


A Nonclinical Safety Evaluation of Cold Atmospheric Plasma for Medical Applications: The Role of Genotoxicity and Mutagenicity Studies

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Atmospheric Nonthermal Plasma (ANTP) Instrument
2.2. Cell Culture
2.3. Exposure of Plasma to the Murine Fibroblast Cells
2.4. Cell Viability Testing
2.5. Colony Formation Assay
2.6. Investigation of Intracellular Reactive Oxygen (ROS) and Nitrogen Species (RNS)
2.7. Quantification of Extracellular ROS and RNS
2.8. Mutagenicity Testing
2.8.1. 8-Hydroxy-2′-deoxyguanosine (8-OHdG) Quantification
2.8.2. DNA Strand Break Analysis
2.8.3. Bacterial Reverse Mutation Assay
2.9. Statistical Analysis
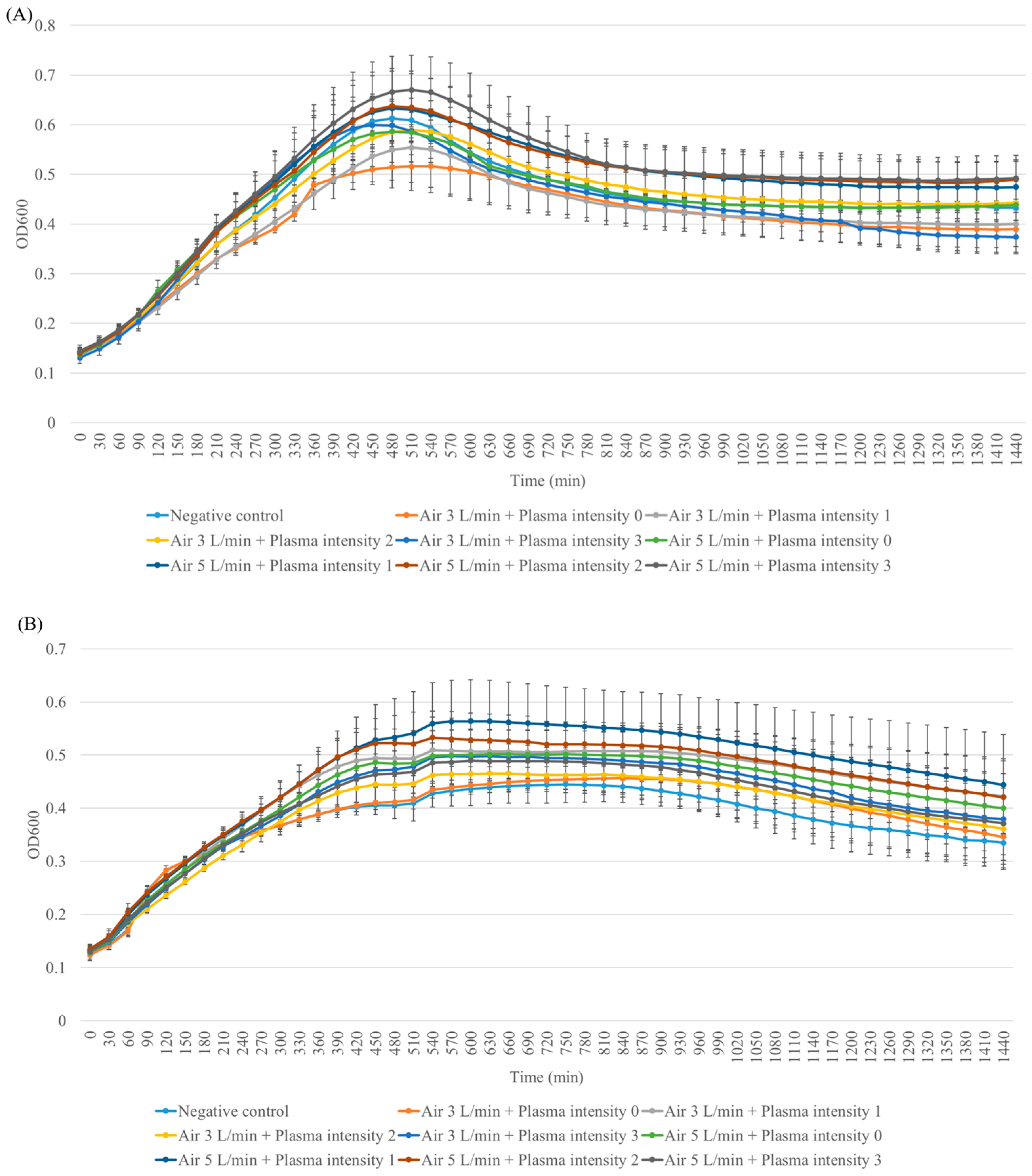
3. Results
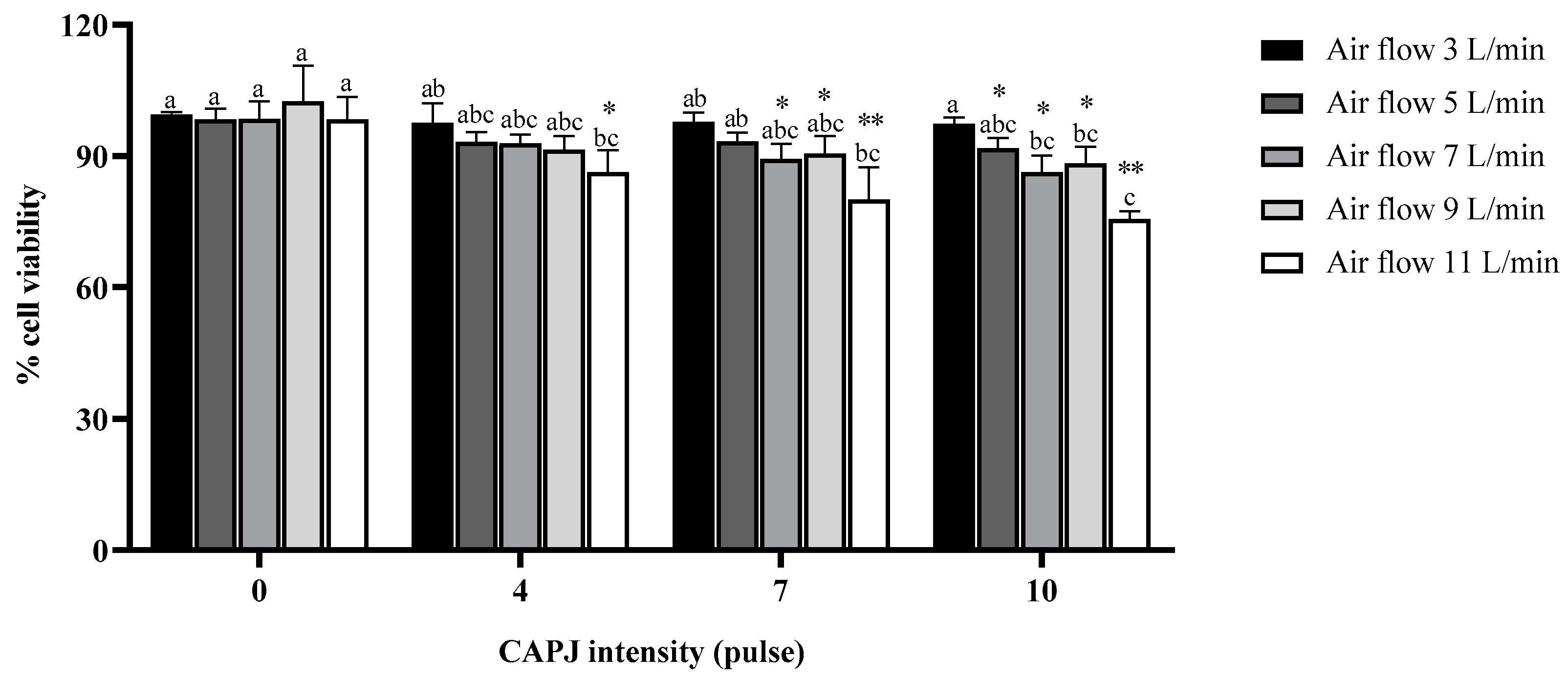
3.1. Fibroblast Cell Viability
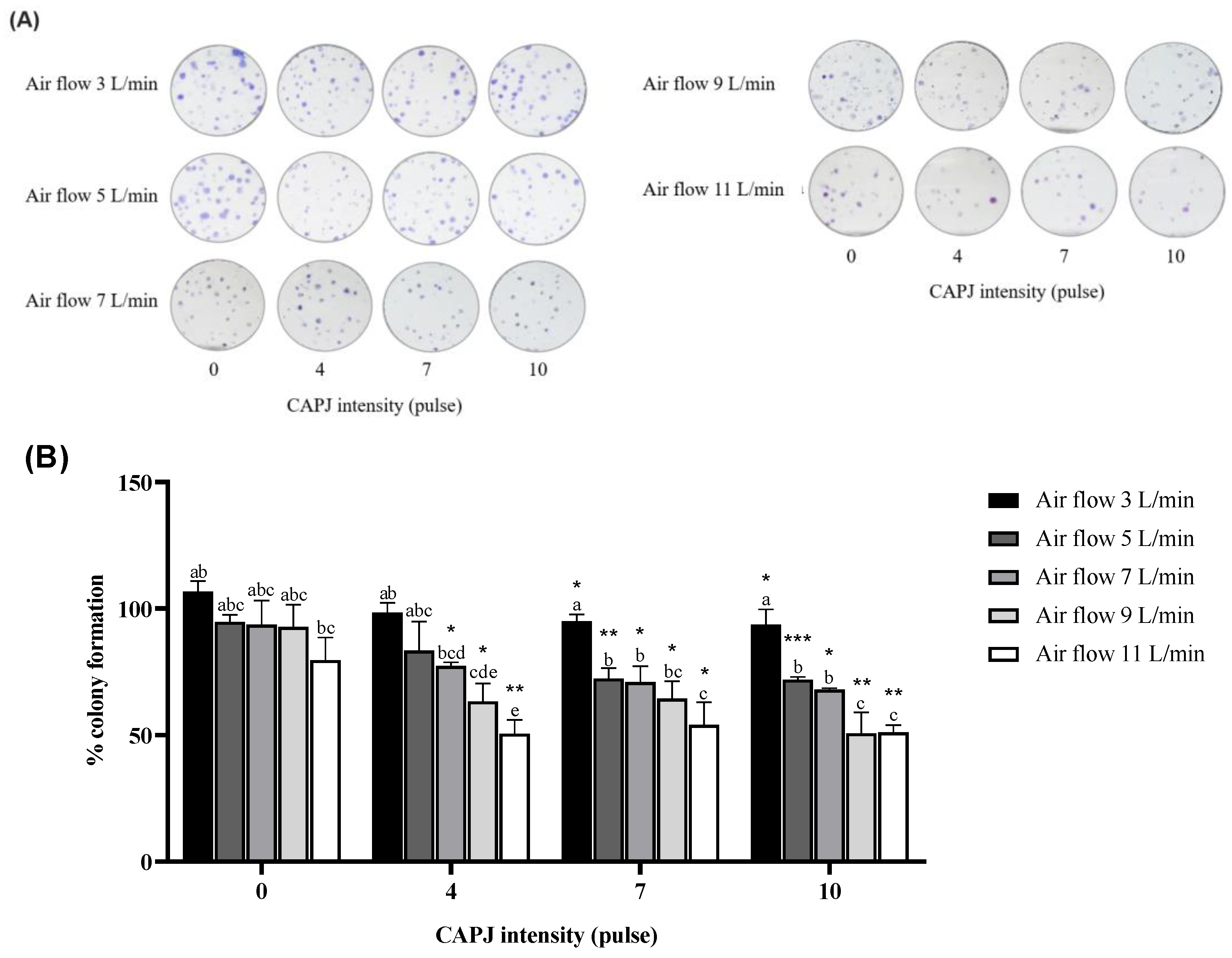
3.2. Suppression of Cell Proliferation
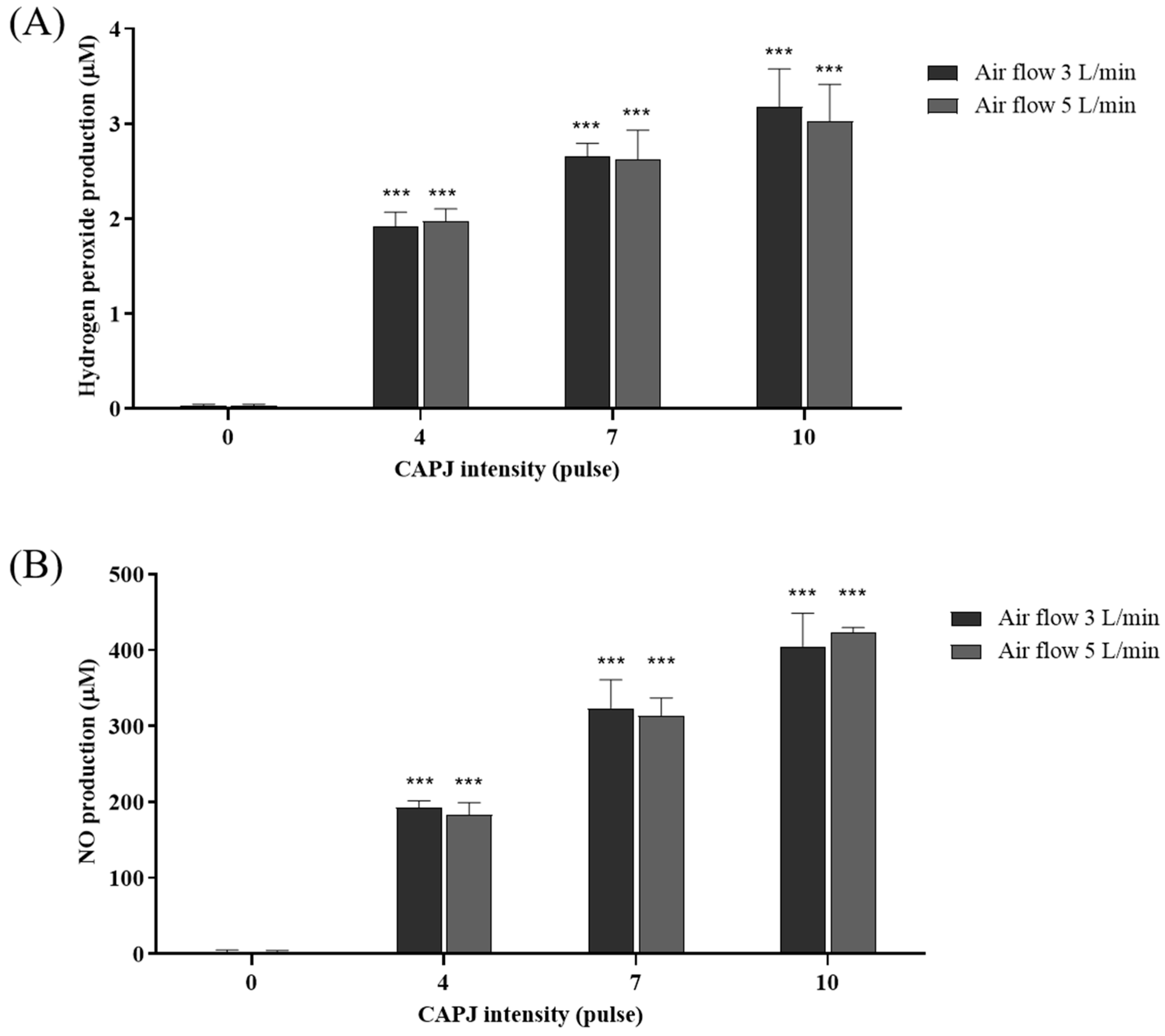
3.3. Production of Intracellular Reactive Oxygen (ROS) and Nitrogen Species (RNS)
3.4. Generation of Extracellular Reactive Oxygen (ROS) and Nitrogen Species (RNS)
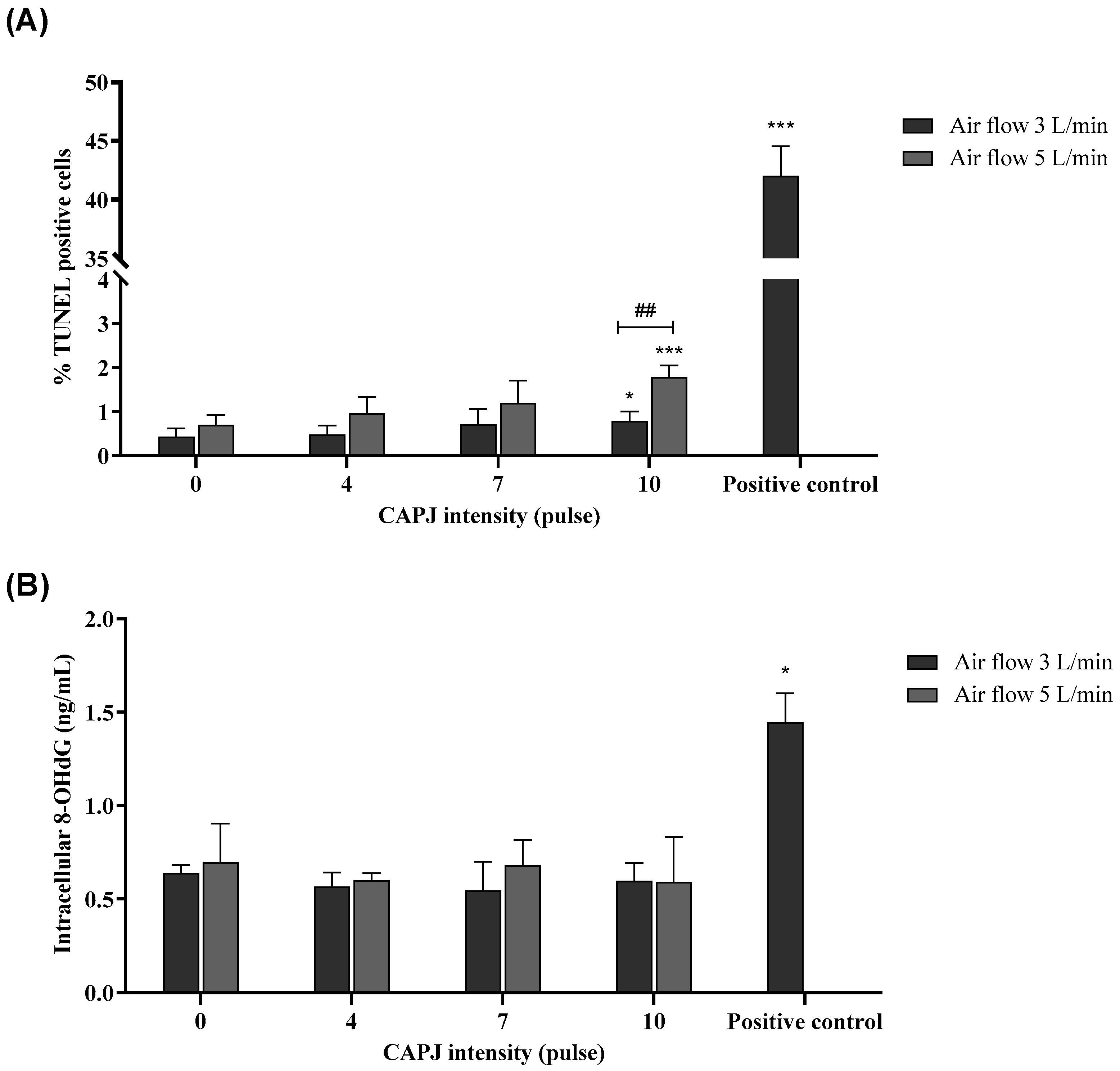
3.5. Induction of DNA Damage
3.6. Mutagenicity Potential of Plasma
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haertel, B.; von Woedtke, T.; Weltmann, K.-D.; Lindequist, U. Non-thermal atmospheric-pressure plasma possible application in wound healing. Biomol. Ther. 2014, 22, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, T.; Semmler, M.L.; Schäfer, M.; Bekeschus, S.; Emmert, S.; Boeckmann, L. Plasma Medicine: Applications of Cold Atmospheric Pressure Plasma in Dermatology. Oxidative Med. Cell. Longev. 2019, 2019, 3873928. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Zhang, S.; Poorun, D.; Liu, D.; Lu, X.; He, M.; Duan, X.; Chen, H. Medical applications of nonthermal atmospheric pressure plasma in dermatology. J. Dtsch. Dermatol. Ges. 2018, 16, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Ziuzina, D.; Los, A.; Bourke, P. Chapter 12—Inactivation of Staphylococcus aureus in Foods by Thermal and Nonthermal Control Strategies. In Staphylococcus aureus; Fetsch, A., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 235–255. [Google Scholar] [CrossRef]
- Kalghatgi, S.; Kelly, C.M.; Cerchar, E.; Torabi, B.; Alekseev, O.; Fridman, A.; Friedman, G.; Azizkhan-Clifford, J. Effects of non-thermal plasma on mammalian cells. PLoS ONE 2011, 6, e16270. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.J.; Kim, K.I.; Hoan, N.N.; Kim, C.H.; Moon, E.; Choi, K.S.; Yang, S.S.; Lee, J.S. Targeting cancer cells with reactive oxygen and nitrogen species generated by atmospheric-pressure air plasma. PLoS ONE 2014, 9, e86173. [Google Scholar] [CrossRef] [PubMed]
- Joh, H.M.; Kim, S.J.; Chung, T.H.; Leem, S.H. Comparison of the characteristics of atmospheric pressure plasma jets using different working gases and applications to plasma-cancer cell interactions. AIP Adv. 2013, 3, 092128. [Google Scholar] [CrossRef]
- Lou, B.-S.; Hsieh, J.-H.; Chen, C.-M.; Hou, C.-W.; Wu, H.-Y.; Chou, P.-Y.; Lai, C.-H.; Lee, J.-W. Helium/Argon-Generated Cold Atmospheric Plasma Facilitates Cutaneous Wound Healing. Front. Bioeng. Biotechnol. 2020, 8, 683. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.U.; Choi, J.W.; Chang, J.W.; Kim, K.i.; Kim, Y.S.; Park, J.K.; Kim, Y.E.; Lee, Y.S.; Yang, S.S.; Kim, C.-H. N2 non-thermal atmospheric pressure plasma promotes wound healing in vitro and in vivo: Potential modulation of adhesion molecules and matrix metalloproteinase-9. Exp. Dermatol. 2017, 26, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.M.; Xu, G.M.; Zhang, G.J.; Liu, J.R.; Wu, Y.M.; Gao, L.G.; Yang, Y.; Chang, Z.S.; Yao, C.W. Low-temperature Plasma Promotes Fibroblast Proliferation in Wound Healing by ROS-activated NF-κB Signaling Pathway. Curr. Med. Sci. 2018, 38, 107–114. [Google Scholar] [CrossRef]
- Isbary, G.; Morfill, G.; Schmidt, H.U.; Georgi, M.; Ramrath, K.; Heinlin, J.; Karrer, S.; Landthaler, M.; Shimizu, T.; Steffes, B.; et al. A first prospective randomized controlled trial to decrease bacterial load using cold atmospheric argon plasma on chronic wounds in patients. Br. J. Dermatol. 2010, 163, 78–82. [Google Scholar] [CrossRef]
- Isbary, G.; Stolz, W.; Shimizu, T.; Monetti, R.; Bunk, W.; Schmidt, H.U.; Morfill, G.E.; Klämpfl, T.G.; Steffes, B.; Thomas, H.M.; et al. Cold atmospheric argon plasma treatment may accelerate wound healing in chronic wounds: Results of an open retrospective randomized controlled study in vivo. Clin. Plasma Med. 2013, 1, 25–30. [Google Scholar] [CrossRef]
- Laroussi, M.; Lu, X.; Keidar, M. Perspective: The physics, diagnostics, and applications of atmospheric pressure low temperature plasma sources used in plasma medicine. J. Appl. Phys. 2017, 122, 020901. [Google Scholar] [CrossRef]
- Lietz, A.M.; Kushner, M.J. Molecular admixtures and impurities in atmospheric pressure plasma jets. J. Appl. Phys. 2018, 124, 153303. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Harmful and Beneficial Role of ROS. Oxidative Med. Cell. Longev. 2016, 2022, 9873652. [Google Scholar] [CrossRef] [PubMed]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar] [CrossRef]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef]
- Braný, D.; Dvorská, D.; Halašová, E.; Škovierová, H. Cold Atmospheric Plasma: A Powerful Tool for Modern Medicine. Int. J. Mol. Sci. 2020, 21, 2932. [Google Scholar] [CrossRef] [PubMed]
- Boehm, D.; Bourke, P. Safety implications of plasma-induced effects in living cells—A review of in vitro and in vivo findings. Biol. Chem. 2019, 400, 3–17. [Google Scholar] [CrossRef]
- Thana, P.; Kuensaen, C.; Poramapijitwat, P.; Sarapirom, S.; Yu, L.; Boonyawan, D. A compact pulse-modulation air plasma jet for the inactivation of chronic wound bacteria: Bactericidal effects & host safety. Surf. Coat. Technol. 2020, 400, 126229. [Google Scholar] [CrossRef]
- Thana, P.; Wijaikhum, A.; Poramapijitwat, P.; Kuensaen, C.; Meerak, J.; Ngamjarurojana, A.; Sarapirom, S.; Boonyawan, D. A compact pulse-modulation cold air plasma jet for the inactivation of chronic wound bacteria: Development and characterization. Heliyon 2019, 5, e02455. [Google Scholar] [CrossRef]
- Herting, S.M.; Monroe, M.B.B.; Weems, A.C.; Briggs, S.T.; Fletcher, G.K.; Blair, S.E.; Hatch, C.J.; Maitland, D.J. In vitro cytocompatibility testing of oxidative degradation products. J. Bioact. Compat. Polym. 2021, 36, 197–211. [Google Scholar] [CrossRef]
- ISO 10993-5:2009; Biological Evaluation of Medical Devices-Part 5: Tests for In Vitro Cytotoxicity. International Organization for Standardization: Geneva, Switzerland, 2009.
- Guerrero-Preston, R.; Ogawa, T.; Uemura, M.; Shumulinsky, G.; Valle, B.L.; Pirini, F.; Ravi, R.; Sidransky, D.; Keidar, M.; Trink, B. Cold atmospheric plasma treatment selectively targets head and neck squamous cell carcinoma cells. Int. J. Mol. Med. 2014, 34, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Maya, N.I.; Padilla-Vaca, F.; Romero-González, O.E.; Rosales-Castillo, E.A.S.; Rangel-Serrano, Á.; Arias-Negrete, S.; Franco, B. Refinement of the Griess method for measuring nitrite in biological samples. J. Microbiol. Methods 2021, 187, 106260. [Google Scholar] [CrossRef] [PubMed]
- Howland, J.L. Methods in nitric oxide research. Biochem. Educ. 1997, 25, 184–185. [Google Scholar] [CrossRef]
- Jang, T.W.; Choi, J.S.; Park, J.H. Protective and inhibitory effects of acteoside from Abeliophyllum distichum Nakai against oxidative DNA damage. Mol. Med. Rep. 2020, 22, 2076–2084. [Google Scholar] [CrossRef] [PubMed]
- OECD. Test No. 471: Bacterial Reverse Mutation Test; OECD: Paris, France, 2020. [Google Scholar] [CrossRef]
- Patenall, B.L.; Hathaway, H.J.; Laabei, M.; Young, A.E.; Thet, N.T.; Jenkins, A.T.A.; Short, R.D.; Allinson, S.L. Assessment of mutations induced by cold atmospheric plasma jet treatment relative to known mutagens in Escherichia coli. Mutagenesis 2021, 36, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Mortelmans, K.; Zeiger, E. The Ames Salmonella/microsome mutagenicity assay. Mutat. Res. 2000, 455, 29–60. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.V.; Colus, I.M.; Silva, M.A.; Vilegas, W.; Varanda, E.A. Assessment of DNA damage by extracts and fractions of Strychnos pseudoquina, a Brazilian medicinal plant with antiulcerogenic activity. Food Chem. Toxicol. 2006, 44, 1585–1589. [Google Scholar] [CrossRef]
- Zarkovic, N. Roles and Functions of ROS and RNS in Cellular Physiology and Pathology. Cells 2020, 9, 767. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- ISO 10993-3:2014; Biological Evaluation of Medical Devices-Part 3: Tests for Genotoxicity, Carcinogenicity and Reproductive Toxicity. International Organization for Standardization: Geneva, Switzerland, 2003.
- Carter, M.; Shieh, J. Chapter 14—Cell Culture Techniques. In Guide to Research Techniques in Neuroscience, 2nd ed.; Carter, M., Shieh, J., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 295–310. [Google Scholar] [CrossRef]
- Laroussi, M. Effects of PAM on select normal and cancerous epithelial cells. Plasma Res. Express 2019, 1, 025010. [Google Scholar] [CrossRef]
- Lin, A.; Gorbanev, Y.; De Backer, J.; Van Loenhout, J.; Van Boxem, W.; Lemière, F.; Cos, P.; Dewilde, S.; Smits, E.; Bogaerts, A. Non-Thermal Plasma as a Unique Delivery System of Short-Lived Reactive Oxygen and Nitrogen Species for Immunogenic Cell Death in Melanoma Cells. Adv. Sci. 2019, 6, 1802062. [Google Scholar] [CrossRef] [PubMed]
- Brehmer, F.; Haenssle, H.A.; Daeschlein, G.; Ahmed, R.; Pfeiffer, S.; Görlitz, A.; Simon, D.; Schön, M.P.; Wandke, D.; Emmert, S. Alleviation of chronic venous leg ulcers with a hand-held dielectric barrier discharge plasma generator (PlasmaDerm® VU-2010): Results of a monocentric, two-armed, open, prospective, randomized and controlled trial (NCT01415622). J. Eur. Acad. Dermatol. Venereol. 2015, 29, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.M.; Shi, X.M.; Cai, J.F.; Chen, S.L.; Li, P.; Yao, C.W.; Chang, Z.S.; Zhang, G.J. Dual effects of atmospheric pressure plasma jet on skin wound healing of mice. Wound Repair. Regen. 2015, 23, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, B.; Costea, T.C.; Nolte, C.; Hiller, J.; Schmidt, J.; Reindel, J.; Masur, K.; Motz, W.; Timm, J.; Kerner, W.; et al. Effect of Cold Atmospheric Plasma Therapy vs Standard Therapy Placebo on Wound Healing in Patients With Diabetic Foot Ulcers: A Randomized Clinical Trial. JAMA Netw. Open 2020, 3, e2010411. [Google Scholar] [CrossRef] [PubMed]
- Kluge, S.; Bekeschus, S.; Bender, C.; Benkhai, H.; Sckell, A.; Below, H.; Stope, M.B.; Kramer, A. Investigating the Mutagenicity of a Cold Argon-Plasma Jet in an HET-MN Model. PLoS ONE 2016, 11, e0160667. [Google Scholar] [CrossRef] [PubMed]
- Wende, K.; Bekeschus, S.; Schmidt, A.; Jatsch, L.; Hasse, S.; Weltmann, K.D.; Masur, K.; von Woedtke, T. Risk assessment of a cold argon plasma jet in respect to its mutagenicity. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2016, 798–799, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, T.; Uehara, K.; Sasaki, Y.; Miyahara, H.; Matsumura, Y.; Iwasawa, A.; Ito, N.; Azuma, T.; Kohno, M.; Okino, A. Investigation of reactive species using various gas plasmas. RSC Adv. 2014, 4, 39901–39905. [Google Scholar] [CrossRef]
- Dobrynin, D.; Fridman, G.; Friedman, G.; Fridman, A. Physical and biological mechanisms of direct plasma interaction with living tissue. New J. Phys. 2009, 11, 115020. [Google Scholar] [CrossRef]
- Straßenburg, S.; Greim, U.; Bussiahn, R.; Haertel, B.; Wende, K.; von Woedtke, T.; Lindequist, U. Comparison of Biological Effects on Human Keratinocytes Using Different Plasma Treatment Regimes. Plasma Med. 2013, 3, 57–69. [Google Scholar] [CrossRef]
- Lunov, O.; Zablotskii, V.; Churpita, O.; Chánová, E.; Syková, E.; Dejneka, A.; Kubinová, Š. Cell death induced by ozone and various non-thermal plasmas: Therapeutic perspectives and limitations. Sci. Rep. 2014, 4, 7129. [Google Scholar] [CrossRef]
- Kim, K.C.; Piao, M.J.; Madduma Hewage, S.R.; Han, X.; Kang, K.A.; Jo, J.O.; Mok, Y.S.; Shin, J.H.; Park, Y.; Yoo, S.J.; et al. Non-thermal dielectric-barrier discharge plasma damages human keratinocytes by inducing oxidative stress. Int. J. Mol. Med. 2016, 37, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Wende, K.; Straßenburg, S.; Haertel, B.; Harms, M.; Holtz, S.; Barton, A.; Masur, K.; von Woedtke, T.; Lindequist, U. Atmospheric pressure plasma jet treatment evokes transient oxidative stress in HaCaT keratinocytes and influences cell physiology. Cell Biol. Int. 2014, 38, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Heinlin, J.; Isbary, G.; Stolz, W.; Zeman, F.; Landthaler, M.; Morfill, G.; Shimizu, T.; Zimmermann, J.L.; Karrer, S. A randomized two-sided placebo-controlled study on the efficacy and safety of atmospheric non-thermal argon plasma for pruritus. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 324–331. [Google Scholar] [CrossRef]
- Ulrich, C.; Kluschke, F.; Patzelt, A.; Vandersee, S.; Czaika, V.A.; Richter, H.; Bob, A.; Hutten, J.; Painsi, C.; Hüge, R.; et al. Clinical use of cold atmospheric pressure argon plasma in chronic leg ulcers: A pilot study. J. Wound Care 2015, 24, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Heinlin, J.; Zimmermann, J.L.; Zeman, F.; Bunk, W.; Isbary, G.; Landthaler, M.; Maisch, T.; Monetti, R.; Morfill, G.; Shimizu, T.; et al. Randomized placebo-controlled human pilot study of cold atmospheric argon plasma on skin graft donor sites. Wound Repair. Regen. 2013, 21, 800–807. [Google Scholar] [CrossRef]
- Shekhter, A.B.; Serezhenkov, V.A.; Rudenko, T.G.; Pekshev, A.V.; Vanin, A.F. Beneficial effect of gaseous nitric oxide on the healing of skin wounds. Nitric Oxide 2005, 12, 210–219. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (OH/O−) in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Schmidt, A.; Dietrich, S.; Steuer, A.; Weltmann, K.D.; von Woedtke, T.; Masur, K.; Wende, K. Non-thermal plasma activates human keratinocytes by stimulation of antioxidant and phase II pathways. J. Biol. Chem. 2015, 290, 6731–6750. [Google Scholar] [CrossRef]
- Martinez, M.C.; Andriantsitohaina, R. Reactive Nitrogen Species: Molecular Mechanisms and Potential Significance in Health and Disease. Antioxid. Redox Signal. 2009, 11, 669–702. [Google Scholar] [CrossRef] [PubMed]
- Milsom, A.B.; Jones, C.J.; Goodfellow, J.; Frenneaux, M.P.; Peters, J.R.; James, P.E. Abnormal metabolic fate of nitric oxide in Type I diabetes mellitus. Diabetologia 2002, 45, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Steuer, A.; Wolff, C.M.; von Woedtke, T.; Weltmann, K.-D.; Kolb, J.F. Cell stimulation versus cell death induced by sequential treatments with pulsed electric fields and cold atmospheric pressure plasma. PLoS ONE 2018, 13, e0204916. [Google Scholar] [CrossRef] [PubMed]
- Gracanin, M.; Lam, M.A.; Morgan, P.E.; Rodgers, K.J.; Hawkins, C.L.; Davies, M.J. Amino acid, peptide, and protein hydroperoxides and their decomposition products modify the activity of the 26S proteasome. Free Radic. Biol. Med. 2011, 50, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Möller, M.N.; Cuevasanta, E.; Orrico, F.; Lopez, A.C.; Thomson, L.; Denicola, A. Diffusion and Transport of Reactive Species Across Cell Membranes. Adv. Exp. Med. Biol. 2019, 1127, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Sawa, T.; Ohshima, H. Nitrative DNA damage in inflammation and its possible role in carcinogenesis. Nitric Oxide 2006, 14, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Dellino, G.I.; Gambino, V.; Roda, N.; Pelicci, P.G. On the epigenetic role of guanosine oxidation. Redox Biol. 2020, 29, 101398. [Google Scholar] [CrossRef] [PubMed]
- Jena, N.R.; Mishra, P.C. Formation of 8-nitroguanine and 8-oxoguanine due to reactions of peroxynitrite with guanine. J. Comput. Chem. 2007, 28, 1321–1335. [Google Scholar] [CrossRef]
- Kurita, H.; Haruta, N.; Uchihashi, Y.; Seto, T.; Takashima, K. Strand breaks and chemical modification of intracellular DNA induced by cold atmospheric pressure plasma irradiation. PLoS ONE 2020, 15, e0232724. [Google Scholar] [CrossRef]
- Kim, K.C.; Ruwan Kumara, M.H.S.; Kang, K.A.; Piao, M.J.; Oh, M.C.; Ryu, Y.S.; Jo, J.O.; Mok, Y.S.; Shin, J.H.; Park, Y.; et al. Exposure of keratinocytes to non-thermal dielectric barrier discharge plasma increases the level of 8-oxoguanine via inhibition of its repair enzyme. Mol. Med. Rep. 2017, 16, 6870–6875. [Google Scholar] [CrossRef]
- Kang, K.A.; Piao, M.J.; Eom, S.; Yoon, S.-Y.; Ryu, S.; Kim, S.B.; Yi, J.M.; Hyun, J.W. Non-thermal dielectric-barrier discharge plasma induces reactive oxygen species by epigenetically modifying the expression of NADPH oxidase family genes in keratinocytes. Redox Biol. 2020, 37, 101698. [Google Scholar] [CrossRef]
- Loo, D.T. TUNEL Assay. In In Situ Detection of DNA Damage. Methods in Molecular Biology; Didenko, V.V., Ed.; Humana Press: Totowa, NJ, USA, 2002; Volume 203. [Google Scholar] [CrossRef]
- Hornsby, P.J.; Didenko, V.V. In situ ligation: A decade and a half of experience. Methods Mol. Biol. 2011, 682, 49–63. [Google Scholar] [CrossRef]
- Burney, S.; Caulfield, J.L.; Niles, J.C.; Wishnok, J.S.; Tannenbaum, S.R. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat. Res. 1999, 424, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Boxhammer, V.; Li, Y.F.; Köritzer, J.; Shimizu, T.; Maisch, T.; Thomas, H.M.; Schlegel, J.; Morfill, G.E.; Zimmermann, J.L. Investigation of the mutagenic potential of cold atmospheric plasma at bactericidal dosages. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2013, 753, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef]
- Alkawareek, M.Y.; Alshraiedeh, N.a.H.; Higginbotham, S.; Flynn, P.B.; Algwari, Q.T.; Gorman, S.P.; Graham, W.G.; Gilmore, B.F. Plasmid DNA Damage Following Exposure to Atmospheric Pressure Nonthermal Plasma: Kinetics and Influence of Oxygen Admixture. Plasma Med. 2014, 4, 211–219. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, C.; Zhou, Q.Q.; Zhang, X.F.; Wang, L.Y.; Chang, H.B.; Li, H.P.; Oda, Y.; Xing, X.H. Quantitative evaluation of DNA damage and mutation rate by atmospheric and room-temperature plasma (ARTP) and conventional mutagenesis. Appl. Microbiol. Biotechnol. 2015, 99, 5639–5646. [Google Scholar] [CrossRef] [PubMed]
- Blackert, S.; Haertel, B.; Wende, K.; von Woedtke, T.; Lindequist, U. Influence of non-thermal atmospheric pressure plasma on cellular structures and processes in human keratinocytes (HaCaT). J. Dermatol. Sci. 2013, 70, 173–181. [Google Scholar] [CrossRef]
- Estarabadi, H.; Atyabi, S.A.; Tavakkoli, S.; Noormohammadi, Z.; Gholami, M.R.; Ghiaseddin, A.; Irani, S. Cold atmospheric plasma induced genotoxicity and cytotoxicity in esophageal cancer cells. Mol. Biol. Rep. 2021, 48, 1323–1333. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | His+ Revertant Colonies | ||
|---|---|---|---|
| Strain TA98 | Strain TA100 | ||
| Phosphate buffer | 20.7 ± 4.3 | 88.6 ± 3.9 | |
| 2-(2-furyl)-3-(5-nitro-2-furyl)-acrylamide (AF-2) | 246.1 ± 44.4 *** | 511.8 ± 53.3 *** | |
| Air flow 3 L/min | Intensity of 0 pulses | 18.6 ± 3.4 | 86.9 ± 0.1 |
| Intensity of 4 pulses | 17.7 ± 2.5 | 80.8 ± 3.3 | |
| Intensity of 7 pulses | 21.2 ± 4.2 | 84.6 ± 9.6 | |
| Intensity of 10 pulses | 23.8 ± 5.1 | 94.7 ± 2.9 | |
| Air flow 5 L/min | Intensity of 0 pulses | 21.8 ± 4.5 | 84.7 ± 5.7 |
| Intensity of 4 pulses | 20.6 ± 3.9 | 91.4 ± 3.1 | |
| Intensity of 7 pulses | 24.0 ± 4.2 | 92.2 ± 4.1 | |
| Intensity of 10 pulses | 20.8 ± 4.5 | 104.7 ± 4.4 *,# | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yarangsee, P.; Khacha-ananda, S.; Pitchakarn, P.; Intayoung, U.; Sriuan, S.; Karinchai, J.; Wijaikhum, A.; Boonyawan, D. A Nonclinical Safety Evaluation of Cold Atmospheric Plasma for Medical Applications: The Role of Genotoxicity and Mutagenicity Studies. Life 2024, 14, 759. https://doi.org/10.3390/life14060759
Yarangsee P, Khacha-ananda S, Pitchakarn P, Intayoung U, Sriuan S, Karinchai J, Wijaikhum A, Boonyawan D. A Nonclinical Safety Evaluation of Cold Atmospheric Plasma for Medical Applications: The Role of Genotoxicity and Mutagenicity Studies. Life. 2024; 14(6):759. https://doi.org/10.3390/life14060759
Chicago/Turabian StyleYarangsee, Piimwara, Supakit Khacha-ananda, Pornsiri Pitchakarn, Unchisa Intayoung, Sirikhwan Sriuan, Jirarat Karinchai, Apiwat Wijaikhum, and Dheerawan Boonyawan. 2024. "A Nonclinical Safety Evaluation of Cold Atmospheric Plasma for Medical Applications: The Role of Genotoxicity and Mutagenicity Studies" Life 14, no. 6: 759. https://doi.org/10.3390/life14060759
APA StyleYarangsee, P., Khacha-ananda, S., Pitchakarn, P., Intayoung, U., Sriuan, S., Karinchai, J., Wijaikhum, A., & Boonyawan, D. (2024). A Nonclinical Safety Evaluation of Cold Atmospheric Plasma for Medical Applications: The Role of Genotoxicity and Mutagenicity Studies. Life, 14(6), 759. https://doi.org/10.3390/life14060759