseire, F.; Bordier, V.; Budzinska, A.; Weltens, N.; Rehfeld, J.F.; Holst, J.J.; Hartmann, B.; Beglinger, C.; Van Oudenhove, L.; Wolnerhanssen, B.K.; et al. The Role of D-allulose and Erythritol on the Activity of the Gut Sweet Taste Receptor and Gastrointestinal Satiation Hormone Release in Humans: A Randomized, Controlled Trial. J. Nutr. 2022, 152, 1228–1238. [Google Scholar] [CrossRef]
- Ortiz, S.R.; Field, M.S. Mammalian metabolism of erythritol: A predictive biomarker of metabolic dysfunction. Curr. Opin. Clin. Nutr. Metab. Care 2020, 23, 296–301. [Google Scholar] [CrossRef]
- Hootman, K.C.; Trezzi, J.P.; Kraemer, L.; Burwell, L.S.; Dong, X.; Guertin, K.A.; Jaeger, C.; Stover, P.J.; Hiller, K.; Cassano, P.A. Erythritol is a pentose-phosphate pathway metabolite and associated with adiposity gain in young adults. Proc. Natl. Acad. Sci. USA 2017, 114, E4233–E4240. [Google Scholar] [CrossRef]
- Mitsutomi, K.; Masaki, T.; Shimasaki, T.; Gotoh, K.; Chiba, S.; Kakuma, T.; Shibata, H. Effects of a nonnutritive sweetener on body adiposity and energy metabolism in mice with diet-induced obesity. Metabolism 2014, 63, 69–78. [Google Scholar] [CrossRef]
- Alamri, H.S.; Akiel, M.A.; Alghassab, T.S.; Alfhili, M.A.; Alrfaei, B.M.; Aljumaa, M.; Barhoumi, T. Erythritol modulates the polarization of macrophages: Potential role of tumor necrosis factor-alpha and Akt pathway. J. Food Biochem. 2022, 46, e13960. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Ye, K.; Li, M.; Ying, J.; Wang, H.; Han, J.; Shi, L.; Xiao, J.; Shen, Y.; Feng, X.; et al. Xylitol enhances synthesis of propionate in the colon via cross-feeding of gut microbiota. Microbiome 2021, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Salli, K.; Lehtinen, M.J.; Tiihonen, K.; Ouwehand, A.C. Xylitol’s Health Benefits beyond Dental Health: A Comprehensive Review. Nutrients 2019, 11, 1813. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Gerspach, A.C.; Drewe, J.; Verbeure, W.; Roux, C.W.L.; Dellatorre-Teixeira, L.; Rehfeld, J.F.; Holst, J.J.; Hartmann, B.; Tack, J.; Peterli, R.; et al. Effect of the Natural Sweetener Xylitol on Gut Hormone Secretion and Gastric Emptying in Humans: A Pilot Dose-Ranging Study. Nutrients 2021, 13, 174. [Google Scholar] [CrossRef] [PubMed]
- Mahalapbutr, P.; Lee, V.S.; Rungrotmongkol, T. Binding Hotspot and Activation Mechanism of Maltitol and Lactitol toward the Human Sweet Taste Receptor. J. Agric. Food Chem. 2020, 68, 7974–7983. [Google Scholar] [CrossRef]
- Shima, K.; Suda, T.; Nishimoto, K.; Yoshimoto, S. Relationship between molecular structures of sugars and their ability to stimulate the release of glucagon-like peptide-1 from canine ileal loops. Acta Endocrinol 1990, 123, 464–470. [Google Scholar] [CrossRef]
- Chukwuma, C.I.; Ibrahim, M.A.; Islam, M.S. Maltitol inhibits small intestinal glucose absorption and increases insulin mediated muscle glucose uptake ex vivo but not in normal and type 2 diabetic rats. Int. J. Food Sci. Nutr. 2017, 68, 73–81. [Google Scholar] [CrossRef]
- Barbalho, S.M.; Goulart, R.A.; Aranao, A.L.C.; de Oliveira, P.G.C. Inflammatory Bowel Diseases and Fermentable Oligosaccharides, Disaccharides, Monosaccharides, and Polyols: An Overview. J. Med. Food 2018, 21, 633–640. [Google Scholar] [CrossRef]
- Simoes, C.D.; Maganinho, M.; Sousa, A.S. FODMAPs, inflammatory bowel disease and gut microbiota: Updated overview on the current evidence. Eur. J. Nutr. 2022, 61, 1187–1198. [Google Scholar] [CrossRef]
- Shon, W.J.; Song, J.W.; Oh, S.H.; Lee, K.H.; Seong, H.; You, H.J.; Seong, J.K.; Shin, D.M. Gut taste receptor type 1 member 3 is an intrinsic regulator of Western diet-induced intestinal inflammation. BMC Med. 2023, 21, 165. [Google Scholar] [CrossRef]



{kind=link}
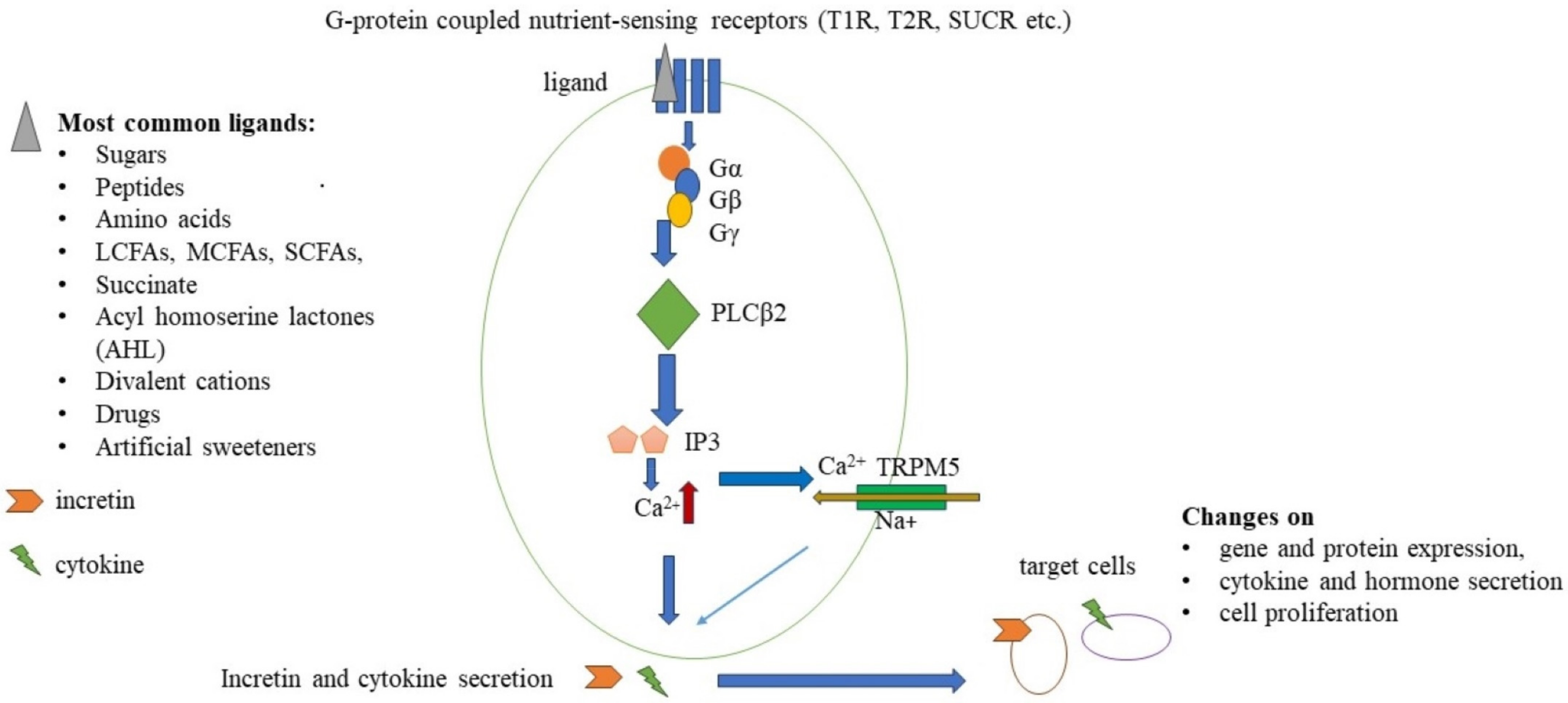{kind=link}
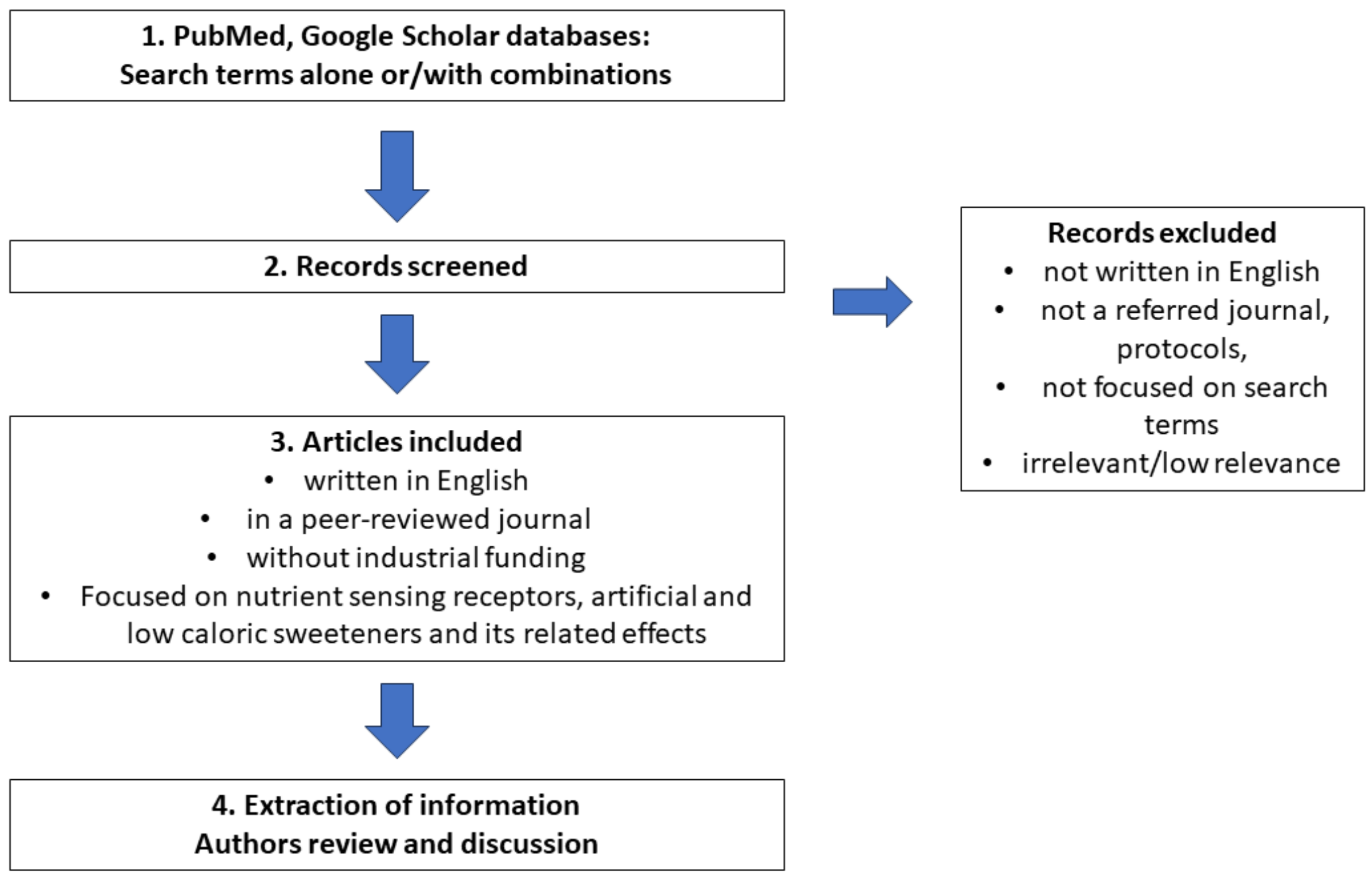{kind=link}
| Enteroendocrine Cell Type | Localization | Secreted Hormones | Taste Receptors | Serum Hormone Level Changes during IBD |
|---|---|---|---|---|
| X/A-cells | stomach | ghrelin, nesfatin1 | sugars, amino acids bitter LCFAs T1R3 T2R FFAR4 CaSR | elevated |
| G-cells | stomach | gastrin | amino acids, sugars peptides T1R3 bitter T2R CaSR GPRC6A GPR92 (LPAR5) | elevated |
| P-cells | stomach | leptin | N/A | |
| Enterochromaffin-like cells | stomach | histamine | N/A | |
| D-cells | stomach, small intestine | somatostatin | N/A | |
| I-cells | small intestine | CCK | Amino acids sugars, bitter, LCFA T1R1/T1R3 T2R CaSR GPR6C GPR120 | |
| K-cells | small intestine | GIP | ||
| L-cells | small intestine, colon | GLP-1, GLP-2 | Sugars, bitter, SCFAs, LCFAs amino acids T1R2/T1R3, T2R FFAR 1/2/3 GPR120 GPR6C | elevated |
| Enterochromaffin cells | colon | 5-HT | decreased |
| Receptor | Ligand | Expression | Function | Health Outcome |
|---|---|---|---|---|
| Sweet taste receptors i.e., T1R3 | sugars saccharin sucralose aspartame acesulfame K amino acids Na-glutamate | HGE neurons (brain) solitary chemosensory cells (upper airway system) chemosensory brush cell (urinary system) neutrophil granulocytes T and B lymphocytes Enteroendocrine cells Tuft cells Paneth cells Pancreas ß-cells | glucose metabolism, blood-brain axis regulation host-pathogen interaction cell migration cell activation incretin secretion glucose absorption Th2 immunity regulation antimicrobial peptides secretion insulin secretion | feeding behavior circadian rhythm regulation allergic, infectious diseases, chronic rhinosinusitis innate immunity glucose metabolism, metabolic syndrome IBD, helminth and viral infections, inflammation IBD infections, inflammation diabetes mellitus |
| Bitter taste receptors i.e., T2R38 | drugs i.e., chloroquine saccharin acesulfame K sucralose bacterial peptides: i.e., acyl-homoserine lactones | placenta myeloid cells macrophages chemosensory cells (upper airway system) chemosensory brush cell (urinary system) Enteroendocrine cells Goblet cells Paneth cells | unknown migration phagocytosis production of antimicrobial peptides glucose metabolism regulation mucin secretion antimicrobial peptide secretion | unknown innate immunity infection, inflammation chronic rhinosinusitis diabetes mellitus, metabolic syndrome, inflammation inflammation, infections inflammation, infections |
| CaSR | aromatic L-amino acids | enteroendocrine cells | calcium homeostasis cytokine secretion | calcium homeostasis gut–kidney axis inflammation, IBD? cancer development |
| GPRC6A | amino acids L-arginine, L-lysine and L-ornithine osteocalcin testosterone | enteroendocrine cells ILC-3 cells | bone metabolism IL-22 secretion tissue repair microbiota balance | bone resorption inflammation, IBD |
| GPR92 (LPAR5) | partially digested proteins | G cells | gastrin secretion | digestion regulation |
| FFAR 1, 4 | n-6 and n-3 PUFAs, DHA | enteroendocrine cells lymphocytes dendritic cells macrophage | antiinflammatory cytokine secretion Treg/Th17 axis regulation insulin sensitivity | inflammation innate immunity antiviral response diabetes mellitus |
| FFAR 2,3 | short-chain fatty acid | enteroendocrine cells innate immune cells: neutrophil granulocytes pancreatic ß cells | epithelial integrity antiinflammatory cytokine secretion NLRP3 inflammasome modulation alpha defensin secretion glucose metabolism | gut permeability microbiome regulation inflammation IBD diabetes mellitus infection diabetes mellitus |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Posta, E.; Fekete, I.; Gyarmati, E.; Stündl, L.; Zold, E.; Barta, Z. The Effects of Artificial Sweeteners on Intestinal Nutrient-Sensing Receptors: Dr. Jekyll or Mr. Hyde? Life 2024, 14, 10. https://doi.org/10.3390/life14010010
Posta E, Fekete I, Gyarmati E, Stündl L, Zold E, Barta Z. The Effects of Artificial Sweeteners on Intestinal Nutrient-Sensing Receptors: Dr. Jekyll or Mr. Hyde? Life. 2024; 14(1):10. https://doi.org/10.3390/life14010010
Chicago/Turabian StylePosta, Edit, Istvan Fekete, Eva Gyarmati, László Stündl, Eva Zold, and Zsolt Barta. 2024. "The Effects of Artificial Sweeteners on Intestinal Nutrient-Sensing Receptors: Dr. Jekyll or Mr. Hyde?" Life 14, no. 1: 10. https://doi.org/10.3390/life14010010
APA StylePosta, E., Fekete, I., Gyarmati, E., Stündl, L., Zold, E., & Barta, Z. (2024). The Effects of Artificial Sweeteners on Intestinal Nutrient-Sensing Receptors: Dr. Jekyll or Mr. Hyde? Life, 14(1), 10. https://doi.org/10.3390/life14010010