
Selected Lark Mitochondrial Genomes Provide Insights into the Evolution of Second Control Region with Tandem Repeats in Alaudidae (Aves, Passeriformes)
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples Collection, DNA Extraction, and Data Download
2.2. PCR Amplification and Sequencing
2.3. Mitogenome Assembly, Annotation, and Mitogenome Analysis
2.4. Phylogenetic Analysis
3. Results
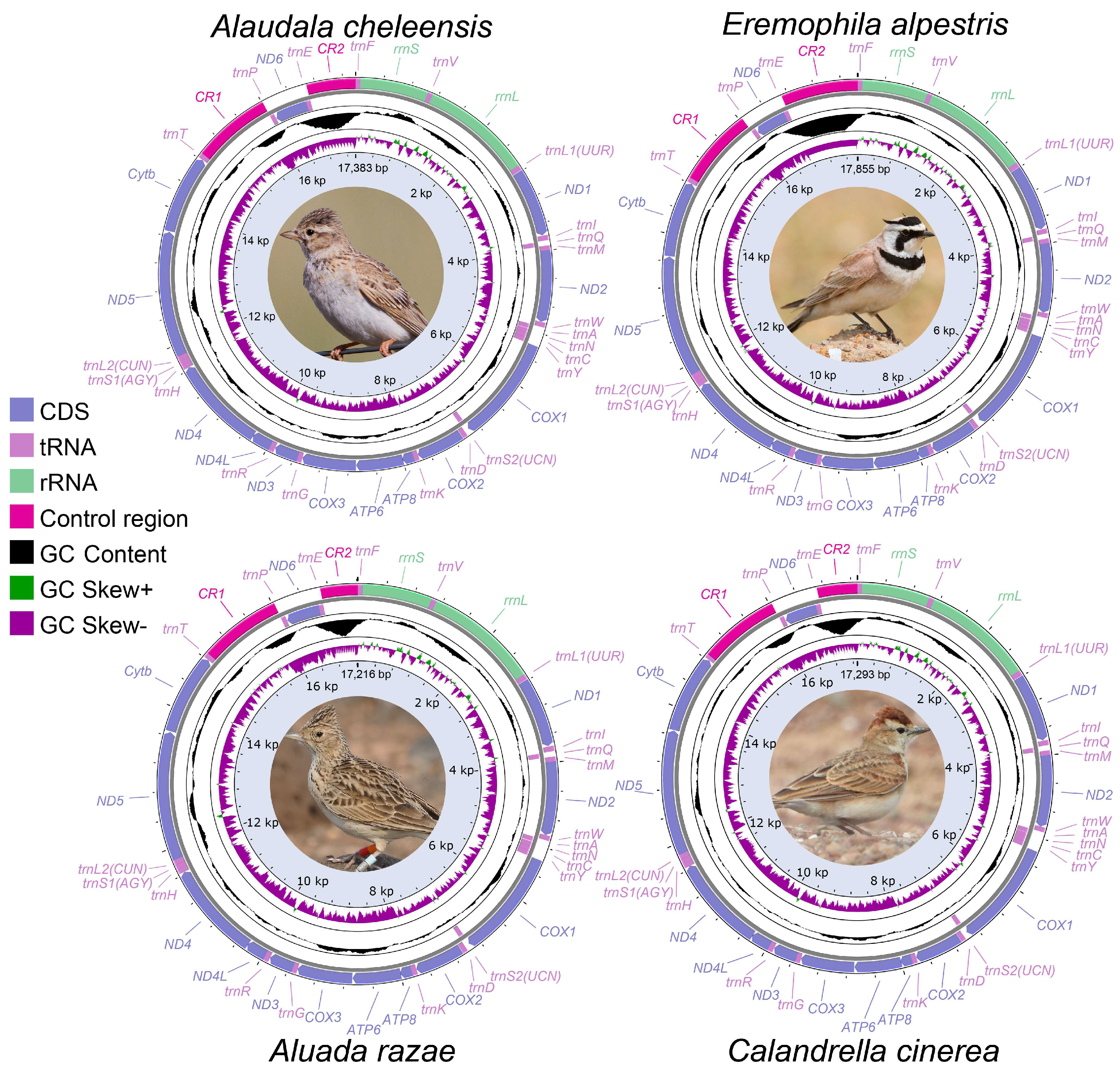
3.1. General Characteristics of Six Mitochondrial Genomes
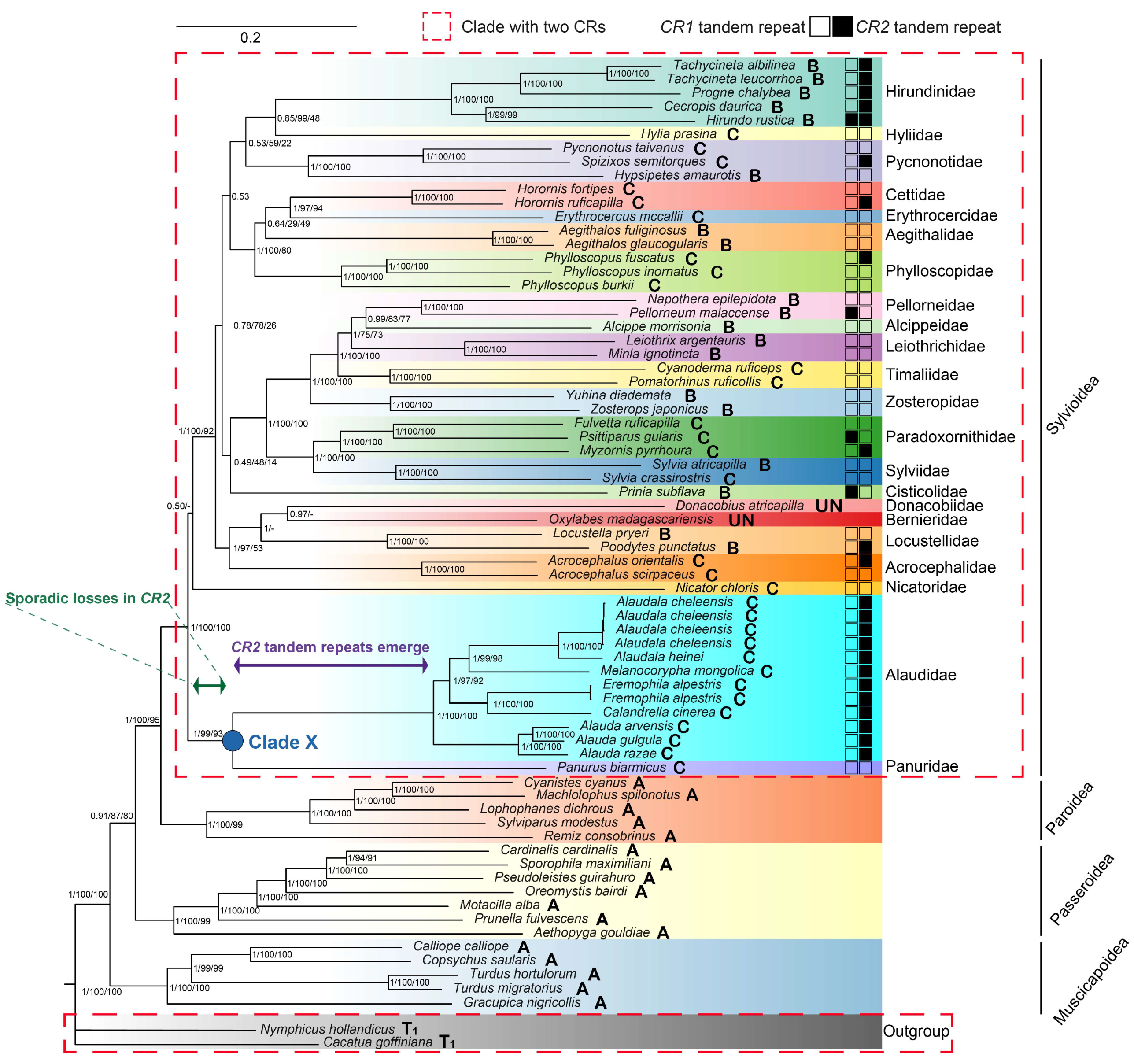
3.2. Phylogenetic Analyses
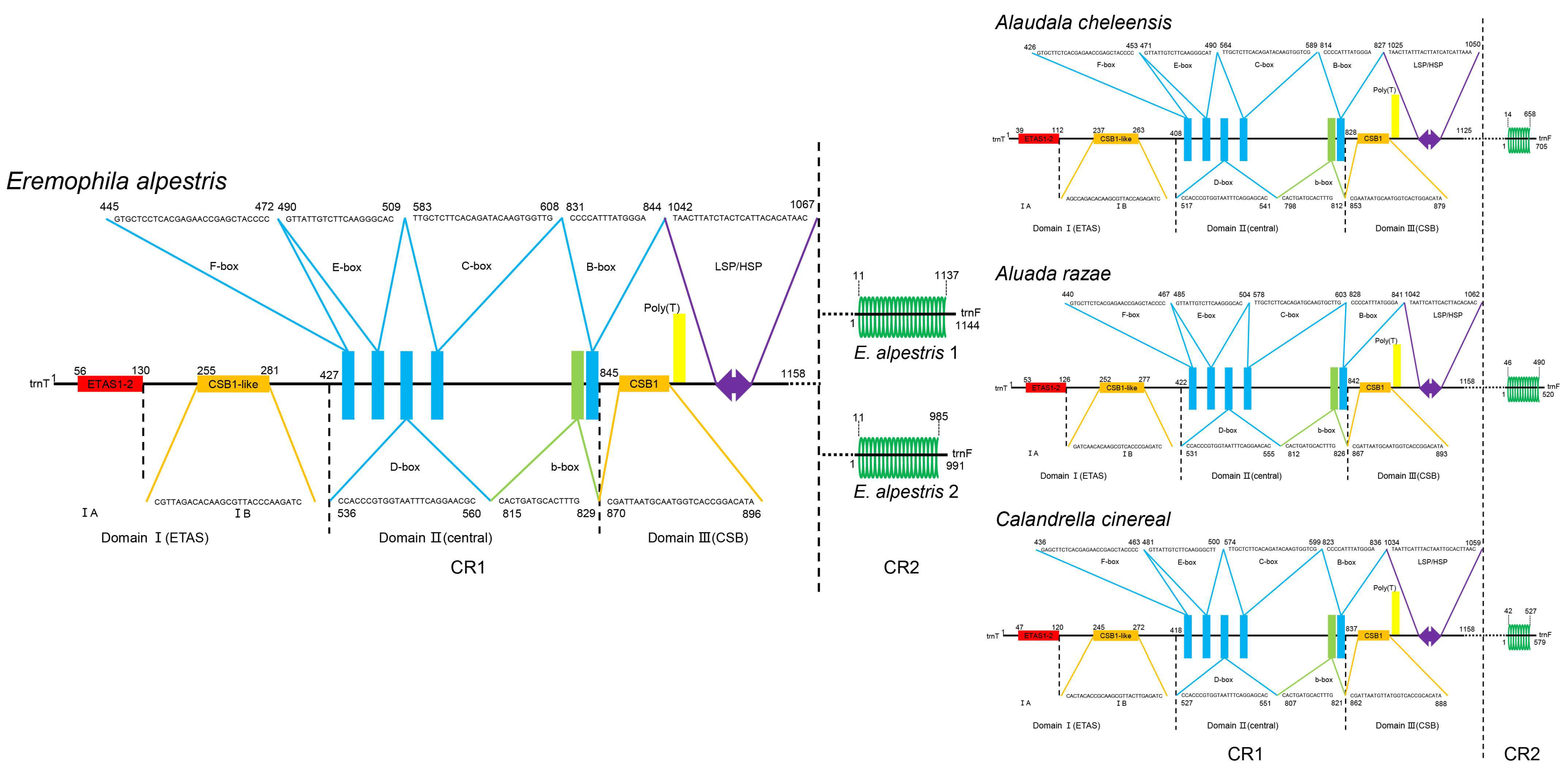
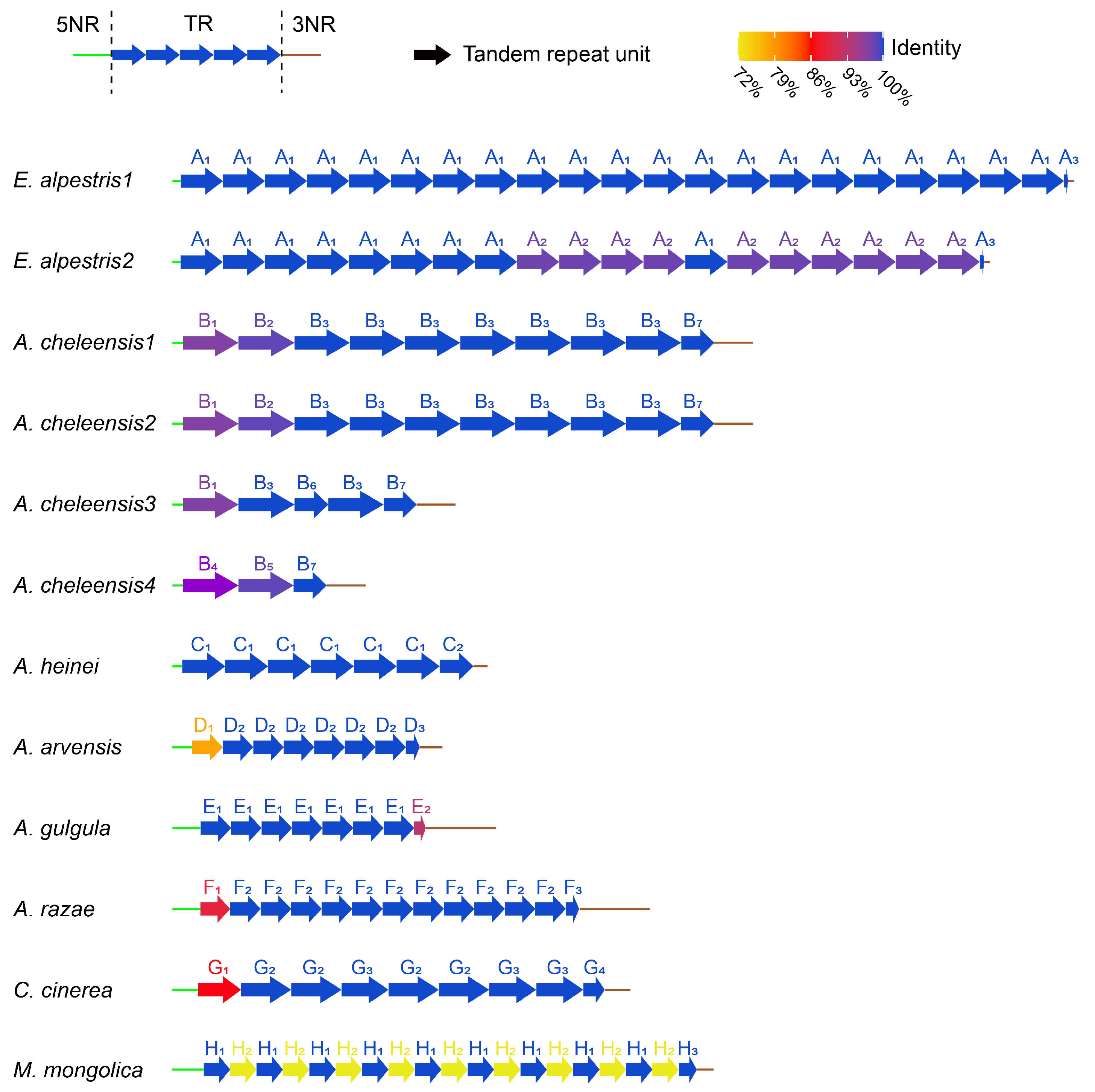
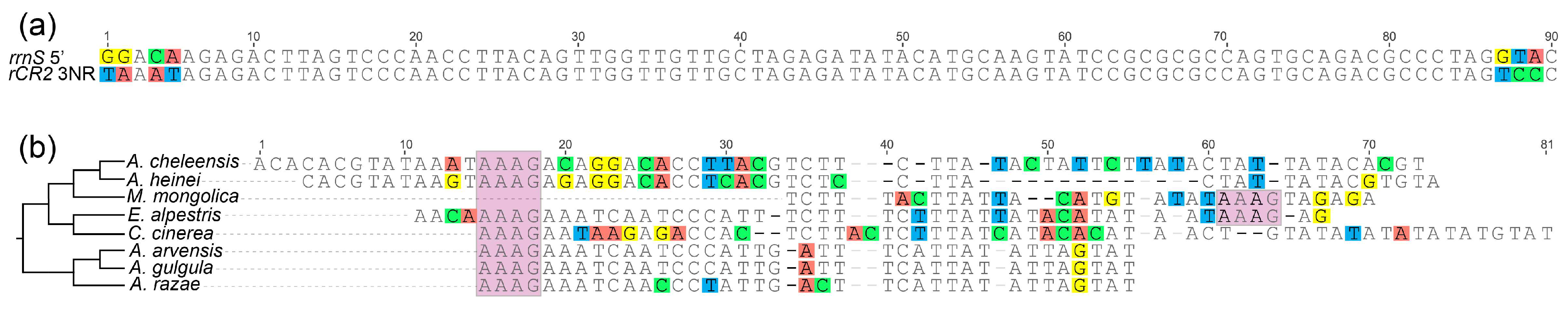
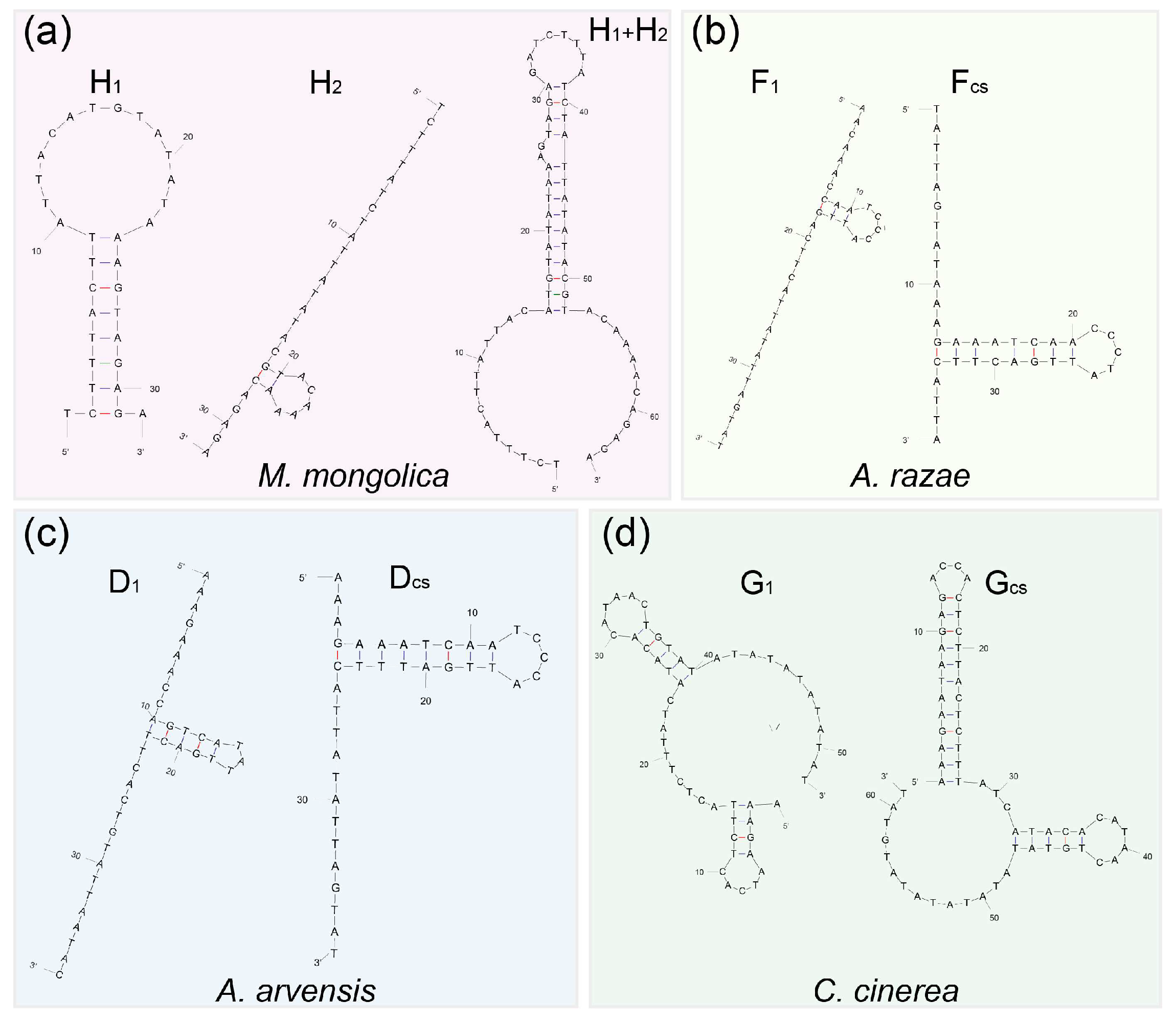
3.3. CR Analysis
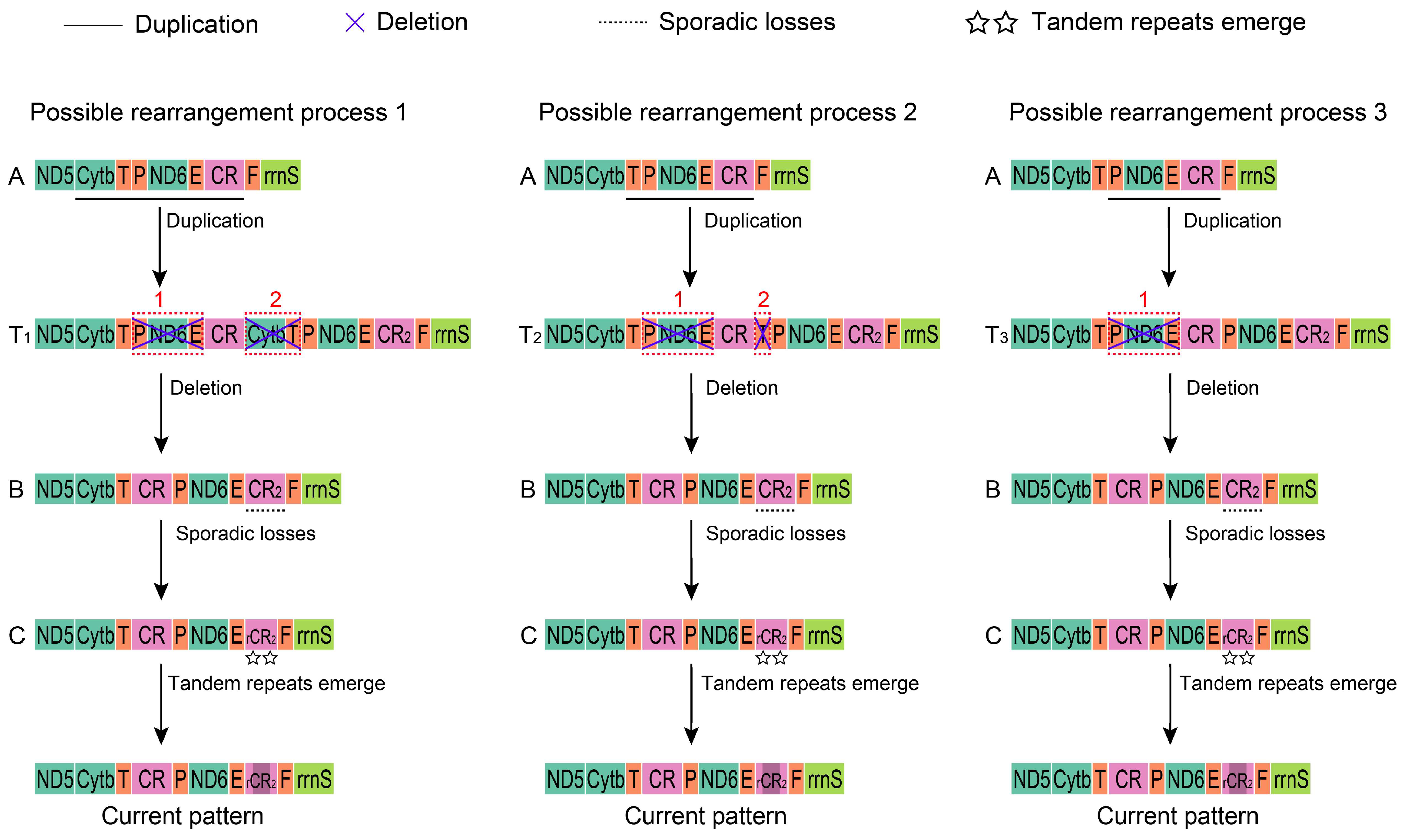
3.4. Gene Rearrangement
4. Discussion
4.1. Phylogenetic Relationships
4.2. Evolutionary Progression of Mt Gene Arrangement in Alaudidae
4.3. Evolutionary Progression of rCR2 in Alaudidae
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moore, W.S. Inferring phylogenies from mtDNA variation: Mitochondrial-gene trees versus nuclear-gene trees. Evol. Int. J. Org. Evol. 1995, 49, 718–726. [Google Scholar] [CrossRef]
- Eberhard, J.R.; Wright, T.F. Rearrangement and evolution of mitochondrial genomes in parrots. Mol. Phylogenet. Evol. 2016, 94, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Li, B.; Ma, X.; Xu, Y. Evolutionary progression of mitochondrial gene rearrangements and phylogenetic relationships in Strigidae (Strigiformes). Gene 2018, 674, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Lin, Q.; Fang, W.; Chen, X. The complete mitochondrial genomes of sixteen ardeid birds revealing the evolutionary process of the gene rearrangements. BMC Genom. 2014, 15, 573. [Google Scholar] [CrossRef] [PubMed]
- Urantowka, A.D.; Kroczak, A.; Strzala, T.; Zaniewicz, G.; Kurkowski, M.; Mackiewicz, P. Mitogenomes of Accipitriformes and Cathartiformes were subjected to ancestral and recent duplications followed by gradual degeneration. Genome Biol. Evol. 2021, 13, evab193. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Nishida, C.; Momose, K.; Onuma, M.; Takami, K.; Masuda, R. Gene duplication and concerted evolution of mitochondrial DNA in crane species. Mol. Phylogenet. Evol. 2017, 106, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Abbott, C.L.; Double, M.C.; Trueman, J.W.H.; Robinson, A.; Cockburn, A. An unusual source of apparent mitochondrial heteroplasmy: Duplicate mitochondrial control regions in Thalassarche albatrosses. Mol. Ecol. 2005, 14, 3605–3613. [Google Scholar] [CrossRef] [PubMed]
- Bensch, S. Mitochondrial genomic rearrangements in songbirds. Mol. Biol. Evol. 2000, 17, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Gibb, G.C.; Kardailsky, O.; Kimball, R.T.; Braun, E.L.; Penny, D. Mitochondrial genomes and avian phylogeny: Complex characters and resolvability without explosive radiations. Mol. Biol. Evol. 2007, 24, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Urantówka, A.D.; Kroczak, A.; Mackiewicz, P. New view on the organization and evolution of Palaeognathae mitogenomes poses the question on the ancestral gene rearrangement in Aves. BMC Genom. 2020, 21, 874. [Google Scholar] [CrossRef] [PubMed]
- Mackiewicz, P.; Urantówka, A.D.; Kroczak, A.; Mackiewicz, D. Resolving phylogenetic relationships within Passeriformes based on mitochondrial genes and inferring the evolution of their mitogenomes in terms of duplications. Genome Biol. Evol. 2019, 11, 2824–2849. [Google Scholar] [CrossRef] [PubMed]
- Caparroz, R.; Rocha, A.V.; Cabanne, G.S.; Tubaro, P.; Aleixo, A.; Lemmon, E.M.; Lemmon, A.R. Mitogenomes of two neotropical bird species and the multiple independent origin of mitochondrial gene orders in Passeriformes. Mol. Biol. Rep. 2018, 45, 279–285. [Google Scholar] [CrossRef]
- Boore, J.L. The duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals. In Comparative Genomics: Empirical and Analytical Approaches to Gene Order Dynamics, Map Alignment and the Evolution of Gene Families; Springer: Dordrecht, The Netherlands, 2000; pp. 133–147. [Google Scholar] [CrossRef]
- Lunt, D.H.; Hyman, B.C. Animal mitochondrial DNA recombination. Nature 1997, 387, 247. [Google Scholar] [CrossRef] [PubMed]
- Skujina, I.; McMahon, R.; Lenis, V.P.E.; Gkoutos, G.V.; Hegarty, M. Duplication of the mitochondrial control region is associated with increased longevity in birds. Aging 2016, 8, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Omote, K.; Nishida, C.; Dick, M.H.; Masuda, R. Limited phylogenetic distribution of a long tandem-repeat cluster in the mitochondrial control region in Bubo (Aves, Strigidae) and cluster variation in Blakiston’s Fish Owl (Bubo blakistoni). Mol. Phylogenet. Evol. 2013, 66, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Mundy, N.I.; Winchell, C.S.; Woodruff, D.S. Tandem repeats and heteroplasmy in the mitochondrial DNA control region of the Loggerhead Shrike (Lanius ludovicianus). J. Hered. 1996, 87, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, N.; Zhang, H.; Yang, X.J.; Huang, Y.; Lei, F. Extreme variation in patterns of tandem repeats in mitochondrial control region of yellow-browed tits (Sylviparus modestus, Paridae). Sci. Rep. 2015, 5, 13227. [Google Scholar] [CrossRef] [PubMed]
- Hasson, J.F.; Mougneau, E.; Cuzin, F.; Yaniv, M. Simian virus 40 illegitimate recombination occurs near short direct repeats. J. Mol. Biol. 1984, 177, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Hoelzel, A.R. Evolution by DNA turnover in the control region of vertebrate mitochondrial DNA. Curr. Opin. Genet. Dev. 1993, 3, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Buroker, N.E.; Brown, J.R.; Gilbert, T.A.; O’Hara, P.J.; Beckenbach, A.T.; Thomas, W.K.; Smith, M.J. Length heteroplasmy of sturgeon mitochondrial DNA: An illegitimate elongation model. Genetics 1990, 124, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Alström, P.; Barnes, K.N.; Olsson, U.; Barker, F.K.; Bloomer, P.; Khan, A.A.; Qureshi, M.A.; Guillaumet, A.; Crochet, P.-A.; Ryan, P.G. Multilocus phylogeny of the avian family Alaudidae (larks) reveals complex morphological evolution, non-monophyletic genera and hidden species diversity. Mol. Phylogenet. Evol. 2013, 69, 1043–1056. [Google Scholar] [CrossRef] [PubMed]
- Alström, P.; Mohammadi, Z.; Enbody, E.D.; Irestedt, M.; Engelbrecht, D.; Crochet, P.-A.; Guillaumet, A.; Rancilhac, L.; Tieleman, B.I.; Olsson, U.; et al. Systematics of the avian family Alaudidae using multilocus and genomic data. Avian Res. 2023, 14, 100095. [Google Scholar] [CrossRef]
- IOC World Bird List (v14.1). Available online: https://www.worldbirdnames.org/new (accessed on 14 May 2024).
- Qian, C.; Wang, Y.; Guo, Z.; Yang, J.; Kan, X. Complete mitochondrial genome of Skylark, Alauda arvensis (Aves: Passeriformes): The first representative of the family Alaudidae with two extensive heteroplasmic control regions. Mitochondrial DNA 2013, 24, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Lu, J.Y.; Yang, M.L.; Kang, H. Complete mitochondrial genome of the Mongolian Lark, Melanocorypha mongolica (Aves: Passeriformes). Mitochondrial DNA Part B 2017, 2, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhao, L.; Wang, Q.; Yuan, H.; Li, X.; Wang, Y. Mitogenome of Alaudala cheleensis (Passeriformes: Alaudidae) and comparative analyses of Sylvioidea mitogenomes. Zootaxa 2021, 4952, 331–353. [Google Scholar] [CrossRef] [PubMed]
- Groth, J.G. Molecular phylogenetics of finches and sparrows: Consequences of character state removal in cytochrome b sequences. Mol. Phylogenet. Evol. 1998, 10, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Barker, F.K.; Cibois, A.; Schikler, P.; Feinstein, J.; Cracraft, J. Phylogeny and diversification of the largest avian radiation. Proc. Natl. Acad. Sci. USA 2004, 101, 11040–11045. [Google Scholar] [CrossRef] [PubMed]
- Ericson, P.G.P.; Klopfstein, S.; Irestedt, M.; Nguyen, J.M.T.; Nylander, J.A.A. Dating the diversification of the major lineages of Passeriformes (Aves). BMC Evol. Biol. 2014, 14, 8. [Google Scholar] [CrossRef]
- Barker, F.K. Mitogenomic data resolve basal relationships among Passeriform and passerida birds. Mol. Phylogenet. Evol. 2014, 79, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Gibb, G.C.; England, R.; Hartig, G.; McLenachan, P.A.; Smith, B.L.T.; McComish, B.J.; Cooper, A.; Penny, D. New Zealand passerines help clarify the diversification of major songbird lineages during the Oligocene. Genome Biol. Evol. 2015, 7, 2983–2995. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bai, Y.; Shi, X.; Sun, L.; Wu, X. The complete mitochondrial genomes of Tarsiger cyanurus and Phoenicurus auroreus: A phylogenetic analysis of Passeriformes. Genes Genom. 2018, 40, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Beresford, P.; Barker, F.K.; Ryan, P.G.; Crowe, T.M. African endemics span the tree of songbirds (Passeri): Molecular systematics of several evolutionary ‘enigmas’. Proc. R. Soc. B-Biol. Sci. 2005, 272, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Claramunt, S.; Cracraft, J. A bew time tree reveals earth history’s imprint on the evolution of modern birds. Sci. Adv. 2015, 1, e1501005. [Google Scholar] [CrossRef] [PubMed]
- Ericson, P.G.P.; Johansson, U.S. Phylogeny of Passerida (Aves: Passeriformes) based on nuclear and mitochondrial sequence data. Mol. Phylogenet. Evol. 2003, 1, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Alström, P.; Ericson, P.G.P.; Olsson, U.; Sundberg, P. Phylogeny and classification of the avian superfamily Sylvioidea. Mol. Phylogenet. Evol. 2006, 38, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, K.A.; Fjeldsa, J. A phylogenetic supertree of oscine passerine birds (Aves: Passeri). Zool. Scr. 2006, 35, 149–186. [Google Scholar] [CrossRef]
- Johansson, U.S.; Fjelds, J.; Bowie, R. Phylogenetic relationships within passerida (Aves: Passeriformes): A review and a new molecular phylogeny based on three nuclear intron markers. Mol. Phylogenet. Evol. 2008, 48, 858–876. [Google Scholar] [CrossRef] [PubMed]
- Fregin, S.; Haase, M.; Olsson, U.; Alström, P. New insights into family relationships within the avian superfamily Sylvioidea (Passeriformes) based on seven molecular markers. BMC Evol. Biol. 2012, 12, 157. [Google Scholar] [CrossRef] [PubMed]
- Alström, P.; Hooper, D.M.; Liu, Y.; Olsson, U.; Mohan, D.; Gelang, M.; Le Manh, H.; Zhao, J.; Lei, F.; Price, T.D. Discovery of a relict lineage and monotypic family of passerine birds. Biol. Lett. 2014, 10, 20131067. [Google Scholar] [CrossRef] [PubMed]
- Selvatti, A.P.; Gonzaga, L.P.; de Moraes Russo, C.A. A Paleogene origin for crown passerines and the diversification of the oscines in the New World. Mol. Phylogenet. Evol. 2015, 88, 1–15. [Google Scholar] [CrossRef]
- Moyle, R.G.; Oliveros, C.H.; Andersen, M.J.; Hosner, P.A.; Benz, B.W.; Manthey, J.D.; Travers, S.L.; Brown, R.M.; Faircloth, B.C. Tectonic collision and uplift of Wallacea triggered the global songbird radiation. Nat. Commun. 2016, 7, 12709. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, C.H.; Field, D.J.; Ksepka, D.T.; Barker, F.K.; Aleixo, A.; Andersen, M.J.; Alstrom, P.; Benz, B.W.; Braun, E.L.; Braun, M.J.; et al. Earth history and the passerine superradiation. Proc. Natl. Acad. Sci. USA 2019, 116, 7916–7925. [Google Scholar] [CrossRef] [PubMed]
- Alstrom, P.; van Linschooten, J.; Donald, P.F.; Sundev, G.; Mohammadi, Z.; Ghorbani, F.; Shafaeipour, A.; van den Berg, A.; Robb, M.; Aliabadian, M.; et al. Multiple species delimitation approaches applied to the avian lark genus Alaudala. Mol. Phylogenet. Evol. 2021, 154, 106994. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, F.; Aliabadian, M.; Zhang, R.; Irestedt, M.; Hao, Y.; Sundev, G.; Lei, F.; Ma, M.; Olsson, U.; Alstrom, P. Densely sampled phylogenetic analyses of the Lesser Short-Toed Lark (Alaudala rufescens)—Sand Lark (A. raytal) species complex (Aves, Passeriformes) reveal cryptic diversity. Zool. Scr. 2020, 49, 427–439. [Google Scholar] [CrossRef]
- Sigeman, H.; Strandh, M.; Proux-Wéra, E.; Kutschera, V.E.; Ponnikas, S.; Zhang, H.; Lundberg, M.; Soler, L.; Bunikis, I.; Tarka, M.; et al. Avian neo-sex chromosomes reveal dynamics of recombination suppression and W degeneration. Mol. Biol. Evol. 2021, 38, 5275–5291. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; dePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Stothard, P.; Grant, J.R.; Van Domselaar, G. Visualizing and comparing circular genomes using the CGView family of tools. Brief. Bioinform. 2019, 20, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovli, I.; Zou, H.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Donath, A.; Jühling, F.; Al-Arab, M.; Bernhart, S.H.; Reinhardt, F.; Stadler, P.F.; Middendorf, M.; Bernt, M. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 2019, 47, 10543–10552. [Google Scholar] [CrossRef] [PubMed]
- Randi, E.; Lucchini, V. Organization and evolution of the mitochondrial DNA control region in the avian genus Alectoris. J. Mol. Evol. 1998, 47, 449–462. [Google Scholar] [CrossRef]
- Ruokonen, M.; Kvist, L. Structure and evolution of the avian mitochondrial control region. Mol. Phylogenet. Evol. 2002, 23, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The european molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef] [PubMed]
- Zardoya, R.; Meyer, A. Phylogenetic performance of mitochondrial protein-coding genes in resolving relationships among vertebrates. Mol. Biol. Evol. 1996, 13, 933–942. [Google Scholar] [CrossRef]
- Xiang, C.Y.; Gao, F.; Jakovlić, I.; Lei, H.P.; Hu, Y.; Zhang, H.; Zou, H.; Wang, G.T.; Zhang, D. Using PhyloSuite for molecular phylogeny and tree-based analyses. iMeta 2023, 2, e87. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and ssability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Ranwez, V.; Douzery, E.J.P.; Cambon, C.; Chantret, N.; Delsuc, F. MACSE v2: Toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evol. 2018, 35, 2582–2584. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in bayesian phylogenetics using tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://birdsoftheworld.org/bow/support/citations-and-references (accessed on 14 May 2024).
- Chen, W.; Miao, K.; Wang, J.; Wang, H.; Sun, W.; Yuan, S.; Luo, S.; Hu, C.; Chang, Q. Five new mitogenomes sequences of Calidridine sandpipers (Ayes: Charadriiformes) and comparative mitogenomics of genus Calidris. Peerj 2022, 10, e13268. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Du, X.; Liu, Y.; Yuan, H.; Wang, Q.; Hou, X.; Gong, H.; Wang, Y.; Huang, Y.; Li, X.; et al. Comparative mitogenomics of the genus Motacilla (Aves, Passeriformes) and its phylogenetic implications. ZooKeys 2022, 1109, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Dong, X.; Wang, Q.; Hou, X.; Yuan, H.; Li, X. Mitochondrial genome characteristics of six Phylloscopus species and their phylogenetic implication. PeerJ 2023, 11, e16233. [Google Scholar] [CrossRef]
- Ding, H.; Bi, D.; Han, S.; Yi, R.; Zhang, S.; Ye, Y.; Gao, J.; Yang, J.; Kan, X. Mitogenomic codon usage patterns of superfamily Certhioidea (Aves, Passeriformes): Insights into asymmetrical bias and phylogenetic implications. Animals 2023, 13, 96. [Google Scholar] [CrossRef] [PubMed]
- Montana-Lozano, P.; Balaguera-Reina, S.A.; Prada-Quiroga, C.F. Comparative analysis of codon usage of mitochondrial genomes provides evolutionary insights into reptiles. Gene 2023, 851, 146999. [Google Scholar] [CrossRef] [PubMed]
- Berlin, S.; Smith, N.G.C.; Ellegren, H. Do avian mitochondria recombine? J. Mol. Evol. 2004, 58, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Kurabayashi, A.; Sumida, M.; Yonekawa, H.; Glaw, F.; Vences, M.; Hasegawa, M. Phylogeny, recombination, and mechanisms of stepwise mitochondrial genome reorganization in Mantellid Frogs from Madagascar. Mol. Biol. Evol. 2008, 25, 874–891. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Nakada, K.; Akimoto, M.; Ishikawa, K.; Ono, T.; Shitara, H.; Yonekawa, H.; Hayashi, J.L. Rare creation of recombinant mtDNA haplotypes in mammalian tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 6057–6062. [Google Scholar] [CrossRef]
- Broughton, R.E.; Dowling, T.E. Length variation in mitochondrial DNA of the minnow Cyprinella spiloptera. Genetics 1994, 138, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Cadahía, L.; Pinsker, W.; Negro, J.J.; Pavlicev, M.; Urios, V.; Haring, E. Repeated sequence homogenization between the control and pseudo-control regions in the mitochondrial genomes of the subfamily Aquilinae. J. Exp. Zoolog. B Mol. Dev. Evol. 2009, 312, 171–185. [Google Scholar] [CrossRef]
- Chen, J.M.; Cooper, D.N.; Chuzhanova, N.; Ferec, C.; Patrinos, G.P. Gene conversion: Mechanisms, evolution and human disease. Nat. Rev. Genet. 2007, 8, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Rand, D.M.; Harrison, R.G. Molecular population genetics of mtDNA size variation in crickets. Genetics 1989, 121, 551–569. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Haplotype | Sequence (5′–3′) | Similarity among Haplotype | Free Energy (kcal/mol) | |
|---|---|---|---|---|---|
| One Unit | Two Unit | ||||
| E. alpestris | ACS | AACAAAAGAAATCAATCCCATTTCTTTCTTTATTATACATATAATAAAGAG | 98.0~100.0% | −9.04 | −18.26~−18.08 |
| A1 | ................................................... | ||||
| A2 | ..................T................................ | ||||
| A3 | .....---------------------------------------------- | ||||
| A. cheleensis | BCS | ACACACGTATAAATAAAGACAGGACACCTT-ACGTCTTCTTATACTATTCTTATACTATTATACACGT | 94.1~100.0% | −3.23~−2.63 | −8.17 |
| B1 | ......A.....G.................-..................................... | ||||
| B2 | ..............................T..................................... | ||||
| B3 | ..............................-..................................... | ||||
| B4 | ......A.....G.................-..........................G.......... | ||||
| B5 | ..............................-..........................G.......... | ||||
| B6 | .........---------------------------................................ | ||||
| B7 | ..............................-...........-------------------------- | ||||
| A. heinei | CCS | CACGTATAAGTAAAGAGAGGACACCTCACGTCTCCTTACTATTATACGTGTA | 100% | −8.30~−7.54 | −18.48~−17.72 |
| C1 | .................................................... | ||||
| C2 | ............................................-------- | ||||
| A. arvensis | DCS | AAAGAAATCAATCCCATTGATTTCATTATATTAGTAT | 100.0% | −6.81~−5.93 | −15.36 |
| D1 | .......C..G..ATA....C....C.G.....A..C | −2.58~−2.49 | |||
| D2 | ..................................... | ||||
| D3 | .................-------------------- | ||||
| A. gulgula | ECS | AAAGAAATCAATCCCATTGATTTCATTATATTAGTAT | 92.9% | −6.81~−5.93 | −15.36 |
| E1 | ..................................... | ||||
| E2 | ....G.........----------------------- | ||||
| A. razae | FCS | AAAGAAATCAACCCTATTGACTTCATTATATTAGTAT | 100% | −3.10~−2.22 | −7.94 |
| F1 | ..CA..CCA.T...-...................... | −0.90~−0.10 | |||
| F2 | ..................................... | ||||
| F3 | ................--------------------- | ||||
| C. cinerea | GCS | AAAGAATAAGAGACCACTCTTACTCTTTATCATACACATAACTGTATATATATATATGTAT | 100% | −7.70~−6.82 | −19.60 |
| G1 | .......---------.A..CTTA..C.T.ATC.T...C.TAAC.G..........TA... | −2.92~−2.31 | |||
| G2 | ............................................................. | ||||
| G3 | ............................................----............. | ||||
| G4 | .......................... | ||||
| M. mongolica | H1 | TCTTTACTTATTACATGTATATAAAGTAGAGA | −2.03 | ||
| H2 | ......TC.....T..ACG..C...AC..... | 0.10 | |||
| H3 | ......................---------- | ||||
| H1 + H2 | TCTTTACTTATTACATGTATATAAAGTAGAGATCTTTATCTATTATATACGTACAAAACAGAGA | 100.0% | −3.65 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, C.; Kang, H.; Zhou, Y.; Zhu, W.; Zhao, X.; Mohamed, N.; Li, B. Selected Lark Mitochondrial Genomes Provide Insights into the Evolution of Second Control Region with Tandem Repeats in Alaudidae (Aves, Passeriformes). Life 2024, 14, 881. https://doi.org/10.3390/life14070881
Jiang C, Kang H, Zhou Y, Zhu W, Zhao X, Mohamed N, Li B. Selected Lark Mitochondrial Genomes Provide Insights into the Evolution of Second Control Region with Tandem Repeats in Alaudidae (Aves, Passeriformes). Life. 2024; 14(7):881. https://doi.org/10.3390/life14070881
Chicago/Turabian StyleJiang, Chuan, Hui Kang, Yang Zhou, Wenwen Zhu, Xilong Zhao, Nassoro Mohamed, and Bo Li. 2024. "Selected Lark Mitochondrial Genomes Provide Insights into the Evolution of Second Control Region with Tandem Repeats in Alaudidae (Aves, Passeriformes)" Life 14, no. 7: 881. https://doi.org/10.3390/life14070881
APA StyleJiang, C., Kang, H., Zhou, Y., Zhu, W., Zhao, X., Mohamed, N., & Li, B. (2024). Selected Lark Mitochondrial Genomes Provide Insights into the Evolution of Second Control Region with Tandem Repeats in Alaudidae (Aves, Passeriformes). Life, 14(7), 881. https://doi.org/10.3390/life14070881