
Genetic Diversity and Population Structure of the Endangered Salamander Hynobius yiwuensis Inferred from Mitochondrial DNA Sequences
Abstract
:1. Introduction
2. Materials and Methods
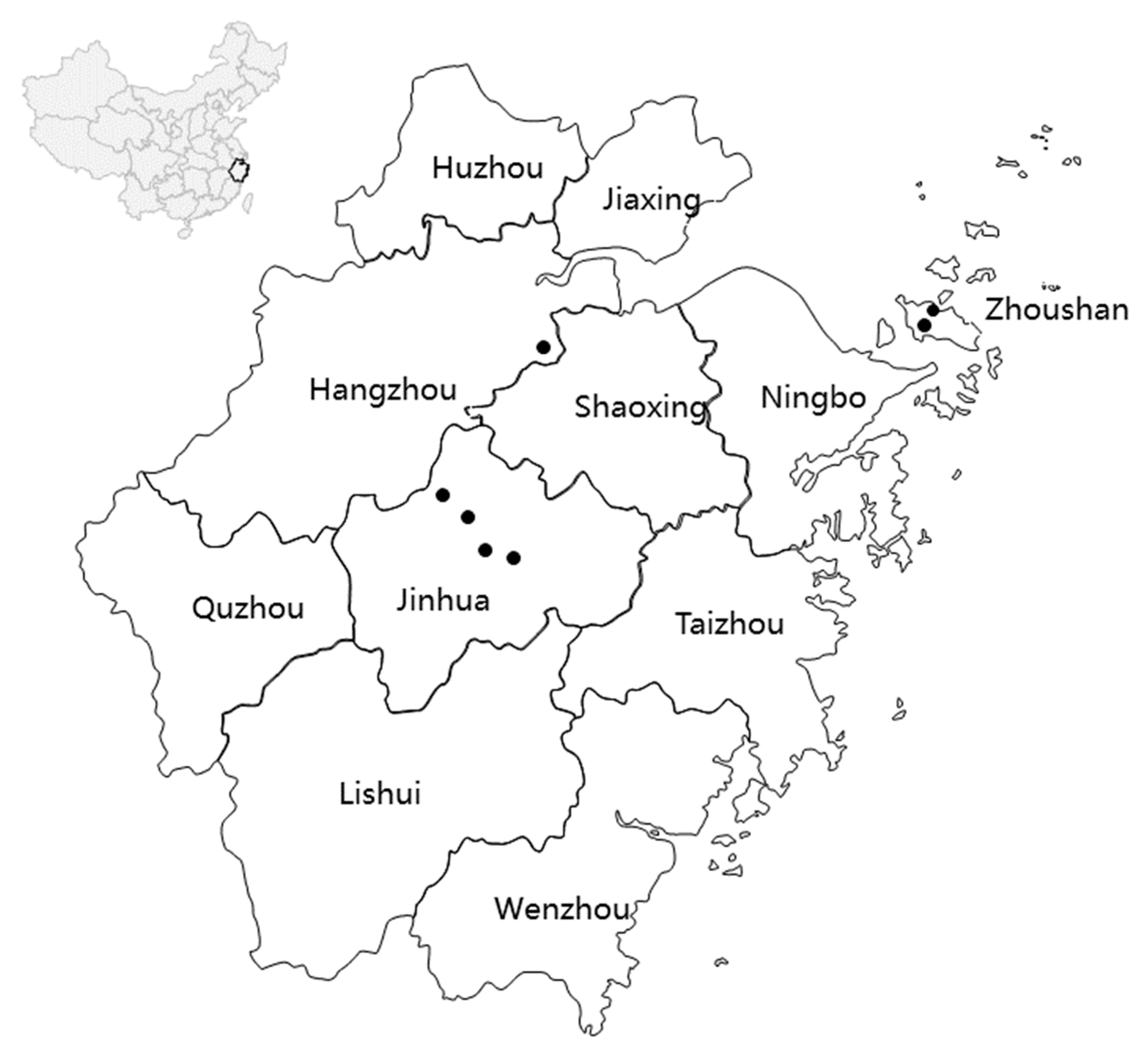
2.1. Sample Collection and DNA Extraction
2.2. mtDNA Sequencing and Analysis
3. Results
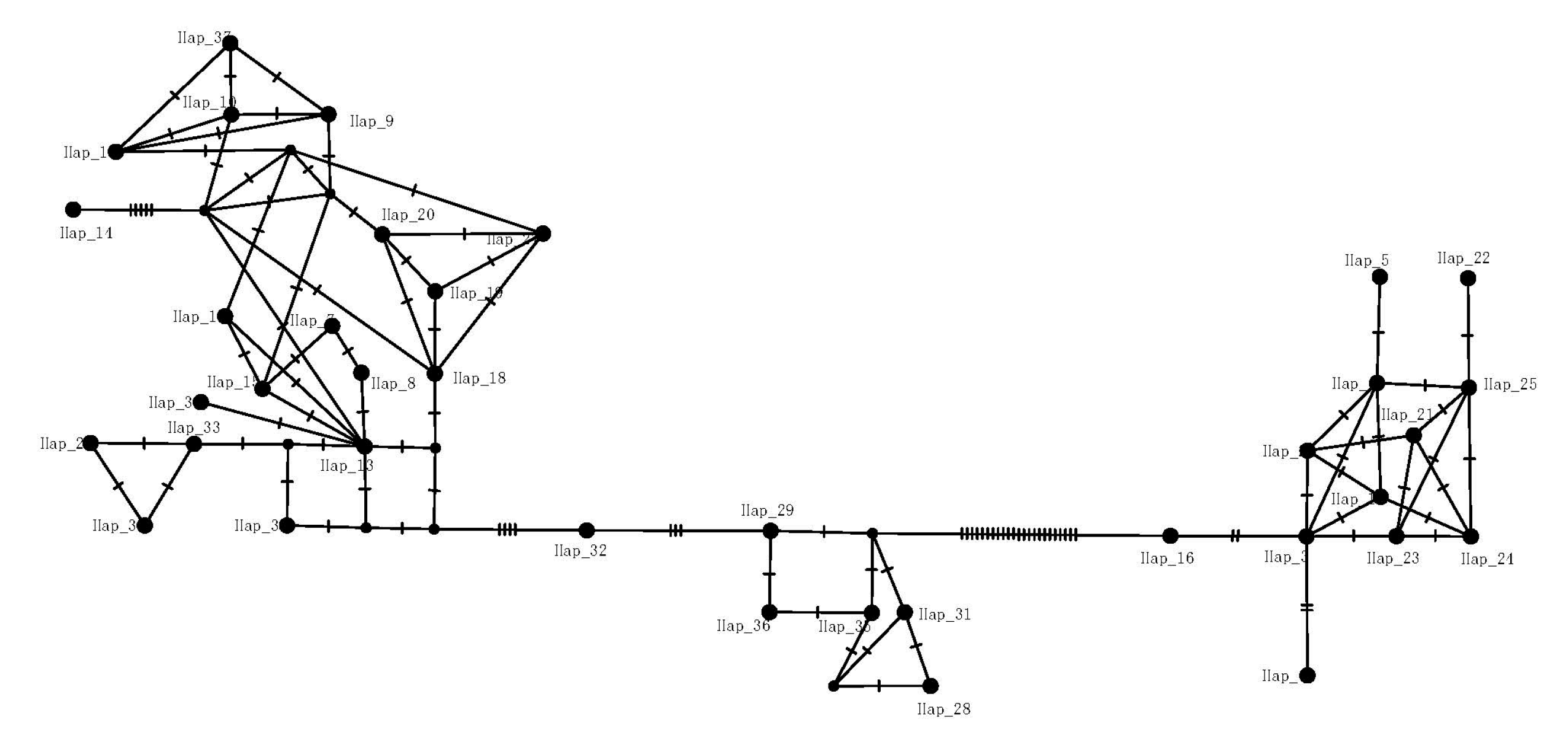
3.1. Population Genetic Diversity and Differentiation of mtDNA
3.2. Population Genetic Diversity and Differentiation of mtDNA
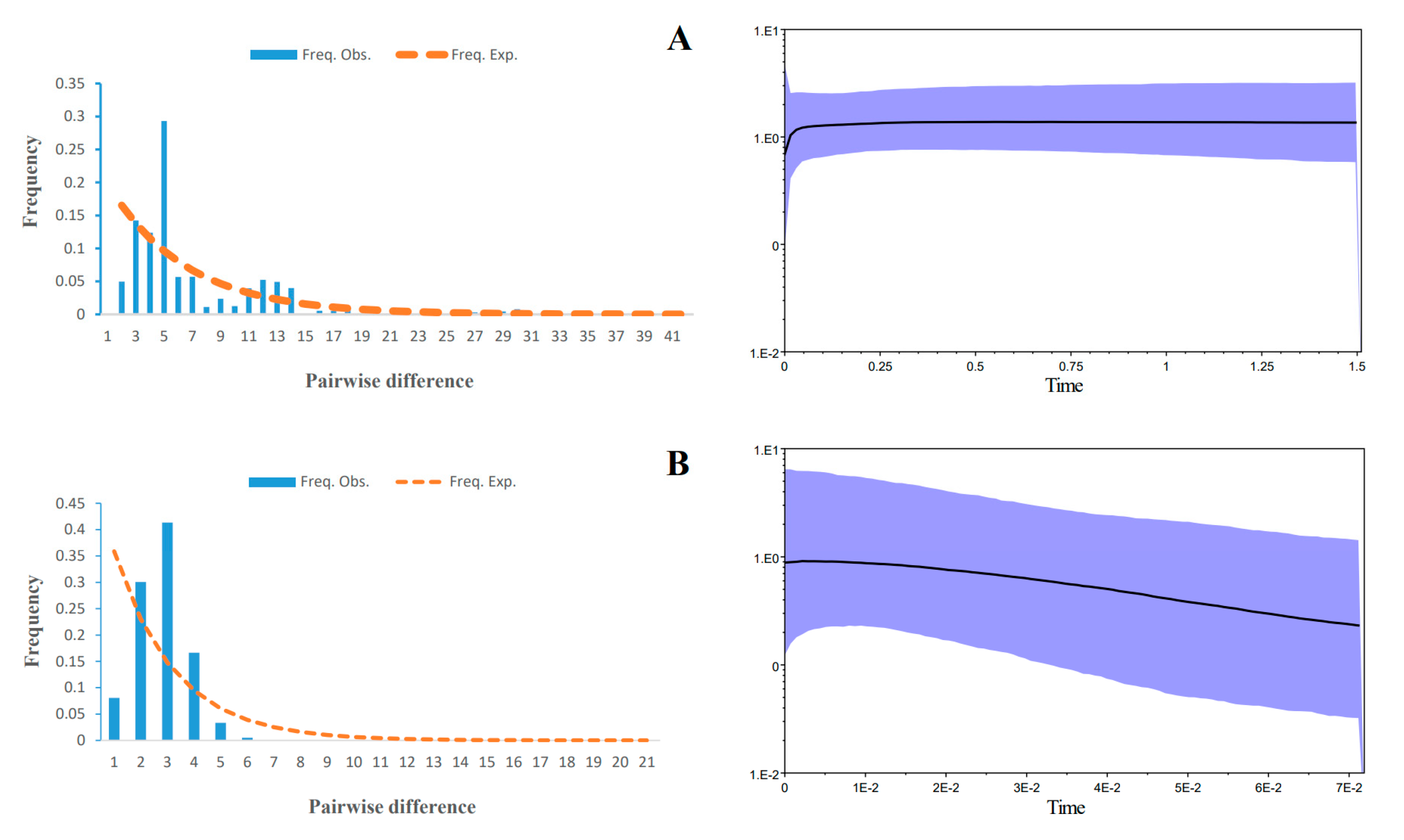
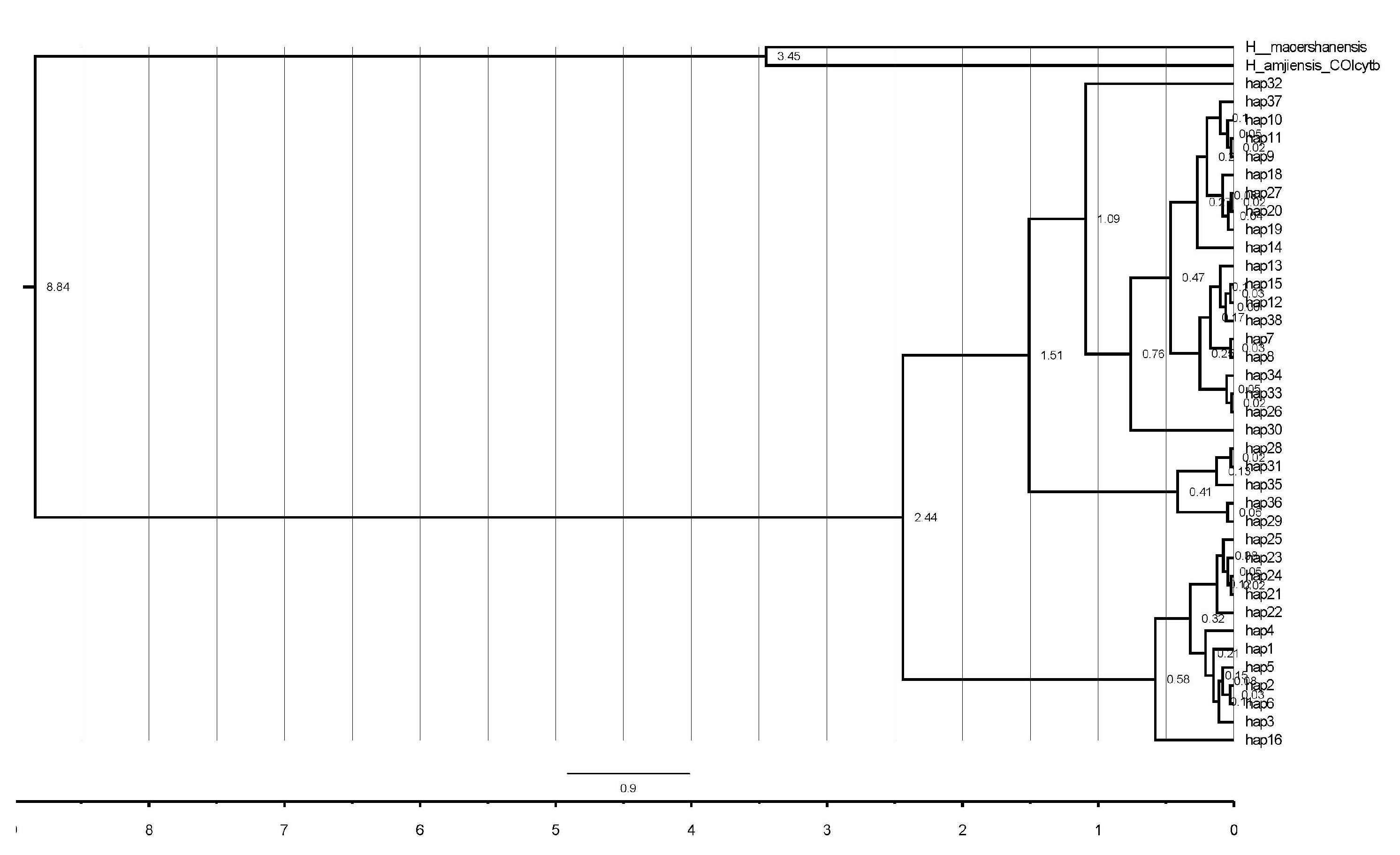
3.3. Demographic History and Divergence Time
4. Discussion
4.1. Genetic Diversity
4.2. Phylogenetic Inference and Genetic Structure
4.3. Phylogeographic Inference
4.4. Conservation Implications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mi, C.; Huettmann, F.; Li, X.; Jiang, Z.; Du, W.; Sun, B. Effects of climate and human activity on the current distribution of amphibians in China. Conserv. Biol. 2022, 36, e13964. [Google Scholar] [CrossRef] [PubMed]
- Dodd, C.K.; Smith, L.L. Habitat destruction and alteration: Historical trends and future prospects for amphibians. In Amphibian Conservation; Semlitsch, R.D., Ed.; Smithsonian Institution Press: Washington, DC, USA, 2003; pp. 94–112. [Google Scholar]
- Fei, L.; Ye, C.; Jiang, J. Colored Atlas of Chinese Amphibians and Their Distributions; Sichuan Publishing House of Science and Technology: Chengdu, China, 2012; pp. 1–620. [Google Scholar]
- IUCN SSC Amphibian Specialist Group. Hynobius yiwuensis. The IUCN Red List of Threatened Species. 2020, p. e.T59106A63858761. Available online: https://doi.org/10.2305/IUCN.UK.2020-2.RLTS.T59106A63858761.en (accessed on 1 January 2024).
- Max, S. Salamanders of the Old World: The Salamanders of Europe, Asia and Northern Africa; Brill: Leiden, The Netherlands, 2014. [Google Scholar]
- Shan, S.; Kan, X.; Gong, J.; Shao, C.; Wang, Y. Development and characterization of 36 SNP markers for Hynobius yiwuensis. Conserv. Genet. Resour. 2022, 14, 15–18. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, J.; Wang, Y.; Zhang, E.; Zhang, Y.; Li, L.; Xie, F.; Cai, B.; Cao, L.; Zheng, G. Red list of China’s vertebrates. Biodivers. Sci. 2016, 24, 500. [Google Scholar]
- Fei, X. Hynobius yiwuensis Population Resources Investigation, Embryonic Development and Genetic Diversity Research. Master’s Thesis, Zhejiang Normal University, Jinhua, China, 2020. [Google Scholar]
- Ikebe, C.; Gu, H.; Ruan, R.-S.; Kohno, S.-I. Chromosomes of Hynobius chinensis Günther and Hynobius amjiensis Gu from China, and Comparison with Those of 19 Other Hynobius Species. Zool. Sci. 1998, 15, 981–987. [Google Scholar] [CrossRef]
- Li, S. Cytotaxonomy of Amphibian in China; Science Press: Beijing, China, 2007. [Google Scholar]
- Chen, Q.; Qing, L.; Zeng, X. Review of systematic study on Hynobius. Sichuan J. Zool. 2008, 27, 468–477. [Google Scholar]
- Fu, J.; Hayes, M.; Liu, Z.; Zeng, X. Genetic divergence of the southeastern Chinese salamanders of the genus Hynobius. Acta Zool. Sin. 2003, 49, 585–591. [Google Scholar]
- Liu, P. Analysis of the Genetic Diversity of Lesser Salamander in Zhoushan Area. Master’s Thesis, Zhejiang Normal University, Jinhua, China, 2017. [Google Scholar]
- Paz-Vinas, I.; Loot, G.; Hermoso, V.; Veyssiere, C.; Poulet, N.; Grenouillet, G.; Blanchet, S. Systematic conservation planning for intraspecific genetic diversity. Proc. R. Soc. 2018, 285, 20172746. [Google Scholar] [CrossRef] [PubMed]
- Sudhir, K.; Glen, S.; Koichiro, T. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar]
- Julio, R.; Albert, F.; Juan, C.S.; Sara, G.; Pablo, L.; Sebastián, E.R.; Alejandro, S. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Jessica, W.L.; David, B. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Natsuhiko, Y.; Masafumi, M.; Kanto, N.; Jong-Bum, K.; Alexei, K. Phylogenetic relationships and biogeography of the Japanese clawed salamander, Onychodactylus japonicus (Amphibia: Caudata: Hynobiidae), and its congener inferred from the mitochondrial cytochrome b gene. Mol. Phylogenet. Evol. 2008, 49, 249–259. [Google Scholar]
- Andrew, R.; Alexei, J.D.; Dong, X.; Guy, B.; Marc, A.S. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar]
- Sunny, A.; Duarte-deJesus, L.; Aguilera-Hernández, A.; Ramírez-Corona, F.; Suárez-Atilano, M.; Percino-Daniel, R.; Manjarrez, J.; Monroy-Vilchis, O.; González-Fernández, A. Genetic diversity and demography of the critically endangered Roberts’ false brook salamander (Pseudoeurycea robertsi) in Central Mexico. Genetica 2019, 147, 149–164. [Google Scholar] [CrossRef]
- Salehi, T.; Akmali, V.; Sharifi, M. Population genetic structure of the endangered yellow spotted mountain newt (Neurergus derjugini: Amphibia, Caudata) inferred from mitochondrial DNA sequences. Herpetol. J. 2019, 29, 37. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, Y.; Li, X. Molecular phylogeography and population genetic structure of an endangered species Pachyhynobius shangchengensis (hynobiid Salamander) in a fragmented habitat of southeastern China. PLoS ONE 2013, 8, e78064. [Google Scholar] [CrossRef]
- Yang, J.; Chen, C.; Chen, S.; Ding, P.; Fan, Z.; Lu, Y.; Yu, L.; Lin, H.J.G.M.R. Population genetic structure of critically endangered salamander (Hynobius amjiensis) in China: Recommendations for conservation. Genet. Mol. Res. 2016, 15, 15027733. [Google Scholar] [CrossRef]
- Suk, H.Y.; Bae, H.-G.; Kim, D.-Y.; Won, H.; Baek, H.J.; Lee, C.H.; Kim, D.Y.; Go, Y.M.; Song, J.-Y.; Lee, H.J.G.; et al. Genetic and phylogenetic structure of Hynobius quelpaertensis, an endangered endemic salamander species on the Korean Peninsula. Genes Genom. 2020, 42, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R. Do island populations have less genetic variation than mainland populations? Heredity 1997, 78, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Gu, H. Studies on distribution and population number of Hynobius chinensis on the Zhoushan island. Sichuan J. Zool. 1999, 18, 107–108. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locality | Site | N | nHT | Hd | π |
|---|---|---|---|---|---|
| LM | 119°51′ E, 29°19′ N | 14 | 6 | 0.736 | 0.0055 |
| WD | 119°55′ E, 29°18′ N | 21 | 13 | 0.933 | 0.0046 |
| LH | 119°46′ E, 29°40′ N | 20 | 8 | 0.884 | 0.0023 |
| ZS | 119°79′ E, 29°24′ N | 18 | 9 | 0.908 | 0.0035 |
| HT | 120°18′ E, 29°57′ N | 3 | 2 | 0.667 | 0.0011 |
| Mainland | 76 | 27 | 0.950 | 0.0043 | |
| GH | 122°05′ E, 30°06′ N | 13 | 8 | 0.910 | 0.0014 |
| MH | 122°06′ E, 30°08′ N | 22 | 9 | 0.879 | 0.0012 |
| Island | 35 | 15 | 0.919 | 0.0015 | |
| Total | 111 | 37 | 0.965 | 0.0130 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Sechi, P.; Xie, J.; Dong, R.; Zheng, R. Genetic Diversity and Population Structure of the Endangered Salamander Hynobius yiwuensis Inferred from Mitochondrial DNA Sequences. Life 2024, 14, 769. https://doi.org/10.3390/life14060769
Zhang L, Sechi P, Xie J, Dong R, Zheng R. Genetic Diversity and Population Structure of the Endangered Salamander Hynobius yiwuensis Inferred from Mitochondrial DNA Sequences. Life. 2024; 14(6):769. https://doi.org/10.3390/life14060769
Chicago/Turabian StyleZhang, Liangliang, Pierfrancesco Sechi, Jingbei Xie, Rui Dong, and Rongquan Zheng. 2024. "Genetic Diversity and Population Structure of the Endangered Salamander Hynobius yiwuensis Inferred from Mitochondrial DNA Sequences" Life 14, no. 6: 769. https://doi.org/10.3390/life14060769