
The Role of Human Papilloma Virus (HPV) in Primary Lung Cancer Development: State of the Art and Future Perspectives
 , , ,
, , ,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Role of HPV in Lung Cancer Development
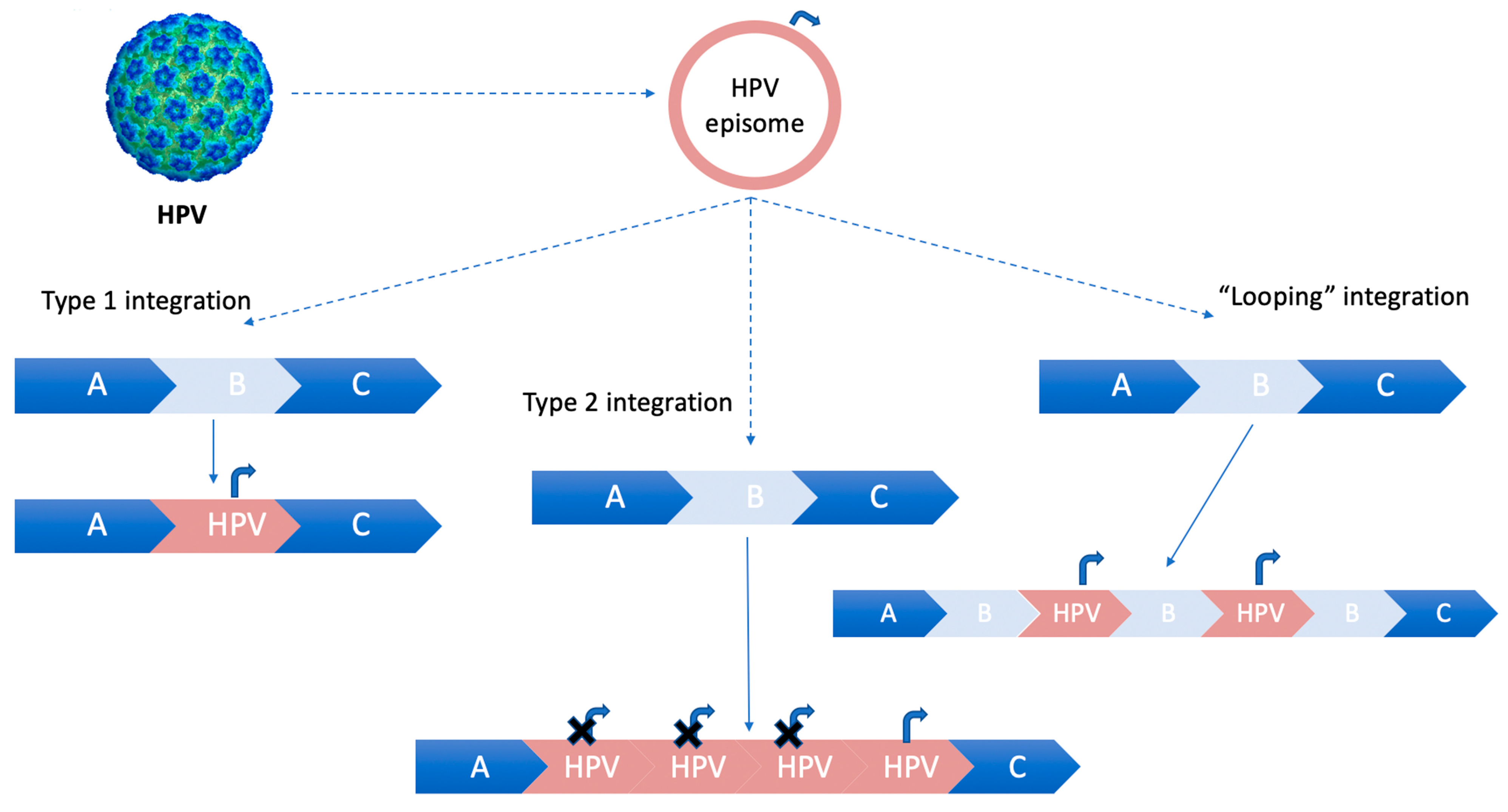
2.1. HPV Genotypes, Integration Sites in Host Genome, and Oncogenetic Activity
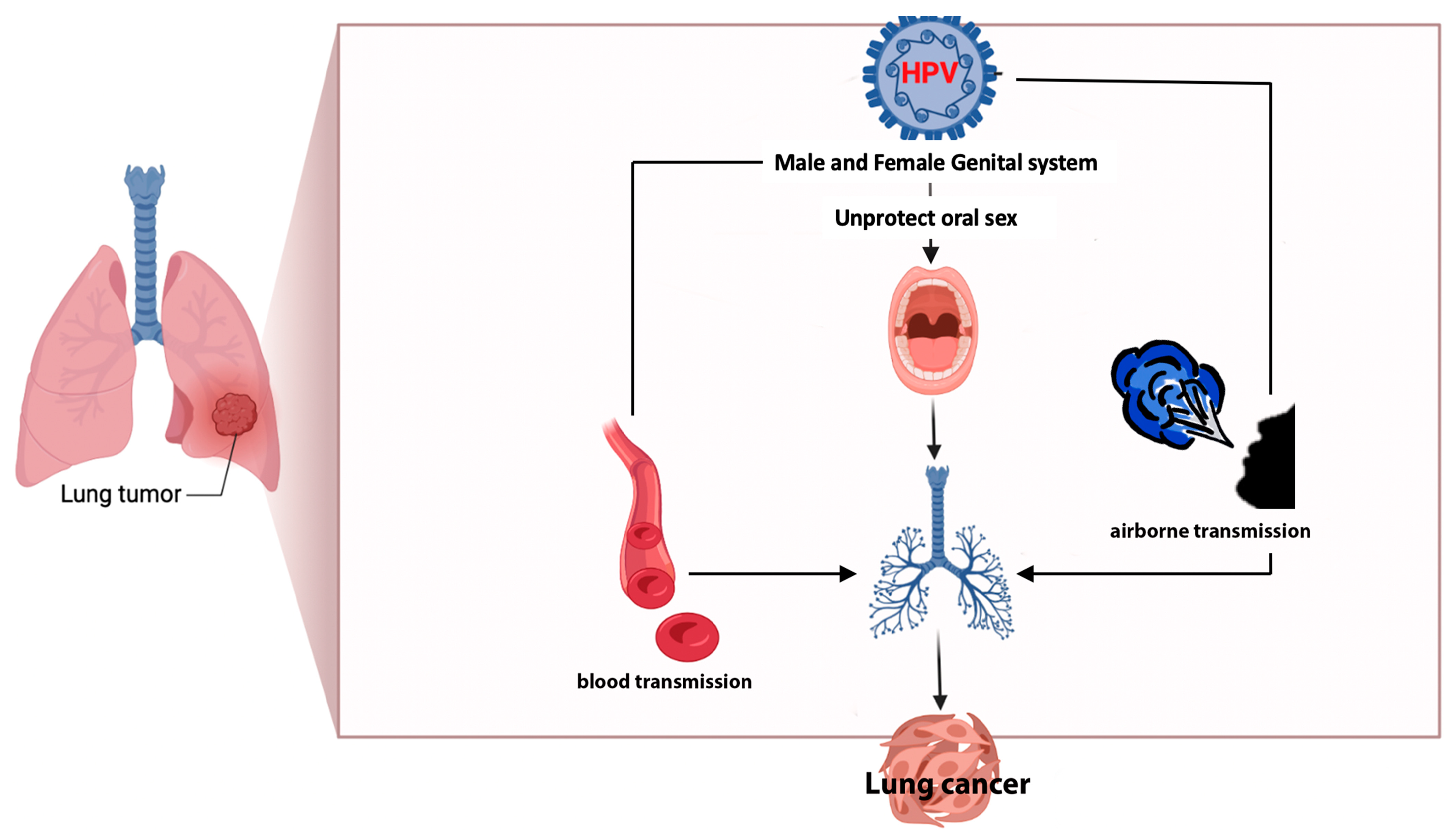
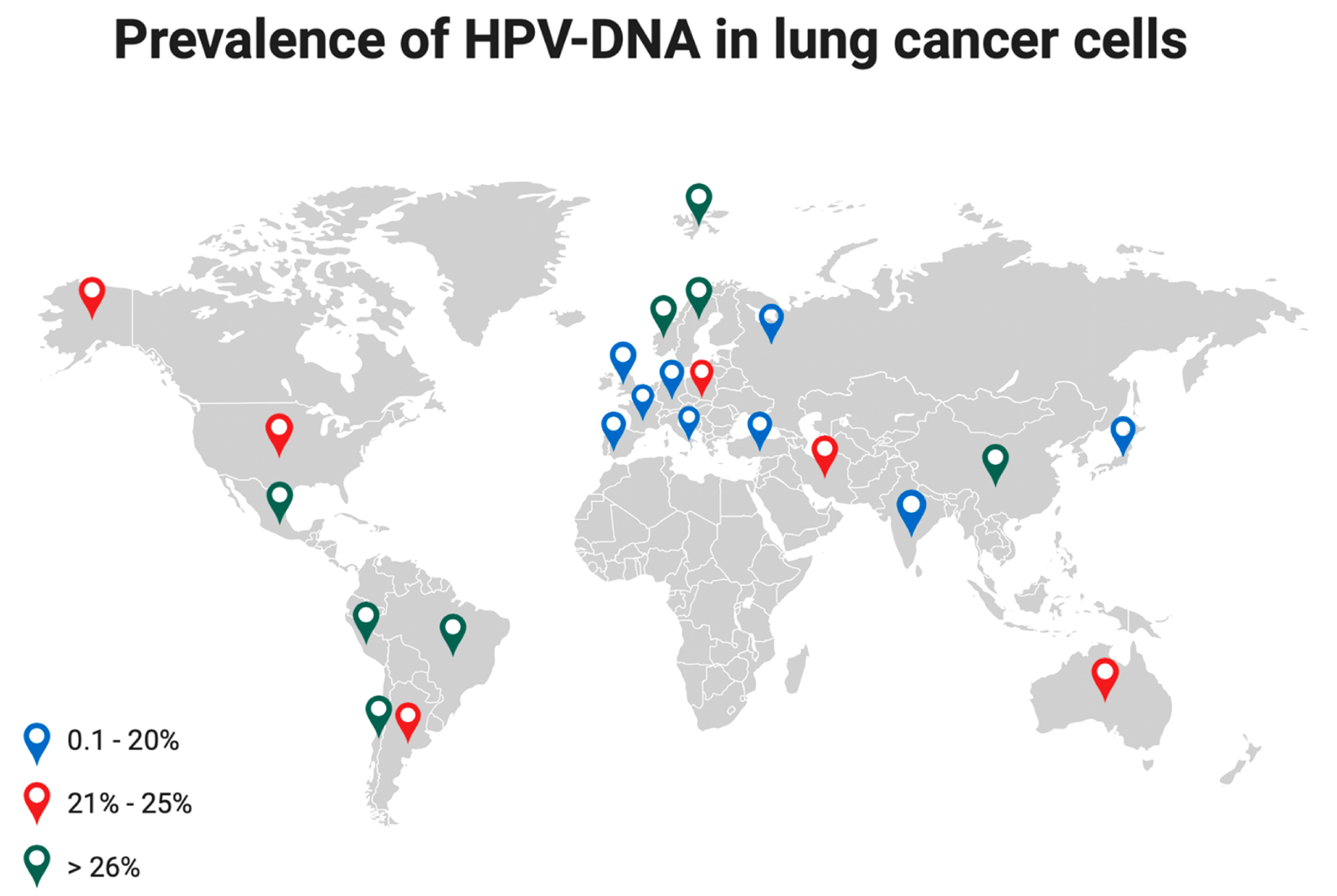
2.2. HPV Infection of Lung Cells: Pathogenesis, Prevalence, and Carcinogenesis
2.3. Second Primary Lung Cancer in Previous HPV-Related Tumors
2.4. Diagnostic Tools to Discriminate Lung Metastases from SPLCs
2.5. Prognostic Aspects of HPV Infection in NSCLC
2.6. New Frontiers in NSCLC Treatments
3. Future Perspective
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Rosell, R.; Cardona, A.F.; Martín, C.; Zatarain-Barrón, Z.L.; Arrieta, O. Lung cancer in never smokers: The role of different risk factors other than tobacco smoking. Crit. Rev. Oncol. Hematol. 2020, 148, 102895. [Google Scholar] [CrossRef] [PubMed]
- Syrjänen, K.J. Condylomatous changes in neoplastic bronchial epithelium. Rep. A Case Respir. 1979, 38, 299–304. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Xiong, W.; Yu, F.; Xiao, R.; Ye, H.; Li, W.; Liu, Z.; Hu, Z.; Cai, L. Human papillomavirus infection maybe not associated with primary lung cancer in the Fujian population of China. Thorac. Cancer 2020, 11, 561–569. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, T.H.A.; do Amaral, C.M.; de França São Marcos, B.; Nascimento, K.C.G.; de Miranda Rios, A.C.; Quixabeira, D.C.A.; Muniz, M.T.C.; Silva Neto, J.D.C.; de Freitas, A.C. Presence and activity of HPV in primary lung cancer. J. Cancer Res. Clin. Oncol. 2018, 144, 2367–2376. [Google Scholar] [CrossRef] [PubMed]
- Tsyganov, M.M.; Pevzner, A.M.; Ibragimova, M.K.; Deryusheva, I.V.; Litviakov, N.V. Human papillomavirus and lung cancer: An overview and a meta-analysis. J. Cancer Res. Clin. Oncol. 2019, 145, 1919–1937. [Google Scholar] [CrossRef]
- Bjørge, T.; Hennig, E.M.; Skare, G.B.; Søreide, O.; Thoresen, S. Second primary cancers in patients with carcinoma in situ of the uterine cervix. The Norwegian experience 1970–1992. Int. J. Cancer 1995, 62, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Hennig, E.M.; Suo, Z.; Karlsen, F.; Holm, R.; Thoresen, S.; Nesland, J.M. HPV positive bronchopulmonary carcinomas in women with previous high-grade cervical intraepithelial neoplasia (CIN III). Acta Oncol. 1999, 38, 639–647. [Google Scholar] [CrossRef]
- Donin, N.M.; Kwan, L.; Lenis, A.T.; Drakaki, A.; Chamie, K. Second primary lung cancer in United States Cancer Survivors, 1992–2008. Cancer Causes Control 2019, 30, 465–475. [Google Scholar] [CrossRef]
- Srinivasan, M.; Taioli, E.; Ragin, C.C. Human papillomavirus type 16 and 18 in primary lung cancers—A meta-analysis. Carcinogenesis 2009, 30, 1722–1728. [Google Scholar] [CrossRef]
- Drokow, E.K.; Effah, C.Y.; Agboyibor, C.; Budu, J.T.; Arboh, F.; Kyei-Baffour, P.A.; Xiao, Y.; Zhang, F.; Wu, I.X. Microbial infections as potential risk factors for lung cancer: Investigating the role of human papillomavirus and chlamydia pneumoniae. AIMS Public Health 2023, 10, 627–646. [Google Scholar] [CrossRef]
- Working Group on the Evaluation of Carcinogenic Risks to Humans. Human Papillomaviruses. Lyon (FR): International Agency for Research on Cancer. (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, No. 90.) 1, Human Papillomavirus (HPV) Infection. 2007. Available online: https://www.ncbi.nlm.nih.gov/books/NBK321770/ (accessed on 10 September 2022).
- Bzhalava, D.; Guan, P.; Franceschi, S.; Dillner, J.; Clifford, G. A systematic review of the prevalence of mucosal and cutaneous human papillomavirus types. Virology 2013, 445, 22431. [Google Scholar] [CrossRef] [PubMed]
- Ault, K.A. Epidemiology and natural history of human papillomavirus infections in the female genital tract. Infect. Dis. Obstet. Gynecol. 2006, 2006, 40470. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef]
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV–Associated cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar] [CrossRef]
- Groves, I.J.; Coleman, N. Human. papillomavirus genome integration in squamous carcinogenesis: What have next-generation sequencing studies taught us? J. Pathol. 2018, 245, 9–18. [Google Scholar] [CrossRef]
- Kamal, M.; Lameiras, S.; Deloger, M.; Morel, A.; Vacher, S.; Lecerf, C.; Dupain, C.; Jeannot, E.; Girard, E.; Baulande, S.; et al. Human papilloma virus (HPV) integration signature in Cervical Cancer: Identification of MACROD2 gene as HPV hot spot integration site. Br. J. Cancer 2021, 124, 777–785. [Google Scholar] [CrossRef]
- Zhang, R.; Shen, C.; Zhao, L.; Wang, J.; McCrae, M.; Chen, X.; Lu, F. Dysregulation of host cellular genes targeted by human papillomavirus (HPV) integration contributes to HPV-related cervical carcinogenesis. Int. J. Cancer 2016, 138, 1163–1174. [Google Scholar] [CrossRef]
- Zhang, Y.; Koneva, L.A.; Virani, S.; Arthur, A.E.; Virani, A.; Hall, P.B.; Warden, C.D.; Carey, T.E.; Chepeha, D.B.; Prince, M.E.; et al. Subtypes of HPV-positive head and neck cancers are associated with HPV characteristics, copy number alterations, PIK3CA mutation, and pathway signatures. Clin. Cancer Res. 2016, 22, 4735–4745. [Google Scholar] [CrossRef]
- Koneva, L.A.; Zhang, Y.; Virani, S.; Hall, P.B.; McHugh, J.B.; Chepeha, D.B.; Wolf, G.T.; Carey, T.E.; Rozek, L.S.; Sartor, M.A. HPV integration in HNSCC correlates with survival outcomes, immune response signatures, and candidate drivers. Mol. Cancer Res. 2018, 16, 90–102. [Google Scholar] [CrossRef]
- Münger, K.; Howley, P.M. Human papillomavirus immortalization and transformation functions. Virus Res. 2002, 89, 213–228. [Google Scholar] [CrossRef]
- de Freitas, A.C.; Gurgel, A.P.; de Lima, E.G.; de França São Marcos, B.; do Amaral, C.M. Human papillomavirus and lung cancinogenesis: An overview. J. Cancer Res. Clin. Oncol. 2016, 142, 2415–2427. [Google Scholar] [CrossRef]
- von Knebel Doeberitz, M.; Prigge, E.S. Role of DNA methylation in HPV associated lesions. Papillomavirus Res. 2019, 7, 180–183. [Google Scholar] [CrossRef]
- Syganov, M.M.; Ibragimova, M.K.; Rodionov, E.O.; Cheremisina, O.V.; Miller, S.V.; Tuzikov, S.A.; Litvyakov, N.V. Human Papillomavirus in Non-Small Cell Lung Carcinoma: Assessing Virus Presence in Tumor and Normal Tissues and Its Clinical Relevance. Microorganisms 2023, 11, 212. [Google Scholar] [CrossRef]
- Osorio, J.C.; Candia-Escobar, F.; Corvalán, A.H.; Calaf, G.M.; Aguayo, F. High-Risk Human Papillomavirus Infection in Lung Cancer: Mechanisms and Perspectives. Biology 2022, 11, 1691. [Google Scholar] [CrossRef]
- Nie, Z.; Zhang, K.; Li, Z.; Bing, X.; Jin, S.; Li, M. Human papillomavirus 16 E6 promotes angiogenesis of lung cancer via SNHG1. Cell Biochem. Biophys. 2023, 81, 325–336. [Google Scholar] [CrossRef]
- Liu, J.; Huang, B.; Xiu, Z.; Zhou, Z.; Liu, J.; Li, X.; Tang, X. PI3K/Akt/HIF-1α signaling pathway mediates HPV-16 oncoprotein-induced expression of EMT-related transcription factors in non-small cell lung cancer cells. J. Cancer 2018, 9, 3456–3466, Erratum in J. Cancer 2023, 14, 2025–2026. [Google Scholar] [CrossRef]
- Harabajsa, S.; Šefčić, H.; Klasić, M.; Milavić, M.; Židovec Lepej, S.; Grgić, I.; Zajc Petranović, M.; Jakopović, M.; Smojver-Ježek, S.; Korać, P. Infection with human cytomegalovirus, Epstein-Barr virus, and high-risk types 16 and 18 of human papillomavirus in EGFR-mutated lung adenocarcinoma. Croat. Med. J. 2023, 64, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xiang, J.; An, Y.; Xu, J.; Xiong, Y.; Wang, S.; Xia, Q. Unveiling the Association between HPV and Pan-Cancers: A Bidirectional Two-Sample Mendelian Randomization Study. Cancers 2023, 15, 5147. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Tsai, Y.C.; Chen, Y.C.; Christiani, D.C. Human papilloma virus and female lung adenocarcinoma. Semin. Oncol. 2009, 36, 542–552. [Google Scholar] [CrossRef]
- Zhai, K.; Ding, J.; Shi, H.Z. HPV and lung cancer risk: A meta-analysis. J. Clin. Virol. 2015, 63, 84–90. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Ando, M.; Kubo, A.; Isa, S.; Yamamoto, S.; Tsujino, K.; Kurata, T.; Ou, S.H.; Takada, M.; Kawaguchi, T. Human papilloma virus in non-small cell lung cancer in never smokers: A systematic review of the literature. Lung Cancer 2014, 83, 8–13. [Google Scholar] [CrossRef]
- Ragin, C.; Obikoya-Malomo, M.; Kim, S.; Chen, Z.; Flores-Obando, R.; Gibbs, D.; Koriyama, C.; Aguayo, F.; Koshiol, J.; Caporaso, N.E.; et al. HPV-associated lung cancers: An international pooled analysis. Carcinogenesis 2014, 35, 1267–1275. [Google Scholar] [CrossRef]
- Rojas, L.; Mayorga, D.; Ruiz-Patiño, A.; Rodríguez, J.; Cardona, A.; Archila, P.; Avila, J.; Bravo, M.; Ricaurte, L.; Sotelo, C.; et al. Human papillomavirus infection and lung adenocarcinoma: Special benefit is observed in patients treated with immune checkpoint inhibitors. ESMO Open 2022, 7, 100500. [Google Scholar] [CrossRef]
- Karnosky, J.; Dietmaier, W.; Knuettel, H.; Freigang, V.; Koch, M.; Koll, F.; Zeman, F.; Schulz, C. HPV and lung cancer: A systematic review and meta-analysis. Cancer Rep. 2021, 4, e1350. [Google Scholar] [CrossRef]
- Burger, M.P.; Hollema, H.; Gouw, A.S.; Pieters, W.J.; Quint, W.G. Cigarette smoking and human papillomavirus in patients with reported cervical cytological abnormality. BMJ 1993, 306, 749–752. [Google Scholar] [CrossRef]
- Peña, N.; Carrillo, D.; Muñoz, J.P.; Chnaiderman, J.; Urzúa, U.; León, O.; Tornesello, M.L.; Corvalán, A.H.; Soto-Rifo, R.; Aguayo, F. Tobacco smoke activates human papillomavirus 16 p97 promoter and cooperates with high-risk E6/E7 for oxidative DNA damage in lung cells. PLoS ONE 2015, 10, e0123029. [Google Scholar] [CrossRef]
- Szymonowicz, K.A.; Chen, J. Biological and clinical aspects of HPV-related cancers. Cancer Biol. Med. 2020, 17, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Tung, M.C.; Lin, P.L.; Cheng, Y.W.; Wu, D.W.; Yeh, S.D.; Chen, C.Y.; Lee, H. Reduction of microRNA-184 by E6 oncoprotein confers cisplatin resistance in lung cancer via increasing Bcl-2. Oncotarget 2016, 7, 32362–32374. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Wang, Y.C.; Wang, L.; Shen, C.J.; Chen, C.Y.; Lee, H. PD-L1 expressed from tumor cells promotes tumor growth and invasion in lung cancer via modulating TGF-β1/SMAD4 expression. Thorac. Cancer 2022, 13, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Viarisio, D.; Müller-Decker, K.; Accardi, R.; Robitaille, A.; Dürst, M.; Beer, K.; Jansen, L.; Flechtenmacher, C.; Bozza, M.; Harbottle, R.; et al. Beta HPV38 oncoproteins act with a hit-and-run mechanism in ultraviolet radiation-induced skin carcinogenesis in mice. PLoS Pathog. 2018, 14, e1006783. [Google Scholar] [CrossRef] [PubMed]
- Kostov, S.; Dzhenkov, D.; Metodiev, D.; Kornovski, Y.; Slavchev, S.; Ivanova, Y.; Yordanov, A. A case of human papillomavirus infection and vulvar cancer in a young patient—“hit and run” theory. Gynecol. Oncol. Rep. 2021, 36, 100760. [Google Scholar] [CrossRef] [PubMed]
- Hussen, B.M.; Ahmadi, G.; Marzban, H.; Fard Azar, M.E.; Sorayyayi, S.; Karampour, R.; Nahand, J.S.; Hidayat, H.J.; Moghoofei, M. The role of HPV gene expression and selected cellular MiRNAs in lung cancer development. Microb. Pathog. 2021, 150, 104692. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C.; Hill, W.; Lim, E.; Lee, C.; Weeden, C.; Augustine, M.; Chen, K.; Kuan, F.-C.; Marongiu, F.; Rodrigues, F.; et al. Mechanism of Action and an Actionable Inflammatory Axis for Air Pollution Induced Non-Small Cell Lung Cancer: Towards Molecular Cancer Prevention. ESMO Congress 2022, LBA 1. Ann. Oncol. 2022, 33, S1413. [Google Scholar] [CrossRef]
- Wu, Y.; Yin, Q.; Zhou, Y.L.; He, L.; Zou, Z.Q.; Dai, X.Y.; Xia, W. Evaluation of microRNAs as potential biomarkers in circulating HPV-DNA-positive non-small cell lung cancer patients. Cancer Biol. Ther. 2021, 22, 136–148. [Google Scholar] [CrossRef]
- Jayaprakash, V.; Cheng, C.; Reid, M.; Dexter, E.U.; Nwogu, C.E.; Hicks, W.; Sullivan, M.; Demmy, T.L.; Yendamuri, S. Previous head and neck cancers portend poor prognoses in lung cancer patients. Ann. Thorac. Surg. 2011, 92, 1056–1060, discussion 1060–1061. [Google Scholar] [CrossRef]
- Martel, M.; Alemany, L.; Taberna, M.; Mena, M.; Tous, S.; Bagué, S.; Castellsagué, X.; Quer, M.; León, X. The role of HPV on the risk of second primary neoplasia in patients with oropharyngeal carcinoma. Oral. Oncol. 2017, 64, 37–43. [Google Scholar] [CrossRef]
- Chang, S.Y.; Keeney, M.; Law, M.; Donovan, J.; Aubry, M.C.; Garcia, J. Detection of human papillomavirus in non-small cell carcinoma of the lung. Hum. Pathol. 2015, 46, 1592–1597. [Google Scholar] [CrossRef]
- Cioffi-Lavina, M.; Chapman-Fredricks, J.; Gomez-Fernandez, C.; Ganjei-Azar, P.; Manoharan, M.; Jorda, M. P16 expression in squamous cell carcinomas of cervix and bladder. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 344–347. [Google Scholar] [CrossRef]
- Oguejiofor, K.K.; Hall, J.S.; Mani, N.; Douglas, C.; Slevin, N.J.; Homer, J.; Hall, G.; West, C.M. The prognostic significance of the biomarker p16 in oropharyngeal squamous cell carcinoma. Clin. Oncol. 2013, 25, 630–638. [Google Scholar] [CrossRef]
- Weichert, W.; Schewe, C.; Denkert, C.; Morawietz, L.; Dietel, M.; Petersen, I. Molecular HPV typing as a diagnostic tool to discriminate primary from metastatic squamous cell carcinoma of the lung. Am. J. Surg. Pathol. 2009, 33, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ying, C.; Zhao, Z.; Sui, L.; Zhang, X.; Qian, C.; Wang, Q.; Chen, L.; Guo, Q.; Wu, J. Identification of reliable biomarkers of human papillomavirus 16 methylation in cervical lesions based on integration status using high-resolution melting analysis. Clin. Epigenetics. 2018, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.D.; Yau, C.; Bowlby, R.; Liu, Y.; Brennan, K.; Fan, H.; Taylor, A.M.; Wang, C.; Walter, V.; Akbani, R.; et al. Genomic, Pathway Network, and Immunologic Features Distinguishing Squamous Carcinomas. Cell Rep. 2018, 23, 194–212.e6. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.S.; Lundine, D.; Leeman, J.E.; Higginson, D.S. Genomic Signatures in HPV-Associated Tumors. Viruses 2021, 13, 1998. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Lee, W.J.; Fang, C.L.; Hsu, H.L.; Chen, B.J.; Liu, H.E. Human Papillomavirus Oncoproteins Confer Sensitivity to Cisplatin by Interfering with Epidermal Growth Factor Receptor Nuclear Trafficking Related to More Favorable Clinical Survival Outcomes in Non-Small Cell Lung Cancer. Cancers 2022, 14, 5333. [Google Scholar] [CrossRef]
- Wang, J.L.; Fang, C.L.; Wang, M.; Yu, M.C.; Bai, K.J.; Lu, P.C.; Liu, H.E. Human papillomavirus infections as a marker to predict overall survival in lung adenocarcinoma. Int. J. Cancer. 2014, 134, 65–71. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, Y.; Li, L.; Yu, G.; Zhang, Y.; He, Y. Associations of high-risk HPV types and viral load with cervical cancer in China. J. Clin. Virol. 2006, 35, 264–269. [Google Scholar] [CrossRef]
- Gravitt, P.E.; Kovacic, M.B.; Herrero, R.; Schiffman, M.; Bratti, C.; Hildesheim, A.; Morales, J.; Alfaro, M.; Sherman, M.E.; Wacholder, S.; et al. High load for most high risk human papillomavirus genotypes is associated with prevalent cervical cancer precursors but only HPV16 load predicts the development of incident disease. Int. J. Cancer 2007, 121, 2787–2793. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, M.N.; Li, N.; Li, X.G.; Wu, L.Y. Association between human papillomavirus DNA load and development of cervical intraepithelial neoplasia and cervical cancer. Int. J. Gynecol. Cancer 2008, 18, 755–760. [Google Scholar] [CrossRef]
- Kawahara, A.; Azuma, K.; Hattori, S.; Nakashima, K.; Basaki, Y.; Akiba, J.; Takamori, S.; Aizawa, H.; Yanagawa, T.; Izumi, H.; et al. The close correlation between 8-hydroxy-2′-deoxyguanosine and epidermal growth factor receptor activating mutation in non-small cell lung cancer. Hum. Pathol. 2010, 41, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Romano, G.; Sgambato, A.; Mancini, R.; Capelli, G.; Giovagnoli, M.R.; Flamini, G.; Boninsegna, A.; Vecchione, A.; Cittadini, A. 8-hydroxy-2′-deoxyguanosine in cervical cells: Correlation with grade of dysplasia and human papillomavirus infection. Carcinogenesis 2000, 21, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer. 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Ma, J.; Zhang, H.; Shen, J.; Chen, J.; Hong, J.; Xu, Y.; Qian, C. Prophylactic and Therapeutic HPV Vaccines: Current Scenario and Perspectives. Front. Cell Infect. Microbiol. 2022, 12, 909223. [Google Scholar] [CrossRef]
- Lin, Y.; Lin, W.Y.; Lin, T.W.; Tseng, Y.J.; Wang, Y.C.; Yu, J.R.; Chung, C.R.; Wang, H.Y. Trend of HPV Molecular Epidemiology in the Post-Vaccine Era: A 10-Year Study. Viruses 2023, 15, 2015. [Google Scholar] [CrossRef]
- Ganti, A.K.; Klein, A.B.; Cotarla, I.; Seal, B.; Chou, E. Update of Incidence, Prevalence, Survival, and Initial Treatment in Patients With Non-Small Cell Lung Cancer in the US. JAMA Oncol. 2021, 7, 1824–1832. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nachira, D.; Congedo, M.T.; D’Argento, E.; Meacci, E.; Evangelista, J.; Sassorossi, C.; Calabrese, G.; Nocera, A.; Kuzmych, K.; Santangelo, R.; et al. The Role of Human Papilloma Virus (HPV) in Primary Lung Cancer Development: State of the Art and Future Perspectives. Life 2024, 14, 110. https://doi.org/10.3390/life14010110
Nachira D, Congedo MT, D’Argento E, Meacci E, Evangelista J, Sassorossi C, Calabrese G, Nocera A, Kuzmych K, Santangelo R, et al. The Role of Human Papilloma Virus (HPV) in Primary Lung Cancer Development: State of the Art and Future Perspectives. Life. 2024; 14(1):110. https://doi.org/10.3390/life14010110
Chicago/Turabian StyleNachira, Dania, Maria Teresa Congedo, Ettore D’Argento, Elisa Meacci, Jessica Evangelista, Carolina Sassorossi, Giuseppe Calabrese, Adriana Nocera, Khrystyna Kuzmych, Rosaria Santangelo, and et al. 2024. "The Role of Human Papilloma Virus (HPV) in Primary Lung Cancer Development: State of the Art and Future Perspectives" Life 14, no. 1: 110. https://doi.org/10.3390/life14010110
APA StyleNachira, D., Congedo, M. T., D’Argento, E., Meacci, E., Evangelista, J., Sassorossi, C., Calabrese, G., Nocera, A., Kuzmych, K., Santangelo, R., Rindi, G., & Margaritora, S. (2024). The Role of Human Papilloma Virus (HPV) in Primary Lung Cancer Development: State of the Art and Future Perspectives. Life, 14(1), 110. https://doi.org/10.3390/life14010110