
A Novel COCH p.D544Vfs*3 Variant Associated with DFNA9 Sensorineural Hearing Loss Causes Pathological Multimeric Cochlin Formation

Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Vestibular Function Assessment
2.3. Next-Generation Sequencing and Bioinformatic Analysis
2.4. Molecular Modeling and Binding Ligand Prediction
2.5. Cell Culture and Plasmid Transfection
2.6. Cell Viability Assay
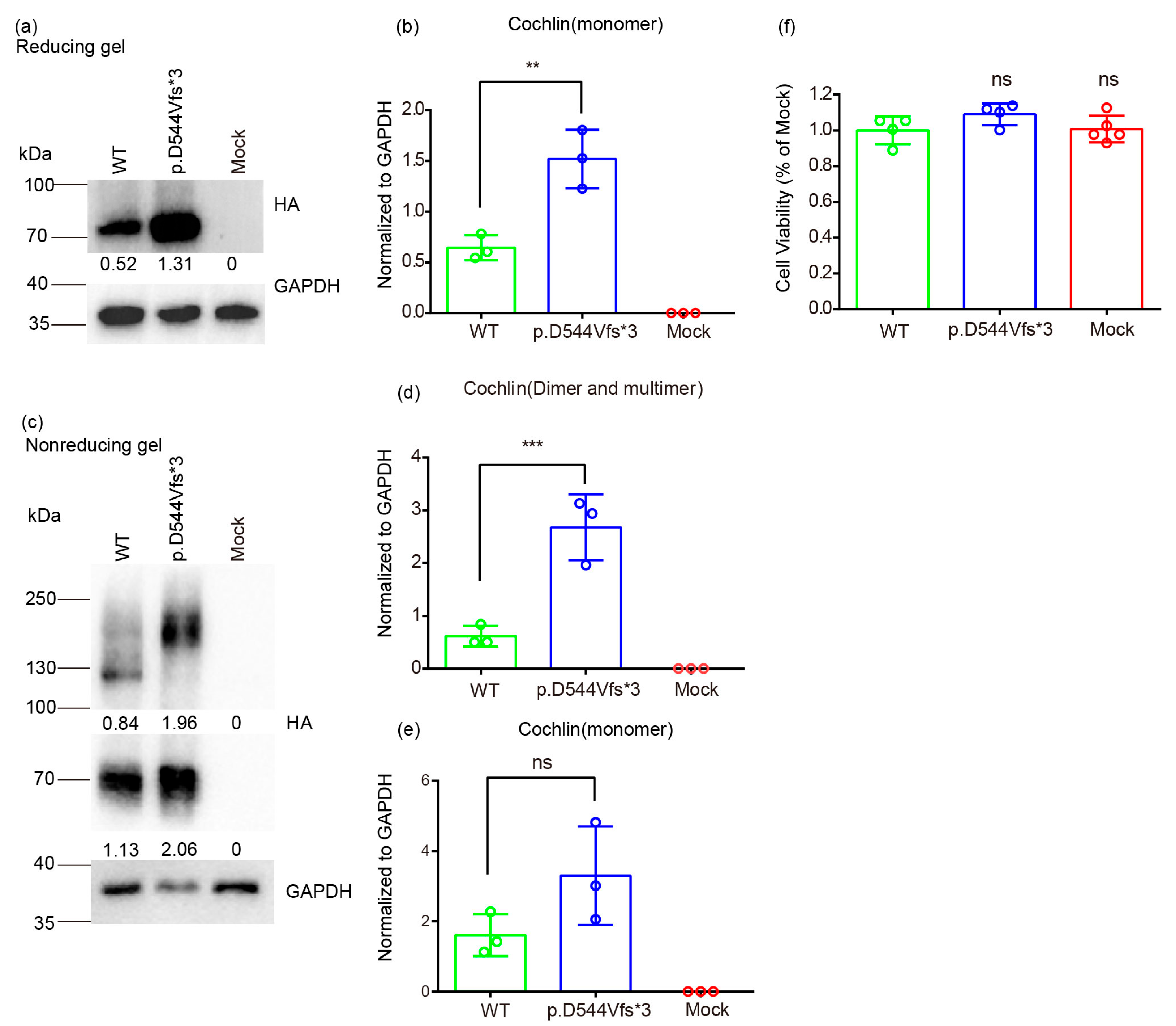
2.7. Protein Extraction and Western Blot
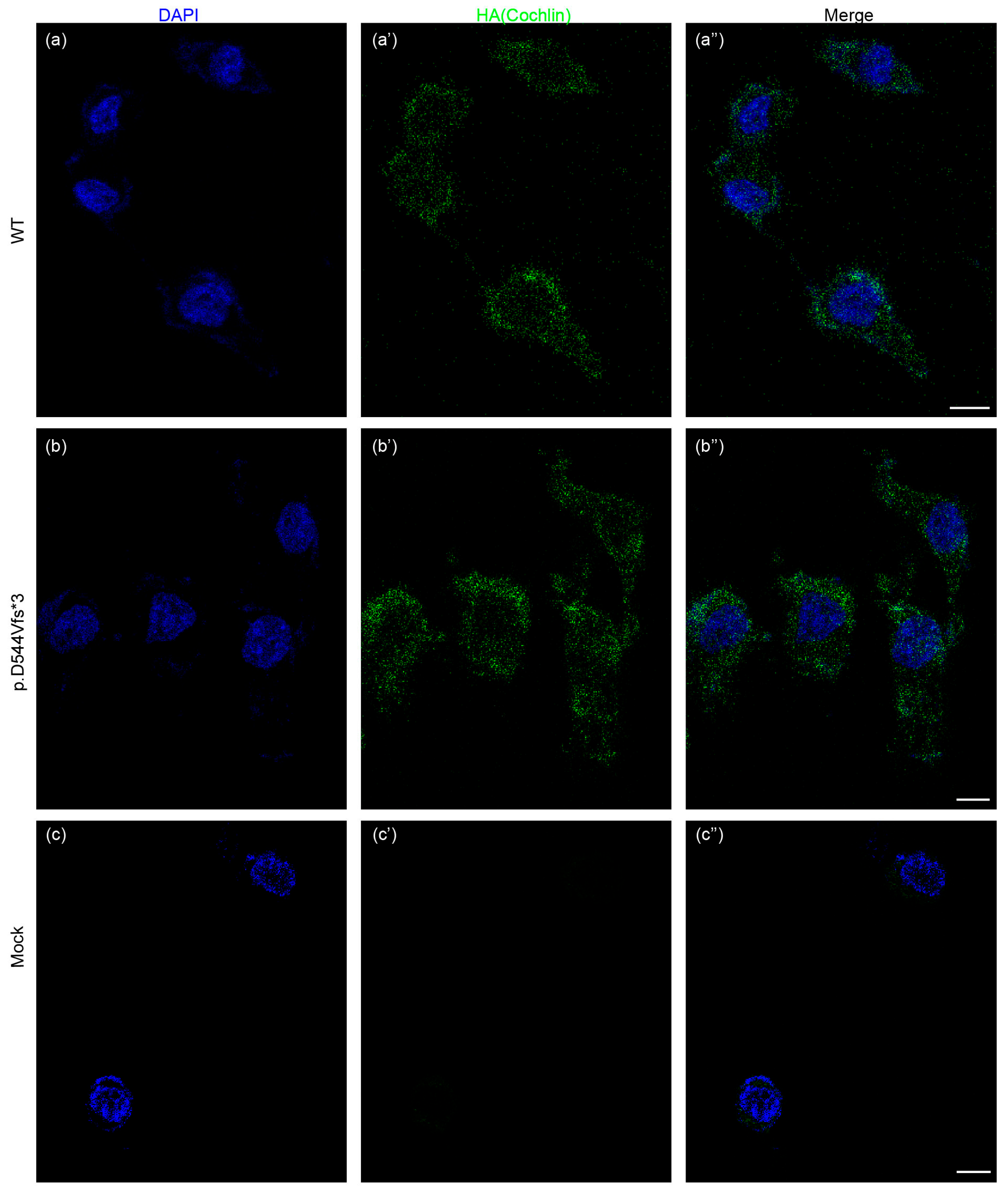
2.8. Immunofluorescence
2.9. Statistical Analysis
3. Results
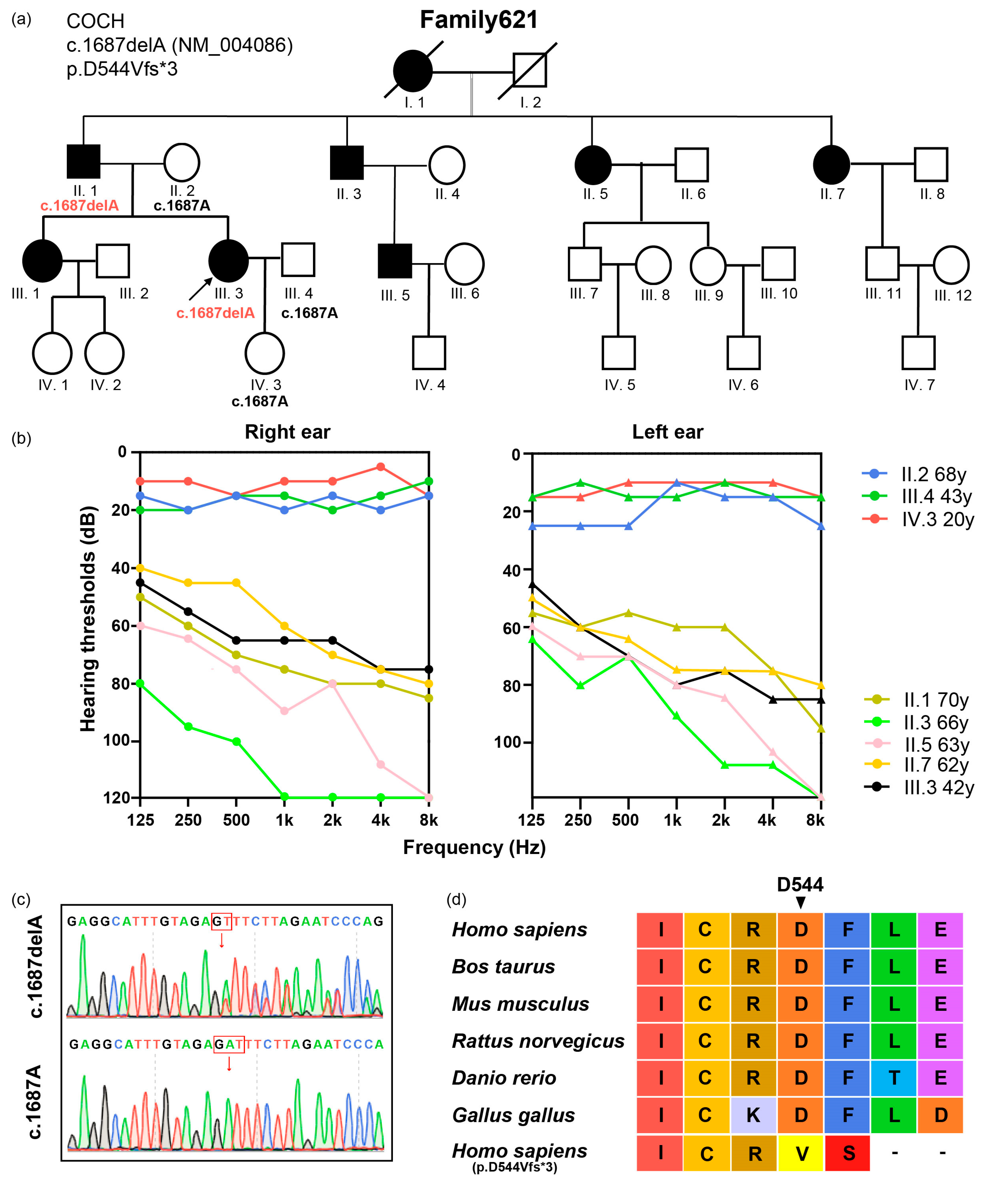
3.1. Subjects and Variant Identification
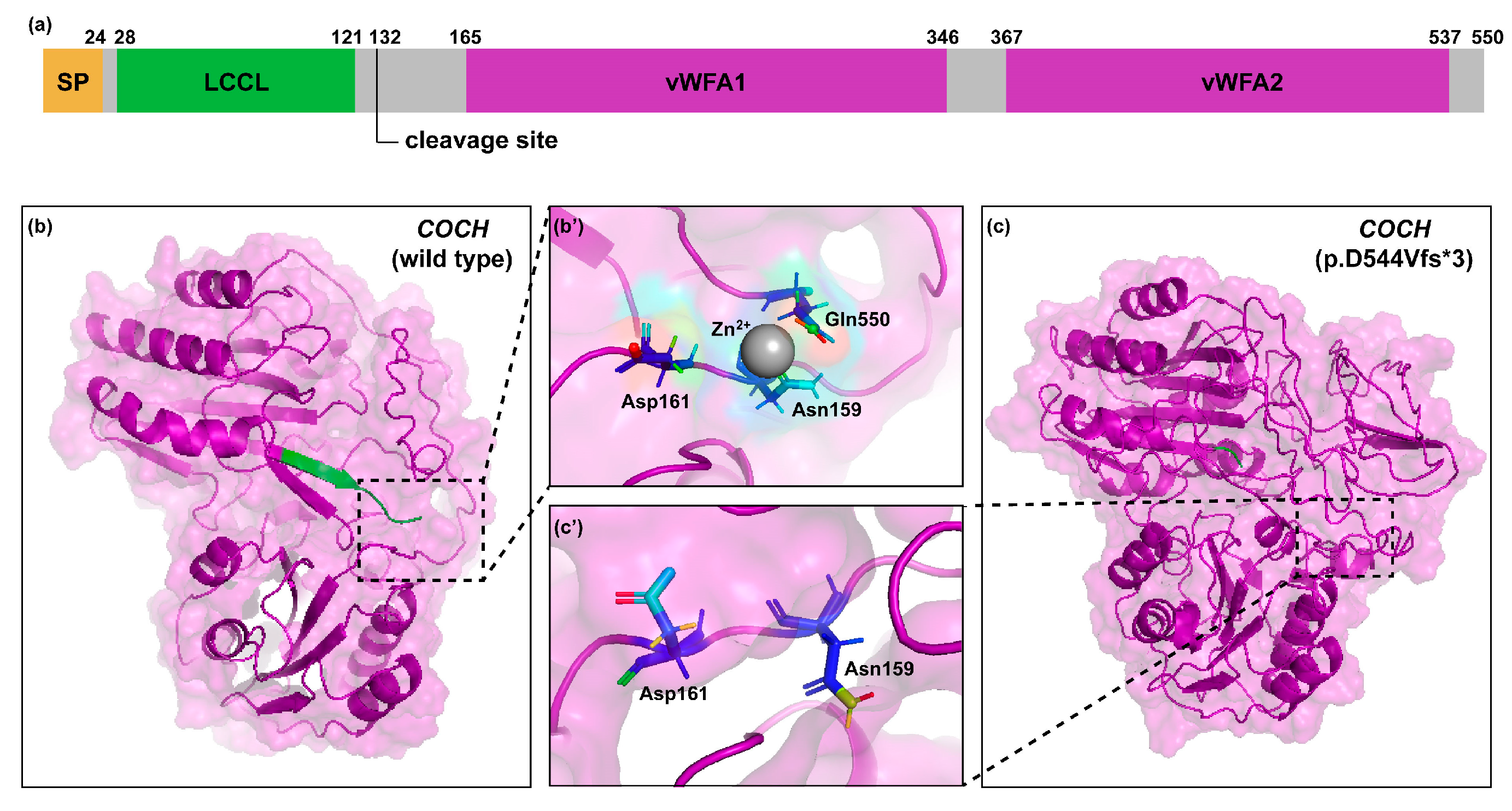
3.2. 3D Protein Modeling
3.3. Molecular Basis of Genotype-Phenotype Correlation in COCH
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tesolin, P.; Morgan, A.; Notarangelo, M.; Ortore, R.P.; Concas, M.P.; Notarangelo, A.; Girotto, G. Non-Syndromic Autosomal Dominant Hearing Loss: The First Italian Family Carrying a Mutation in the NCOA3 Gene. Genes 2021, 12, 1043. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, L.L.; Tucci, D.L. Hearing Loss in Adults. N. Engl. J. Med. 2017, 377, 2465–2473. [Google Scholar] [CrossRef] [PubMed]
- Van Camp G, S.R. Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org (accessed on 6 October 2023).
- Yasukawa, R.; Moteki, H.; Nishio, S.Y.; Ishikawa, K.; Abe, S.; Honkura, Y.; Hyogo, M.; Mihashi, R.; Ikezono, T.; Shintani, T.; et al. The Prevalence and Clinical Characteristics of TECTA-Associated Autosomal Dominant Hearing Loss. Genes 2019, 10, 744. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.S.; Walls, D.; Joo, S.Y.; Kim, J.A.; Yoo, J.E.; Koh, Y.I.; Kim, D.H.; Rim, J.H.; Choi, H.J.; Kim, H.Y.; et al. COCH-related autosomal dominant nonsyndromic hearing loss: A phenotype-genotype study. Hum. Genet. 2022, 141, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Robijn, S.M.M.; Smits, J.J.; Sezer, K.; Huygen, P.L.M.; Beynon, A.J.; van Wijk, E.; Kremer, H.; de Vrieze, E.; Lanting, C.P.; Pennings, R.J.E. Genotype-Phenotype Correlations of Pathogenic COCH Variants in DFNA9: A HuGE Systematic Review and Audiometric Meta-Analysis. Biomolecules 2022, 12, 220. [Google Scholar] [CrossRef] [PubMed]
- Gommeren, H.; Bosmans, J.; Moyaert, J.; Mertens, G.; Cras, P.; Engelborghs, S.; Van Ombergen, A.; Gilles, A.; Fransen, E.; van de Berg, R.; et al. Accelerated Cognitive Decline Associated With Hearing Loss and Bilateral Vestibulopathy: Insights From a Prospective Cross-Sectional Study Using the Repeatable Battery for the Assessment of Neuropsychological Status Adjusted for the Hearing Impaired in the DFNA9 Population. Ear Hear. 2023, 44, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Verdoodt, D.; Van Camp, G.; Ponsaerts, P.; Van Rompaey, V. On the pathophysiology of DFNA9: Effect of pathogenic variants in the COCH gene on inner ear functioning in human and transgenic mice. Hear. Res. 2021, 401, 108162. [Google Scholar] [CrossRef] [PubMed]
- Danial-Farran, N.; Chervinsky, E.; Nadar-Ponniah, P.T.; Cohen Barak, E.; Taiber, S.; Khayat, M.; Avraham, K.B.; Shalev, S.A. Homozygote loss-of-function variants in the human COCH gene underlie hearing loss. Eur. J. Hum. Genet. 2021, 29, 338–342. [Google Scholar] [CrossRef]
- Robertson, N.G.; O’Malley, J.T.; Ong, C.A.; Giersch, A.B.; Shen, J.; Stankovic, K.M.; Morton, C.C. Cochlin in normal middle ear and abnormal middle ear deposits in DFNA9 and Coch (G88E/G88E) mice. J. Assoc. Res. Otolaryngol. 2014, 15, 961–974. [Google Scholar] [CrossRef]
- Liepinsh, E.; Trexler, M.; Kaikkonen, A.; Weigelt, J.; Bányai, L.; Patthy, L.; Otting, G. NMR structure of the LCCL domain and implications for DFNA9 deafness disorder. EMBO J. 2001, 20, 5347–5353. [Google Scholar] [CrossRef]
- Trexler, M.; Bányai, L.; Patthy, L. The LCCL module. Eur. J. Biochem. 2000, 267, 5751–5757. [Google Scholar] [CrossRef] [PubMed]
- Seist, R.; Landegger, L.D.; Robertson, N.G.; Vasilijic, S.; Morton, C.C.; Stankovic, K.M. Cochlin Deficiency Protects Against Noise-Induced Hearing Loss. Front. Mol. Neurosci. 2021, 14, 670013. [Google Scholar] [CrossRef] [PubMed]
- Kommareddi, P.K.; Nair, T.S.; Raphael, Y.; Telian, S.A.; Kim, A.H.; Arts, H.A.; El-Kashlan, H.K.; Carey, T.E. Cochlin isoforms and their interaction with CTL2 (SLC44A2) in the inner ear. J. Assoc. Res. Otolaryngol. 2007, 8, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Nagy, I.; Trexler, M.; Patthy, L. The second von Willebrand type A domain of cochlin has high affinity for type I, type II and type IV collagens. FEBS Lett. 2008, 582, 4003–4007. [Google Scholar] [CrossRef]
- Sadler, J.E. Biochemistry and genetics of von Willebrand factor. Annu. Rev. Biochem. 1998, 67, 395–424. [Google Scholar] [CrossRef] [PubMed]
- Nagy, I.; Horváth, M.; Trexler, M.; Répássy, G.; Patthy, L. A novel COCH mutation, V104del, impairs folding of the LCCL domain of cochlin and causes progressive hearing loss. J. Med. Genet. 2004, 41, e9. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Yoo, J.E.; Choe, Y.H.; Park, S.C.; Lee, H.J.; Lee, H.J.; Noh, B.; Kim, S.H.; Kang, G.Y.; Lee, K.M.; et al. Cleaved Cochlin Sequesters Pseudomonas aeruginosa and Activates Innate Immunity in the Inner Ear. Cell Host Microbe 2019, 25, 513–525.e516. [Google Scholar] [CrossRef]
- Yao, J.; Py, B.F.; Zhu, H.; Bao, J.; Yuan, J. Role of protein misfolding in DFNA9 hearing loss. J. Biol. Chem. 2010, 285, 14909–14919. [Google Scholar] [CrossRef]
- Cho, H.J.; Park, H.J.; Trexler, M.; Venselaar, H.; Lee, K.Y.; Robertson, N.G.; Baek, J.I.; Kang, B.S.; Morton, C.C.; Vriend, G.; et al. A novel COCH mutation associated with autosomal dominant nonsyndromic hearing loss disrupts the structural stability of the vWFA2 domain. J. Mol. Med. 2012, 90, 1321–1331. [Google Scholar] [CrossRef]
- Basu, A.; Boczek, N.J.; Robertson, N.G.; Nasr, S.H.; Jethanamest, D.; McPhail, E.D.; Kurtin, P.J.; Dasari, S.; Butz, M.; Morton, C.C.; et al. First Report of Bilateral External Auditory Canal Cochlin Aggregates (“Cochlinomas”) with Multifocal Amyloid-Like Deposits, Associated with Sensorineural Hearing Loss and a Novel Genetic Variant in COCH Encoding Cochlin. Head Neck Pathol. 2020, 14, 808–816. [Google Scholar] [CrossRef]
- Street, V.A.; Kallman, J.C.; Robertson, N.G.; Kuo, S.F.; Morton, C.C.; Phillips, J.O. A novel DFNA9 mutation in the vWFA2 domain of COCH alters a conserved cysteine residue and intrachain disulfide bond formation resulting in progressive hearing loss and site-specific vestibular and central oculomotor dysfunction. Am. J. Med. Genet. A 2005, 139A, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.J.; Han, D.Y.; Sun, Q.; Yan, D.; Sun, H.J.; Tao, R.; Cheng, J.; Qin, W.; Angeli, S.; Ouyang, X.M.; et al. Novel mutations in the vWFA2 domain of COCH in two Chinese DFNA9 families. Clin. Genet. 2008, 73, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Aldè, M.; Cantarella, G.; Zanetti, D.; Pignataro, L.; La Mantia, I.; Maiolino, L.; Ferlito, S.; Di Mauro, P.; Cocuzza, S.; Lechien, J.R.; et al. Autosomal Dominant Non-Syndromic Hearing Loss (DFNA): A Comprehensive Narrative Review. Biomedicines 2023, 11, 1616. [Google Scholar] [CrossRef] [PubMed]
- WHO. World Report on Hearing; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Hu, M.; Chen, T.; Dong, H.; Wang, W.; Xu, K.; Lin, P. [Clinical values of the sensory organization test in vestibular diseases]. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 2015, 50, 712–717. [Google Scholar] [PubMed]
- Chaudhry, H.; Findley, T.; Quigley, K.S.; Ji, Z.; Maney, M.; Sims, T.; Bukiet, B.; Foulds, R. Postural stability index is a more valid measure of stability than equilibrium score. J. Rehabil. Res. Dev. 2005, 42, 547–556. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S.R.F.; Wilkie, A.O.M.; McVean, G.; Lunter, G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014, 46, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Jia, Y.; Jia, G.; Guo, L.; Song, N.; Wang, Y.M.; Jiang, L.; Shu, Y.; Chen, Y.; Zhu, S.; Li, H.; et al. Novel heterozygous USH1C mutation impacts hair cell mechanotransduction and causes progressive hearing loss. Sci. Bull. 2023; Online ahead of print. [Google Scholar] [CrossRef]
- Antonino, M.; Nicolò, M.; Jerome Renee, L.; Federico, M.; Chiara, V.; Stefano, S.; Maria, S.; Salvatore, C.; Antonio, B.; Calvo-Henriquez, C.; et al. Single-nucleotide polymorphism in chronic rhinosinusitis: A systematic review. Clin. Otolaryngol. 2022, 47, 14–23. [Google Scholar] [CrossRef]
- Vona, B.; Hofrichter, M.A.H.; Schröder, J.; Shehata-Dieler, W.; Nanda, I.; Haaf, T. Hereditary hearing loss SNP-microarray pilot study. BMC Res. Notes 2018, 11, 391. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Roy, A.; Zhang, Y. BioLiP: A semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 2013, 41, D1096–D1103. [Google Scholar] [CrossRef] [PubMed]
- Piker, E.G.; Jacobson, G.P.; Burkard, R.F.; McCaslin, D.L.; Hood, L.J. Effects of age on the tuning of the cVEMP and oVEMP. Ear Hear. 2013, 34, e65–e73. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Cha, C.I.; Jung, T.S.; Park, D.C.; Yeo, S.G. Age-related differences in parameters of vestibular evoked myogenic potentials. Acta Otolaryngol. 2008, 128, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Kim, H.S.; Lee, M.G.; Yang, E.J.; Choi, J.Y. Novel COCH p.V123E Mutation, Causative of DFNA9 Sensorineural Hearing Loss and Vestibular Disorder, Shows Impaired Cochlin Post-Translational Cleavage and Secretion. Hum. Mutat. 2015, 36, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Fei, P.; Gu, H.; Zhang, Y.; Ke, X.; Liu, Y. Different Phenotypes of the Two Chinese Probands with the Same c.889G>A (p.C162Y) Mutation in COCH Gene Verify Different Mechanisms Underlying Autosomal Dominant Nonsyndromic Deafness 9. PLoS ONE 2017, 12, e0170011. [Google Scholar] [CrossRef]
- Tsukada, K.; Ichinose, A.; Miyagawa, M.; Mori, K.; Hattori, M.; Nishio, S.Y.; Naito, Y.; Kitajiri, S.; Usami, S. Detailed hearing and vestibular profiles in the patients with COCH mutations. Ann. Otol. Rhinol. Laryngol. 2015, 124 (Suppl. 1), 100s–110s. [Google Scholar] [CrossRef]
- Sun, Q.; Yang, S.Z.; Kang, D.Y.; Zhang, X.; Cao, J.Y.; Liu, X.; Dai, P.; Yuan, H.J.; Han, D.Y. [Mutation screening of the COCH gene in familial and sporadic patients with late onset nonsyndromic sensorineural hearing loss among Chinese population]. Zhonghua Yi Xue Za Zhi 2007, 87, 3107–3110. [Google Scholar]
- Py, B.F.; Gonzalez, S.F.; Long, K.; Kim, M.S.; Kim, Y.A.; Zhu, H.; Yao, J.; Degauque, N.; Villet, R.; Ymele-Leki, P.; et al. Cochlin produced by follicular dendritic cells promotes antibacterial innate immunity. Immunity 2013, 38, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Robertson, N.G.; Cho, H.J.; Morton, C.C.; Jung, D.J.; Baek, J.I.; Choi, S.Y.; Lee, J.; Lee, K.Y.; Kim, U.K. Identification of pathogenic mechanisms of COCH mutations, abolished cochlin secretion, and intracellular aggregate formation: Genotype-phenotype correlations in DFNA9 deafness and vestibular disorder. Hum. Mutat. 2014, 35, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Fransen, E.; Verstreken, M.; Verhagen, W.I.; Wuyts, F.L.; Huygen, P.L.; D’Haese, P.; Robertson, N.G.; Morton, C.C.; McGuirt, W.T.; Smith, R.J.; et al. High prevalence of symptoms of Menière’s disease in three families with a mutation in the COCH gene. Hum. Mol. Genet. 1999, 8, 1425–1429. [Google Scholar] [CrossRef] [PubMed]
- JanssensdeVarebeke, S.P.F.; Moyaert, J.; Fransen, E.; Bulen, B.; Neesen, C.; Devroye, K.; van de Berg, R.; Pennings, R.J.E.; Topsakal, V.; Vanderveken, O.; et al. Genotype-Phenotype Correlation Study in a Large Series of Patients Carrying the p.Pro51Ser (p.P51S) Variant in COCH (DFNA9) Part II: A Prospective Cross-Sectional Study of the Vestibular Phenotype in 111 Carriers. Ear Hear. 2021, 42, 1525–1543. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Yoo, J.E.; Hong, J.W.; Park, H.R.; Noh, B.; Kim, H.; Kang, M.; Hyun, Y.M.; Gee, H.Y.; Choi, J.Y.; et al. LCCL peptide cleavage after noise exposure exacerbates hearing loss and is associated with the monocyte infiltration in the cochlea. Hear. Res. 2021, 412, 108378. [Google Scholar] [CrossRef] [PubMed]
- Rhyu, H.J.; Bae, S.H.; Jung, J.; Hyun, Y.M. Cochlin-cleaved LCCL is a dual-armed regulator of the innate immune response in the cochlea during inflammation. BMB Rep. 2020, 53, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Burgess, B.J.; O’Malley, J.T.; Kamakura, T.; Kristiansen, K.; Robertson, N.G.; Morton, C.C.; Nadol, J.B., Jr. Histopathology of the Human Inner Ear in the p.L114P COCH Mutation (DFNA9). Audiol. Neurootol. 2016, 21, 88–97. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pedigree | Ethnicity | Age of Onset | Mode | g.DNA | c.DNA a | Protein | MutationTaster Prediction (Prob) b | HL-Specific ACMG Classification | HL-Specific ACMG Components | CADD c | Novelty (Reported PMID) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| YUHL5 | Korean | 37–52 years | AD | 14:31348145T>A | c.424T>A | p.V123E | Disease causing (1.00) | Likely pathogenic | PS3, PM2, PP1, PP3, PP4 | 26.8 | 26256111 [39] |
| Family32, Family208 | Chinese | 12–67 years | AD | 14:31349796G>A | c.541G>A | p.C162Y | Disease causing (1.00) | Likely pathogenic | PS3, PM2, PP1, PP3, PP4 | 28.1 | 28099493 [40] |
| - | Caucasian | childhood | - | 14:31358965A>T | c.1677A>T | p.I541F | Disease causing (1.00) | Likely pathogenic | PS3, PM2, PP4 | 26.3 | 31493294 [21] |
| FAM986 | Japanese | childhood | AD | 14:31358968T>C | c. 1680T>C | p.C542R | Disease causing (1.00) | Likely pathogenic | PM2, PM5, PP1, PP3, PP4 | 24.6 | 25780252 [41] |
| HL3 | European | childhood | AD | 14:31358969G>T | c.1681G>T | p.C542F | Disease causing (1.00) | Likely pathogenic | PS3, PM2, PP1, PP3, PP4 | 24.1 | 16261627 [22] |
| SD-Z001 | Chinese | 15–59 years | AD | 14:31358969G>A | c.1681G>A | p.C542Y | Disease causing (1.00) | Likely pathogenic | PM2, PM5, PP1, PP3, PP4 | 24.0 | 18312449 [23] 18269866 [42] |
| Family621 | Chinese | early 30s | AD | 14:31358975_ 31358975delA | c.1687delA | p.D544Vfs*3 | Disease causing (1.00) | Likely pathogenic | PM2, PM4, PP1, PP4 | - | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Y.; Xiang, M.; Fan, T.; Zhong, X.; Dai, A.; Feng, J.; Guan, P.; Gong, J.; Li, J.; Wang, Y. A Novel COCH p.D544Vfs*3 Variant Associated with DFNA9 Sensorineural Hearing Loss Causes Pathological Multimeric Cochlin Formation. Life 2024, 14, 33. https://doi.org/10.3390/life14010033
Peng Y, Xiang M, Fan T, Zhong X, Dai A, Feng J, Guan P, Gong J, Li J, Wang Y. A Novel COCH p.D544Vfs*3 Variant Associated with DFNA9 Sensorineural Hearing Loss Causes Pathological Multimeric Cochlin Formation. Life. 2024; 14(1):33. https://doi.org/10.3390/life14010033
Chicago/Turabian StylePeng, Yingqiu, Mengya Xiang, Ting Fan, Xiaofang Zhong, Aqiang Dai, Jialing Feng, Pengfei Guan, Jiamin Gong, Jian Li, and Yunfeng Wang. 2024. "A Novel COCH p.D544Vfs*3 Variant Associated with DFNA9 Sensorineural Hearing Loss Causes Pathological Multimeric Cochlin Formation" Life 14, no. 1: 33. https://doi.org/10.3390/life14010033