

Rosmarinic Acid Potentiates Cytotoxicity of Cisplatin against Colorectal Cancer Cells by Enhancing Apoptotic and Ferroptosis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture and Treatments
2.3. Cytotoxicity Assay
2.4. Cell Cycle Analysis and Cell Apoptosis Assay
2.5. Protein Extraction and Western Blot Analysis
2.6. Statistical Analysis
3. Results
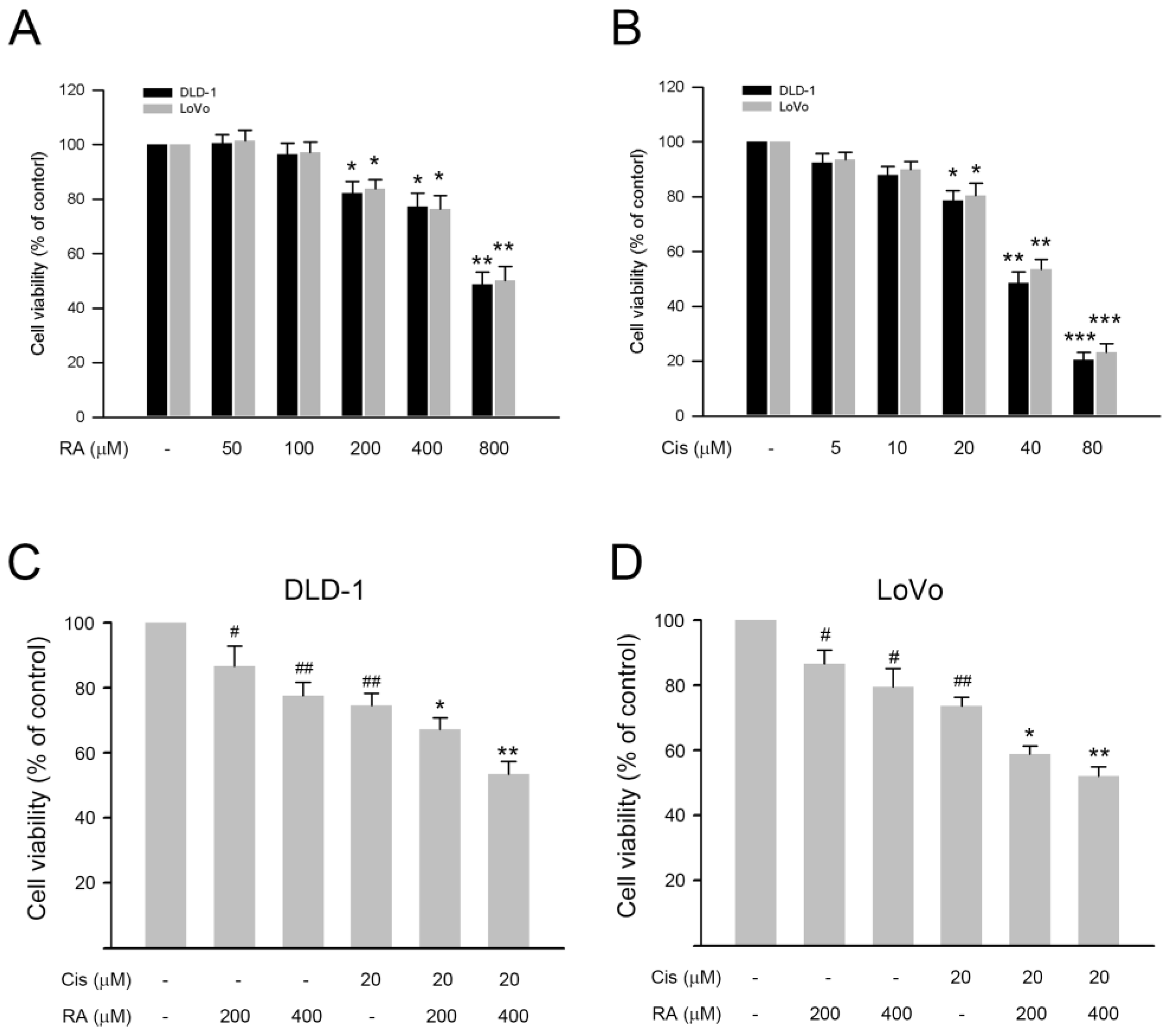
3.1. RA Enhanced the Inhibitory Effect of Cisplatin on Cell Viability of DLD-1 and LoVo Cells
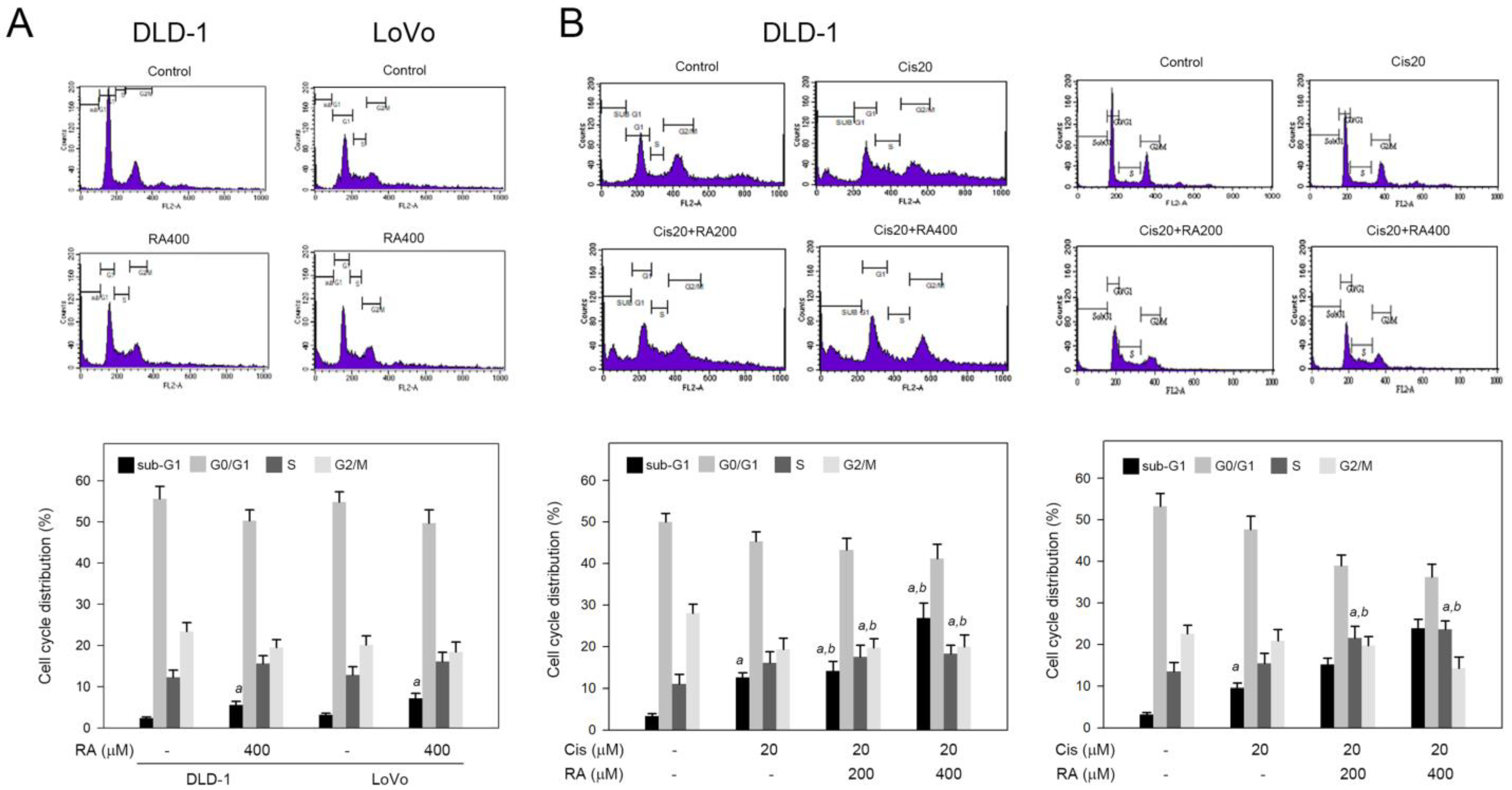
3.2. RA Combined with Cisplatin Promoted Cisplatin-Induced Sub-G1 and S Phase Accumulation in DLD-1 and LoVo Cells
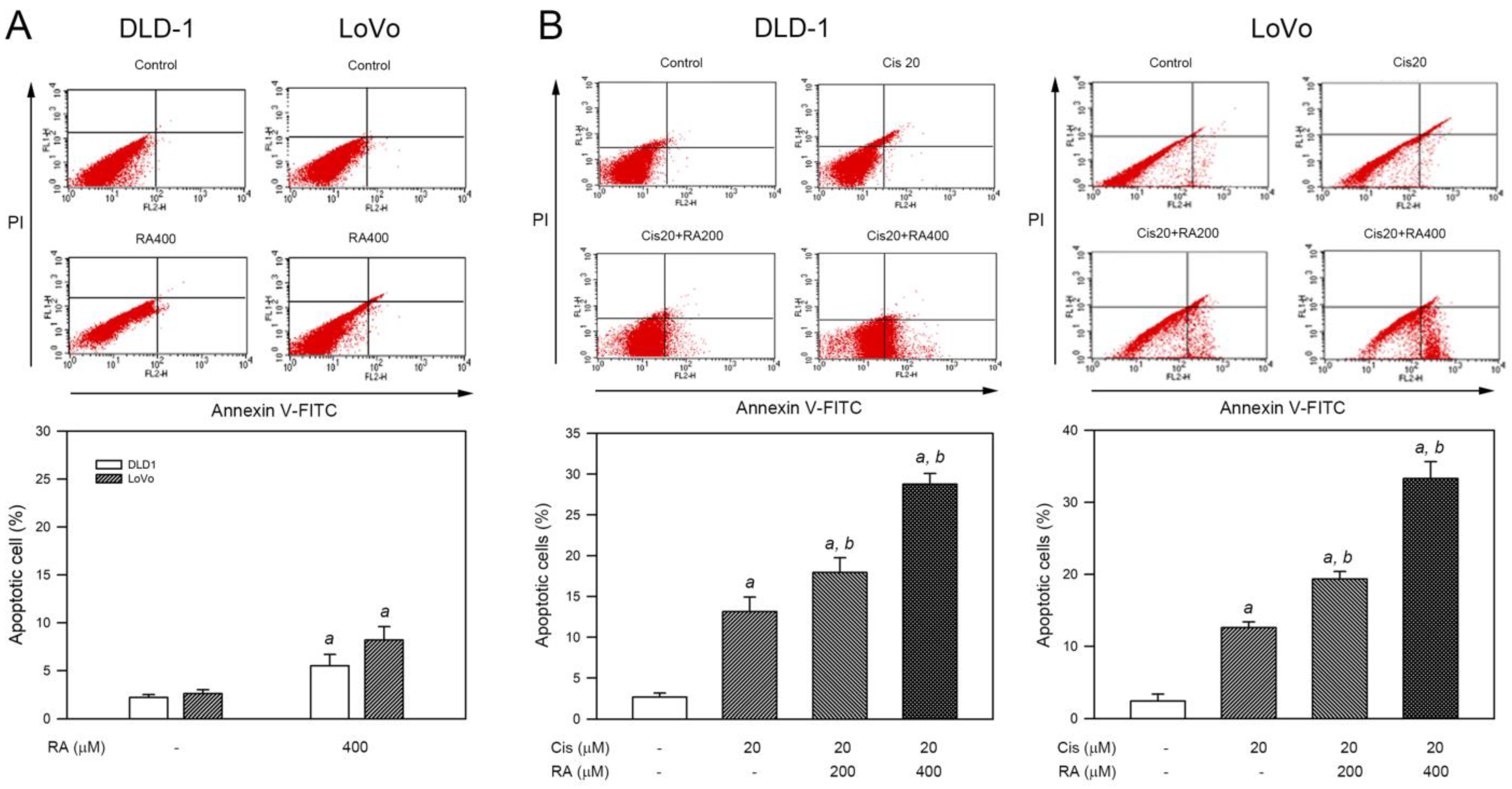
3.3. RA Enhanced Cisplatin-Induced Apoptosis in CRC Cells
3.4. RA Enhanced the Apoptotic Signaling Induced by Cisplatin in DLD-1 Cells
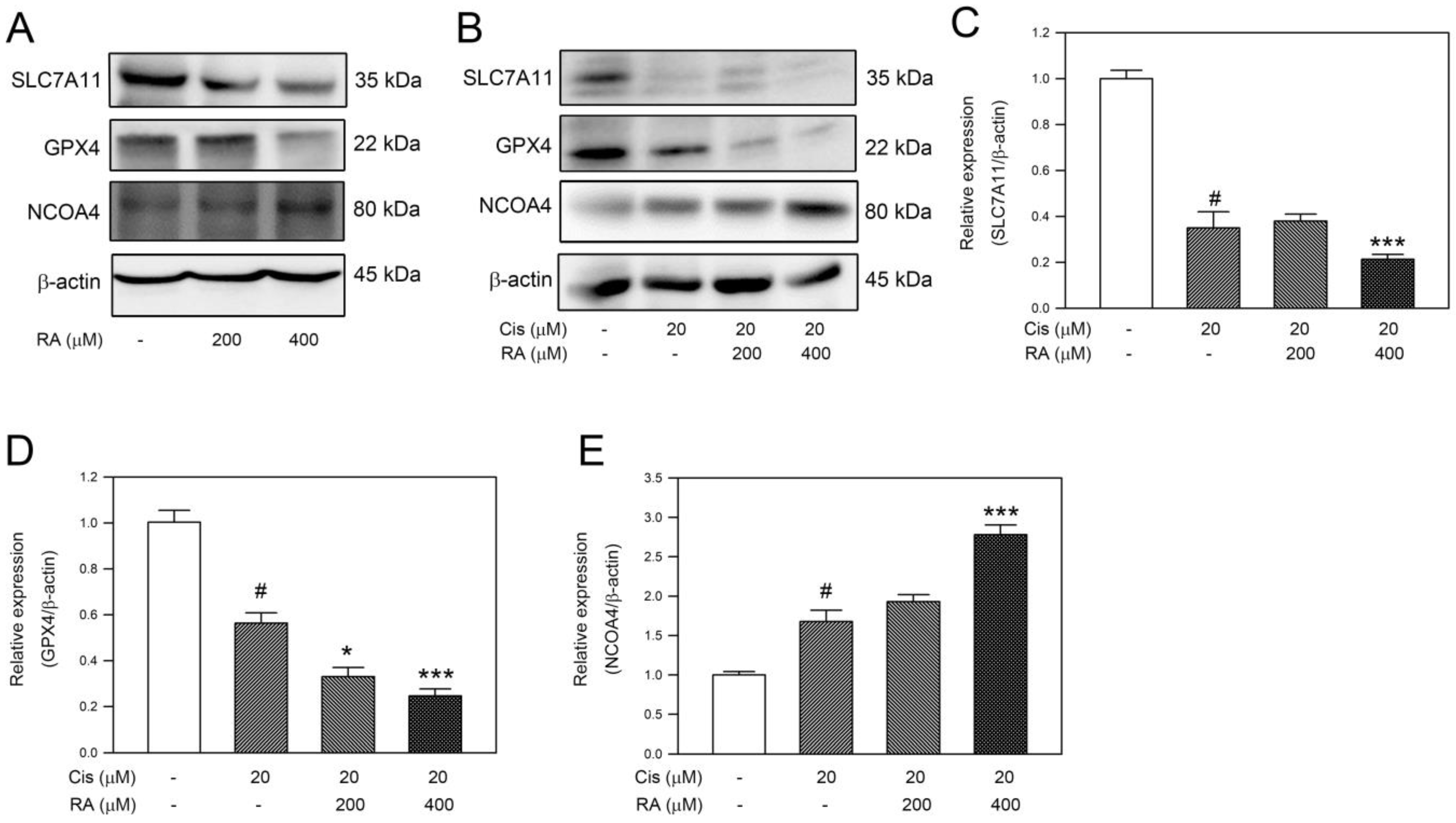
3.5. RA Promoted the Ferroptosis Signaling Induced by Cisplatin in DLD-1 Cells
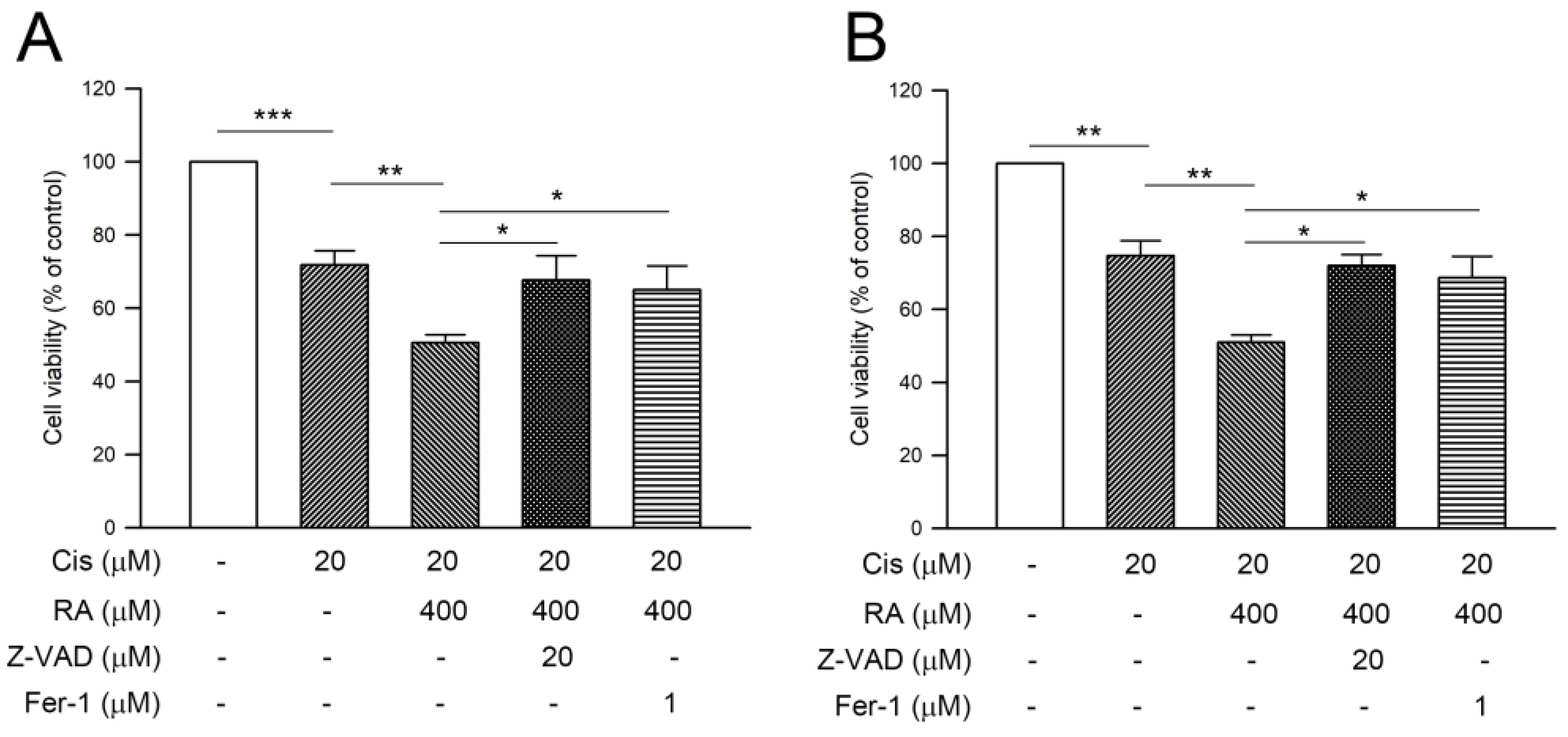
3.6. RA Enhanced Cisplatin-Induced Cytotoxicity through Promoting the Apoptosis and Ferroptosis in CRC Cells
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Qiu, W.; Leibowitz, B.; Zhang, L.; Yu, J. Growth factors protect intestinal stem cells from radiation-induced apoptosis by suppressing PUMA through the PI3K/AKT/p53 axis. Oncogene 2010, 29, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [PubMed]
- Manogaran, P.; Somasundaram, B.; Viswanadha, V.P. Reversal of cisplatin resistance by neferine/isoliensinine and their combinatorial regimens with cisplatin-induced apoptosis in cisplatin-resistant colon cancer stem cells (CSCs). J. Biochem. Mol. Toxicol. 2022, 36, e22967. [Google Scholar] [CrossRef]
- Zhang, T.; Zheng, P.; Shen, X.; Shao, R.; Wang, B.; Shen, H.; Zhang, J.; Xia, Y.; Zou, P. Curcuminoid WZ26, a TrxR1 inhibitor, effectively inhibits colon cancer cell growth and enhances cisplatin-induced cell death through the induction of ROS. Free Radic. Biol. Med. 2019, 141, 93–102. [Google Scholar] [CrossRef]
- Hitl, M.; Kladar, N.; Gavaric, N.; Bozin, B. Rosmarinic Acid-Human Pharmacokinetics and Health Benefits. Planta Med. 2021, 87, 273–282. [Google Scholar] [CrossRef]
- Konstantinou, E.K.; Panagiotopoulos, A.A.; Argyri, K.; Panoutsopoulos, G.I.; Dimitriou, M.; Gioxari, A. Molecular Pathways of Rosmarinic Acid Anticancer Activity in Triple-Negative Breast Cancer Cells: A Literature Review. Nutrients 2023, 16, 2. [Google Scholar] [CrossRef]
- Liao, X.Z.; Gao, Y.; Sun, L.L.; Liu, J.H.; Chen, H.R.; Yu, L.; Chen, Z.Z.; Chen, W.H.; Lin, L.Z. Rosmarinic acid reverses non-small cell lung cancer cisplatin resistance by activating the MAPK signaling pathway. Phytother Res. 2020, 34, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, W.; Li, Z.; Chen, L.; Wen, C.; Ruan, Q.; Xu, Z.; Liu, R.; Xu, J.; Bai, Y.; et al. Rosmarinic Acid Decreases the Malignancy of Pancreatic Cancer Through Inhibiting Gli1 Signaling. Phytomedicine 2022, 95, 153861. [Google Scholar] [CrossRef] [PubMed]
- Orellana, E.A.; Kasinski, A.L. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio Protoc. 2016, 6, e1984. [Google Scholar] [CrossRef] [PubMed]
- Messeha, S.S.; Zarmouh, N.O.; Asiri, A.; Soliman, K.F.A. Rosmarinic acid-induced apoptosis and cell cycle arrest in triple-negative breast cancer cells. Eur. J. Pharmacol. 2020, 885, 173419. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, X.; Tang, H.; Pan, Y.; Hu, B.; Huang, G. Rosmarinic acid inhibits cell proliferation, migration, and invasion and induces apoptosis in human glioma cells. Int. J. Mol. Med. 2021, 47, 67. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Shen, Y.; Zhu, L.; Yao, L.; Wang, X.; Zhang, A.; Li, J.; Wu, J.; Qin, L. Rosmarinic acid, the active component of Rubi Fructus, induces apoptosis of SGC-7901 and HepG2 cells through mitochondrial pathway and exerts anti-tumor effect. Naunyn. Schmiedebergs Arch. Pharmacol. 2023, 396, 3743–3755. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef]
- Yu, H.; Guo, P.; Xie, X.; Wang, Y.; Chen, G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell. Mol. Med. 2017, 21, 648–657. [Google Scholar] [CrossRef]
- Jin, B.R.; Chung, K.S.; Hwang, S.; Hwang, S.N.; Rhee, K.J.; Lee, M.; An, H.J. Rosmarinic acid represses colitis-associated colon cancer: A pivotal involvement of the TLR4-mediated NF-kappaB-STAT3 axis. Neoplasia 2021, 23, 561–573. [Google Scholar] [CrossRef]
- Pagano, K.; Carminati, L.; Tomaselli, S.; Molinari, H.; Taraboletti, G.; Ragona, L. Molecular Basis of the Antiangiogenic Action of Rosmarinic Acid, a Natural Compound Targeting Fibroblast Growth Factor-2/FGFR Interactions. Chembiochem 2021, 22, 160–169. [Google Scholar] [CrossRef]
- Cao, W.; Mo, K.; Wei, S.; Lan, X.; Zhang, W.; Jiang, W. Effects of rosmarinic acid on immunoregulatory activity and hepatocellular carcinoma cell apoptosis in H22 tumor-bearing mice. Korean J. Physiol. Pharmacol. 2019, 23, 501–508. [Google Scholar] [CrossRef]
- Nagasaka, T.; Mishima, H.; Sawaki, A.; Shimokawa, M.; Inukai, M.; Shinozaki, K.; Tanioka, H.; Nasu, J.; Nishina, T.; Hazama, S.; et al. Protocol of a randomised phase III clinical trial of sequential capecitabine or 5-fluorouracil plus bevacizumab (Cape/5-FU-Bmab) to capecitabine or 5-fluorouracil plus oxaliplatin plus bevacizumab (CapeOX/mFOLFOX6-Bmab) versus combination CapeOX/mFOLFOX6-Bmab in advanced colorectal cancer: The C-cubed (C3) study. BMJ Open 2016, 6, e011454. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.-Y.; Hsu, T.-W.; Chen, Y.-R.; Kao, S.-H. Rosmarinic Acid Potentiates Cytotoxicity of Cisplatin against Colorectal Cancer Cells by Enhancing Apoptotic and Ferroptosis. Life 2024, 14, 1017. https://doi.org/10.3390/life14081017
Huang J-Y, Hsu T-W, Chen Y-R, Kao S-H. Rosmarinic Acid Potentiates Cytotoxicity of Cisplatin against Colorectal Cancer Cells by Enhancing Apoptotic and Ferroptosis. Life. 2024; 14(8):1017. https://doi.org/10.3390/life14081017
Chicago/Turabian StyleHuang, Jhen-Yu, Ta-Wen Hsu, Yu-Ru Chen, and Shao-Hsuan Kao. 2024. "Rosmarinic Acid Potentiates Cytotoxicity of Cisplatin against Colorectal Cancer Cells by Enhancing Apoptotic and Ferroptosis" Life 14, no. 8: 1017. https://doi.org/10.3390/life14081017