
The Uncoupling Effect of 17β-Estradiol Underlies the Resilience of Female-Derived Mitochondria to Damage after Experimental TBI

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Animals and Experimental Design
2.2. Estradiol and Vehicle Formulations
2.3. Mouse Model of Traumatic Brain Injury
2.4. Isolation of Mitochondria from Brain Tissue
2.5. Reactive Oxygen Species Production
2.6. Mitochondrial Bioenergetic Measurements
2.7. Electron Transport Chain Complex Activities
2.8. OXPHOS Protein Expression
2.9. β-Oxidation Respiratory Measurement
2.10. Statistical Analysis
3. Results
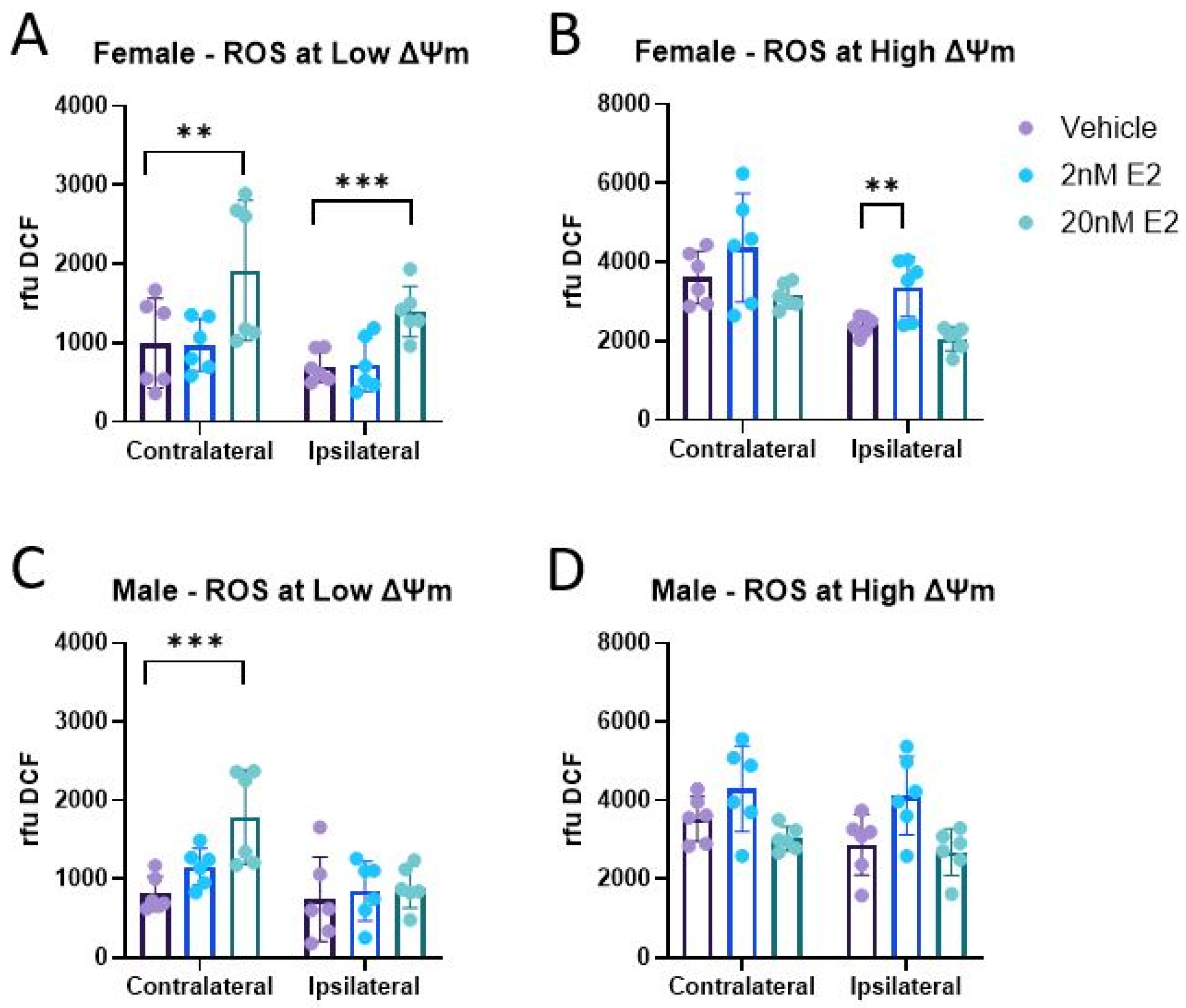
3.1. ROS Production
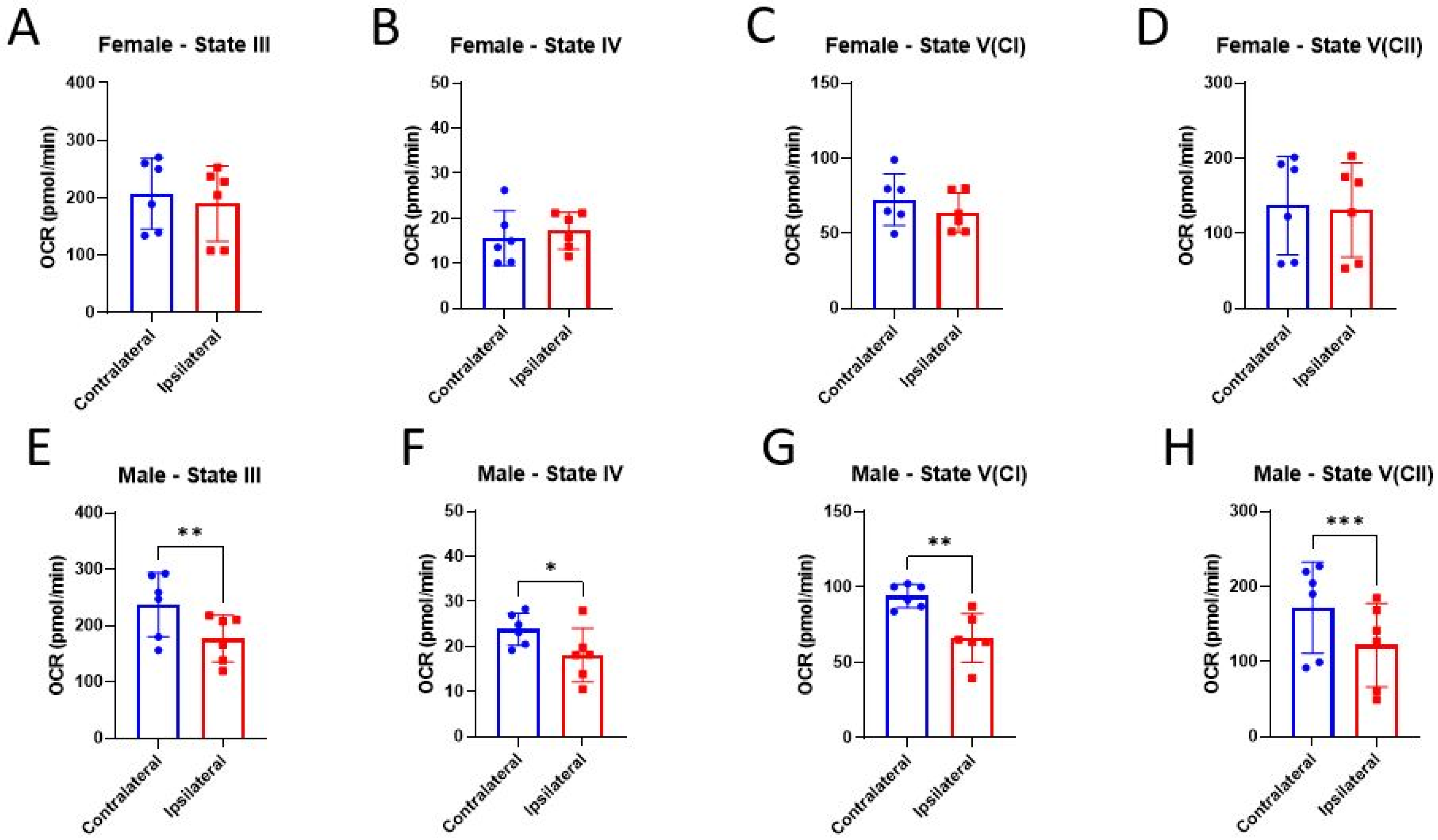
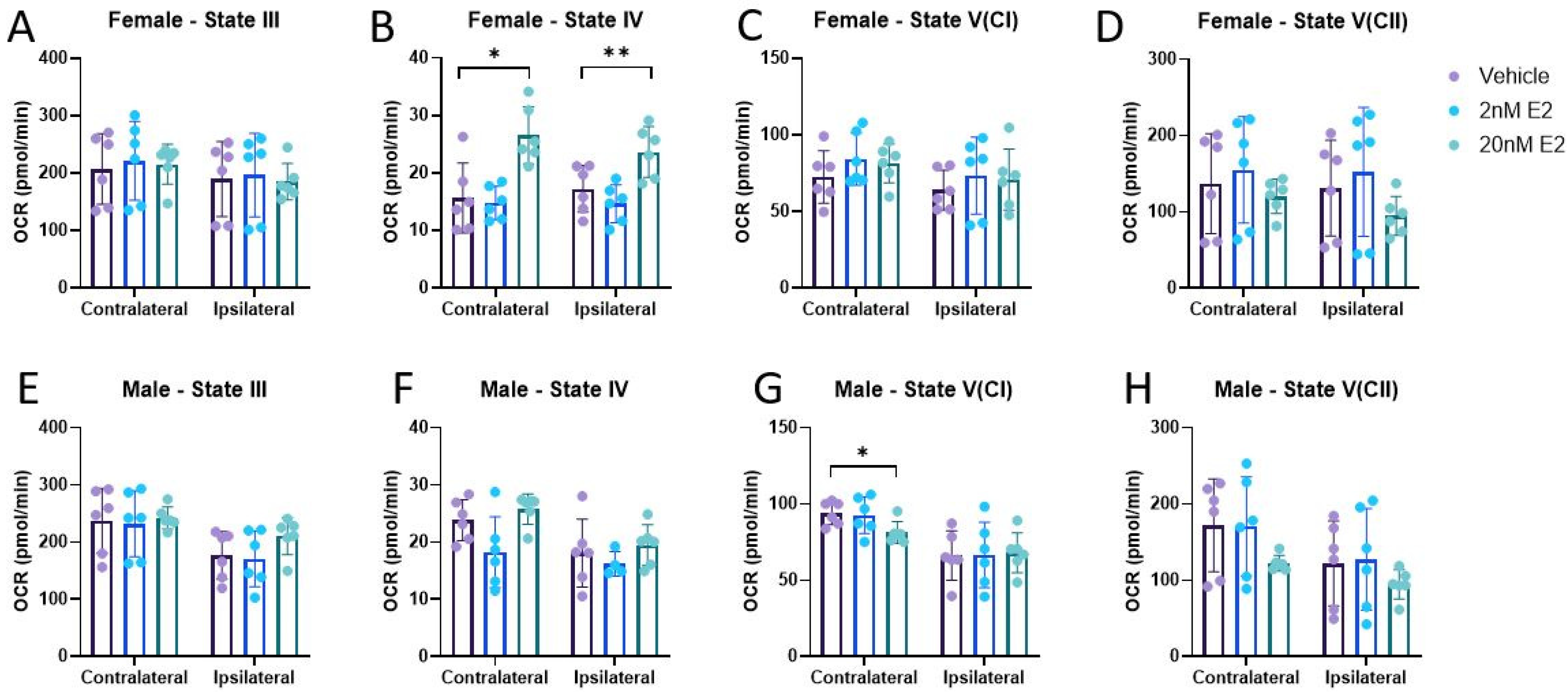
3.2. Bioenergetics
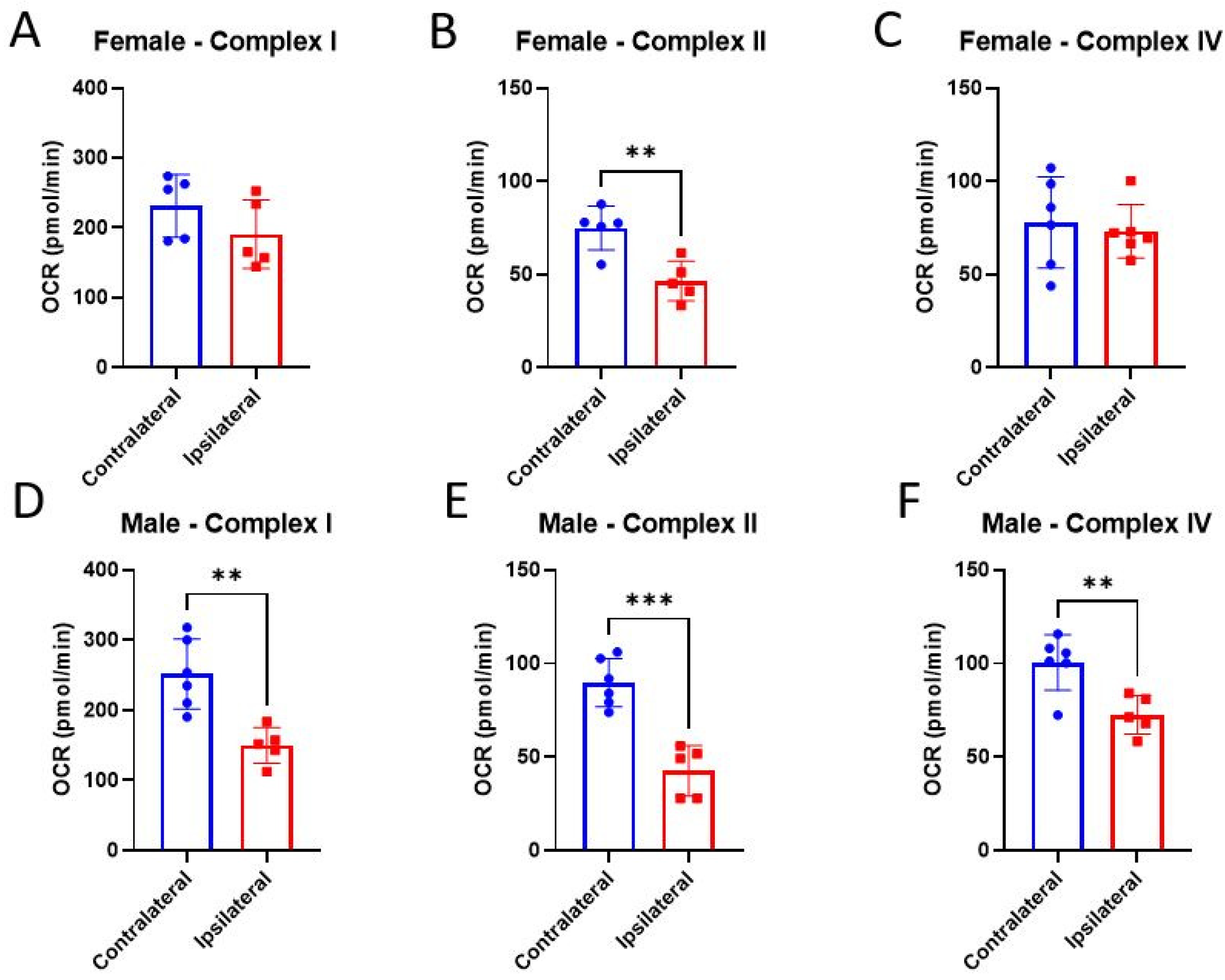
3.3. ETC Complex Activities
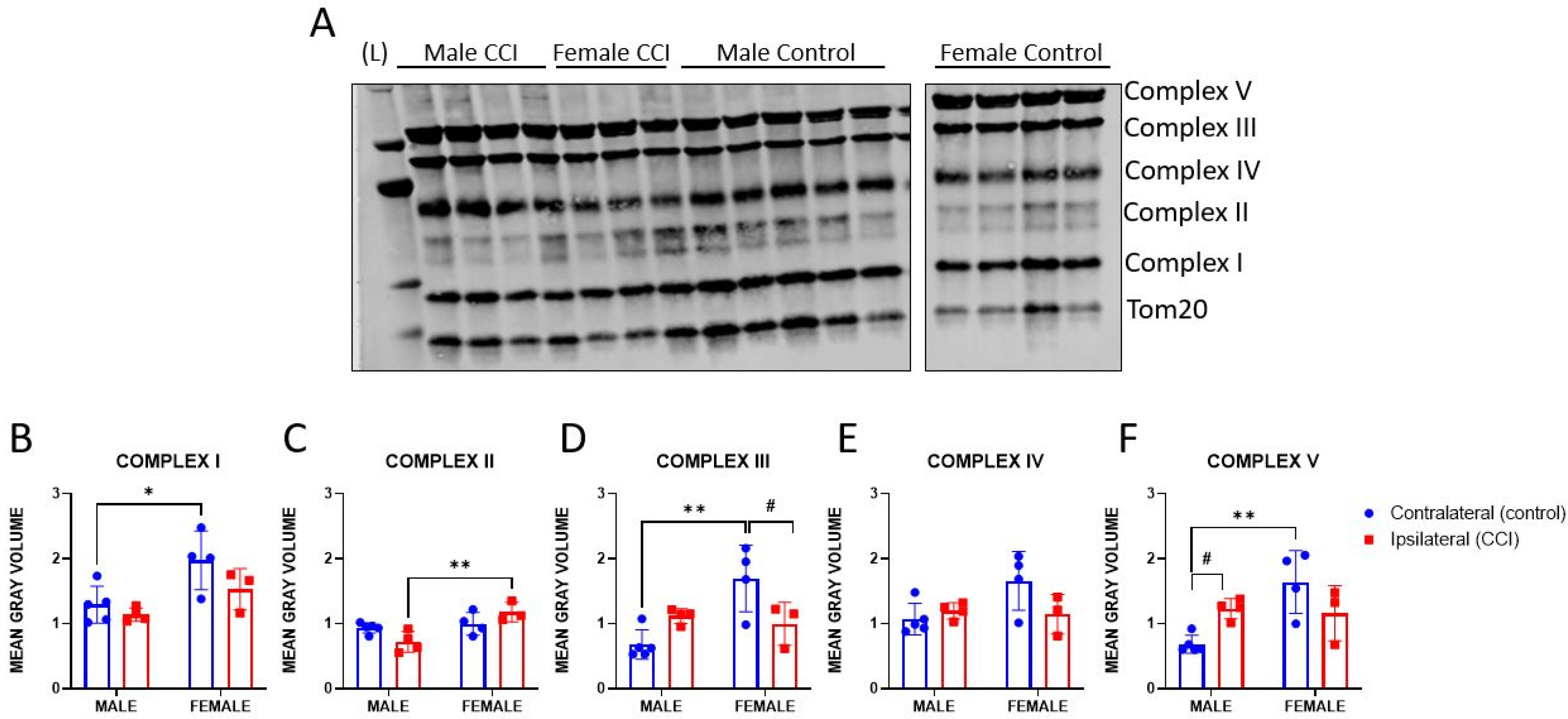
3.4. OXPHOS Protein Expression
3.5. Beta-Oxidation of Palmitoyl Carnitine
4. Discussion/Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ΔΨm | mitochondrial membrane potential |
| ATP | adenosine triphosphate |
| Bcl-2 | B-cell lymphoma 2 |
| CCI | controlled cortical impact |
| DCF-DA | 2′,7′-dichlorodihydroflurorescein diacetate |
| E2 | 17β-estradiol |
| ETC | electron transport chain |
| GPx | glutathione peroxidase |
| MAP | mean arteriole pressure |
| NADH | nicotinamide adenine dinucleotide |
| OCR | oxygen consumption rate |
| OXPHOS | oxidative phosphorylation |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
| TBI | traumatic brain injury |
| TMPD | N,N,N′,N′-tetramethyl-p-phenylenediamine |
References
- Kim, L.H.; Quon, J.L.; Sun, F.W.; Wortman, K.M.; Adamson, M.M.; Harris, O.A. Traumatic brain injury among female veterans: A review of sex differences in military neurosurgery. Neurosurg. Focus 2018, 45, E16. [Google Scholar] [CrossRef] [PubMed]
- Valera, E.M.; Joseph, A.C.; Snedaker, K.; Breiding, M.J.; Robertson, C.L.; Colantonio, A.; Levin, H.; Pugh, M.J.; Yurgelun-Todd, D.; Mannix, R.; et al. Understanding Traumatic Brain Injury in Females: A State-of-the-Art Summary and Future Directions. J. Head Trauma Rehabil. 2021, 36, E1–E17. [Google Scholar] [CrossRef] [PubMed]
- Koerte, I.K.; Schultz, V.; Sydnor, V.J.; Howell, D.R.; Guenette, J.P.; Dennis, E.; Kochsiek, J.; Kaufmann, D.; Sollmann, N.; Mondello, S.; et al. Sex-Related Differences in the Effects of Sports-Related Concussion: A Review. J. Neuroimaging Off. J. Am. Soc. Neuroimaging 2020, 30, 387–409. [Google Scholar] [CrossRef] [PubMed]
- Costello, K.; Greenwald, B.D. Update on Domestic Violence and Traumatic Brain Injury: A Narrative Review. Brain Sci. 2022, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- Gupte, R.; Brooks, W.; Vukas, R.; Pierce, J.; Harris, J. Sex Differences in Traumatic Brain Injury: What We Know and What We Should Know. J. Neurotrauma 2019, 36, 3063–3091. [Google Scholar] [CrossRef] [PubMed]
- Snedaker, K.P. Women with Brain Injury: Past, Present, and Future. Brain Inj. 2020, 17, 8–13. [Google Scholar]
- Bazarian, J.J.; Blyth, B.; Mookerjee, S.; He, H.; McDermott, M.P. Sex differences in outcome after mild traumatic brain injury. J. Neurotrauma 2010, 27, 527–539. [Google Scholar] [CrossRef]
- Stein, D.G.; Wright, D.W. Progesterone in the clinical treatment of acute traumatic brain injury. Expert Opin. Investig. Drugs 2010, 19, 847–857. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zhang, X.; Wei, X.; Li, Y. Progesterone attenuates aquaporin-4 expression in an astrocyte model of ischemia/reperfusion. Neurochem. Res. 2014, 39, 2251–2261. [Google Scholar] [CrossRef]
- Chen, G.; Shi, J.X.; Qi, M.; Wang, H.X.; Hang, C.H. Effects of progesterone on intestinal inflammatory response, mucosa structure alterations, and apoptosis following traumatic brain injury in male rats. J. Surg. Res. 2008, 147, 92–98. [Google Scholar] [CrossRef]
- Sayeed, I.; Stein, D.G. Progesterone as a neuroprotective factor in traumatic and ischemic brain injury. Prog. Brain Res. 2009, 175, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.W.; Kellermann, A.L.; Hertzberg, V.S.; Clark, P.L.; Frankel, M.; Goldstein, F.C.; Salomone, J.P.; Dent, L.L.; Harris, O.A.; Ander, D.S.; et al. ProTECT: A randomized clinical trial of progesterone for acute traumatic brain injury. Ann. Emerg. Med. 2007, 49, 391–402.e2. [Google Scholar] [CrossRef]
- Xiao, G.; Wei, J.; Yan, W.; Wang, W.; Lu, Z. Improved outcomes from the administration of progesterone for patients with acute severe traumatic brain injury: A randomized controlled trial. Crit. Care 2008, 12, R61. [Google Scholar] [CrossRef] [PubMed]
- Skolnick, B.E.; Maas, A.I.; Narayan, R.K.; van der Hoop, R.G.; MacAllister, T.; Ward, J.D.; Nelson, N.R.; Stocchetti, N. A clinical trial of progesterone for severe traumatic brain injury. N. Engl. J. Med. 2014, 371, 2467–2476. [Google Scholar] [CrossRef]
- Roof, R.L.; Hall, E.D. Estrogen-related gender difference in survival rate and cortical blood flow after impact-acceleration head injury in rats. J. Neurotrauma 2000, 17, 1155–1169. [Google Scholar] [CrossRef] [PubMed]
- Dubal, D.B.; Shughrue, P.J.; Wilson, M.E.; Merchenthaler, I.; Wise, P.M. Estradiol modulates bcl-2 in cerebral ischemia: A potential role for estrogen receptors. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 6385–6393. [Google Scholar] [CrossRef] [PubMed]
- Wise, P.M.; Dubal, D.B.; Wilson, M.E.; Rau, S.W. Estradiol is a neuroprotective factor in in vivo and in vitro models of brain injury. J. Neurocytol. 2000, 29, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Bruce-Keller, A.J.; Dimayuga, F.O.; Reed, J.L.; Wang, C.; Angers, R.; Wilson, M.E.; Dimayuga, V.M.; Scheff, S.W. Gender and estrogen manipulation do not affect traumatic brain injury in mice. J. Neurotrauma 2007, 24, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Day, N.L.; Floyd, C.L.; D’Alessandro, T.L.; Hubbard, W.J.; Chaudry, I.H. 17β-estradiol confers protection after traumatic brain injury in the rat and involves activation of G protein-coupled estrogen receptor 1. J. Neurotrauma 2013, 30, 1531–1541. [Google Scholar] [CrossRef]
- Soustiel, J.F.; Palzur, E.; Nevo, O.; Thaler, I.; Vlodavsky, E. Neuroprotective anti-apoptosis effect of estrogens in traumatic brain injury. J. Neurotrauma 2005, 22, 345–352. [Google Scholar] [CrossRef]
- Naderi, V.; Khaksari, M.; Abbasi, R.; Maghool, F. Estrogen provides neuroprotection against brain edema and blood brain barrier disruption through both estrogen receptors α and β following traumatic brain injury. Iran. J. Basic. Med. Sci. 2015, 18, 138–144. [Google Scholar] [PubMed]
- Torres, M.J.; Kew, K.A.; Ryan, T.E.; Pennington, E.R.; Lin, C.T.; Buddo, K.A.; Fix, A.M.; Smith, C.A.; Gilliam, L.A.; Karvinen, S.; et al. 17β-Estradiol Directly Lowers Mitochondrial Membrane Microviscosity and Improves Bioenergetic Function in Skeletal Muscle. Cell Metab. 2018, 27, 167–179.e167. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, H.; Quinn, P.; Halliwell, B. Tamoxifen and related compounds decrease membrane fluidity in liposomes. Mechanism for the antioxidant action of tamoxifen and relevance to its anticancer and cardioprotective actions? FEBS Lett. 1993, 330, 53–56. [Google Scholar] [CrossRef]
- Behl, C.; Skutella, T.; Lezoualc’h, F.; Post, A.; Widmann, M.; Newton, C.J.; Holsboer, F. Neuroprotection against oxidative stress by estrogens: Structure-activity relationship. Mol. Pharmacol. 1997, 51, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Oosthuyse, T.; Strauss, J.A.; Hackney, A.C. Understanding the female athlete: Molecular mechanisms underpinning menstrual phase differences in exercise metabolism. Eur. J. Appl. Physiol. 2023, 123, 423–450. [Google Scholar] [CrossRef] [PubMed]
- Culmsee, C.; Vedder, H.; Ravati, A.; Junker, V.; Otto, D.; Ahlemeyer, B.; Krieg, J.C.; Krieglstein, J. Neuroprotection by estrogens in a mouse model of focal cerebral ischemia and in cultured neurons: Evidence for a receptor-independent antioxidative mechanism. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow. Metab. 1999, 19, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Prokai, L.; Prokai-Tatrai, K.; Perjesi, P.; Zharikova, A.D.; Perez, E.J.; Liu, R.; Simpkins, J.W. Quinol-based cyclic antioxidant mechanism in estrogen neuroprotection. Proc. Natl. Acad. Sci. USA 2003, 100, 11741–11746. [Google Scholar] [CrossRef] [PubMed]
- Shahrokhi, N.; Haddad, M.K.; Joukar, S.; Shabani, M.; Keshavarzi, Z.; Shahozehi, B. Neuroprotective antioxidant effect of sex steroid hormones in traumatic brain injury. Pak. J. Pharm. Sci. 2012, 25, 219–225. [Google Scholar] [PubMed]
- Scheff, S.W.; Ansari, M.A.; Roberts, K.N. Neuroprotective effect of Pycnogenol® following traumatic brain injury. Exp. Neurol. 2013, 239, 183–191. [Google Scholar] [CrossRef]
- Yang, T.; Kong, B.; Gu, J.W.; Kuang, Y.Q.; Cheng, L.; Yang, W.T.; Xia, X.; Shu, H.F. Anti-apoptotic and anti-oxidative roles of quercetin after traumatic brain injury. Cell. Mol. Neurobiol. 2014, 34, 797–804. [Google Scholar] [CrossRef]
- Hicdonmez, T.; Kanter, M.; Tiryaki, M.; Parsak, T.; Cobanoglu, S. Neuroprotective effects of N-acetylcysteine on experimental closed head trauma in rats. Neurochem. Res. 2006, 31, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Cui, Y.; Gao, J.L.; Li, R.; Jiang, X.H.; Tian, Y.X.; Wang, K.J.; Li, M.H.; Zhang, H.A.; Cui, J.Z. Neuroprotective effects of resveratrol against traumatic brain injury in rats: Involvement of synaptic proteins and neuronal autophagy. Mol. Med. Rep. 2016, 13, 5248–5254. [Google Scholar] [CrossRef] [PubMed]
- Prokai, L.; Prokai-Tatrai, K.; Perjesi, P.; Simpkins, J.W. Mechanistic insights into the direct antioxidant effects of estrogens. Drug Dev. Res. 2005, 66, 118–125. [Google Scholar] [CrossRef]
- Davis, C.K.; Vemuganti, R. Antioxidant therapies in traumatic brain injury. Neurochem. Int. 2022, 152, 105255. [Google Scholar] [CrossRef] [PubMed]
- Tarudji, A.W.; Miller, H.A.; Curtis, E.T.; Porter, C.L.; Madsen, G.L.; Kievit, F.M. Sex-based differences of antioxidant enzyme nanoparticle effects following traumatic brain injury. J. Control Release 2023, 355, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Kalimon, O.J.; Sullivan, P.G. Sex Differences in Mitochondrial Function Following a Controlled Cortical Impact Traumatic Brain Injury in Rodents. Front. Mol. Neurosci. 2021, 14, 753946. [Google Scholar] [CrossRef] [PubMed]
- Yonutas, H.M.; Vekaria, H.J.; Sullivan, P.G. Mitochondrial specific therapeutic targets following brain injury. Brain Res. 2016, 1640, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Rabchevsky, A.G.; Waldmeier, P.C.; Springer, J.E. Mitochondrial permeability transition in CNS trauma: Cause or effect of neuronal cell death? J. Neurosci. Res. 2005, 79, 231–239. [Google Scholar] [CrossRef]
- Pandya, J.D.; Nukala, V.N.; Sullivan, P.G. Concentration dependent effect of calcium on brain mitochondrial bioenergetics and oxidative stress parameters. Front. Neuroenergetics 2013, 5, 10. [Google Scholar] [CrossRef]
- Hubbard, W.B.; Spry, M.L.; Gooch, J.L.; Cloud, A.L.; Vekaria, H.J.; Burden, S.; Powell, D.K.; Berkowitz, B.A.; Geldenhuys, W.J.; Harris, N.G.; et al. Clinically relevant mitochondrial-targeted therapy improves chronic outcomes after traumatic brain injury. Brain A J. Neurol. 2021, 144, 3788–3807. [Google Scholar] [CrossRef]
- Fiskum, G. Mitochondrial participation in ischemic and traumatic neural cell death. J. Neurotrauma 2000, 17, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Keller, J.N.; Bussen, W.L.; Scheff, S.W. Cytochrome c release and caspase activation after traumatic brain injury. Brain Res. 2002, 949, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Kulbe, J.R.; Singh, I.N.; Wang, J.A.; Cebak, J.E.; Hall, E.D. Continuous Infusion of Phenelzine, Cyclosporine A, or Their Combination: Evaluation of Mitochondrial Bioenergetics, Oxidative Damage, and Cytoskeletal Degradation following Severe Controlled Cortical Impact Traumatic Brain Injury in Rats. J. Neurotrauma 2018, 35, 1280–1293. [Google Scholar] [CrossRef] [PubMed]
- Greco, T.; Vespa, P.M.; Prins, M.L. Alternative substrate metabolism depends on cerebral metabolic state following traumatic brain injury. Exp. Neurol. 2020, 329, 113289. [Google Scholar] [CrossRef]
- Kalimon, O.J.; Vekaria, H.J.; Velmurugan, G.V.; Hubbard, W.B.; Sullivan, P.G. Characterizing Sex Differences in Mitochondrial Dysfunction After Severe Traumatic Brain Injury in Mice. Neurotrauma Rep. 2023, 4, 627–642. [Google Scholar] [CrossRef]
- Brinton, R.D. The healthy cell bias of estrogen action: Mitochondrial bioenergetics and neurological implications. Trends Neurosci. 2008, 31, 529–537. [Google Scholar] [CrossRef]
- Irwin, R.W.; Yao, J.; Hamilton, R.T.; Cadenas, E.; Brinton, R.D.; Nilsen, J. Progesterone and estrogen regulate oxidative metabolism in brain mitochondria. Endocrinology 2008, 149, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- Simpkins, J.W.; Yang, S.H.; Sarkar, S.N.; Pearce, V. Estrogen actions on mitochondria--physiological and pathological implications. Mol. Cell. Endocrinol. 2008, 290, 51–59. [Google Scholar] [CrossRef]
- Cheng, C.M.; Cohen, M.; Wang, J.; Bondy, C.A. Estrogen augments glucose transporter and IGF1 expression in primate cerebral cortex. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2001, 15, 907–915. [Google Scholar] [CrossRef]
- Morselli, E.; Santos, R.S.; Gao, S.; Ávalos, Y.; Criollo, A.; Palmer, B.F.; Clegg, D.J. Impact of estrogens and estrogen receptor-α in brain lipid metabolism. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E7–E14. [Google Scholar] [CrossRef]
- Gaignard, P.; Savouroux, S.; Liere, P.; Pianos, A.; Thérond, P.; Schumacher, M.; Slama, A.; Guennoun, R. Effect of Sex Differences on Brain Mitochondrial Function and Its Suppression by Ovariectomy and in Aged Mice. Endocrinology 2015, 156, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, J.; Diaz Brinton, R. Mechanism of estrogen-mediated neuroprotection: Regulation of mitochondrial calcium and Bcl-2 expression. Proc. Natl. Acad. Sci. USA 2003, 100, 2842–2847. [Google Scholar] [CrossRef]
- Khalifa, A.R.; Abdel-Rahman, E.A.; Mahmoud, A.M.; Ali, M.H.; Noureldin, M.; Saber, S.H.; Mohsen, M.; Ali, S.S. Sex-specific differences in mitochondria biogenesis, morphology, respiratory function, and ROS homeostasis in young mouse heart and brain. Physiol. Rep. 2017, 5, e13125. [Google Scholar] [CrossRef]
- Borrás, C.; Gambini, J.; López-Grueso, R.; Pallardó, F.V.; Viña, J. Direct antioxidant and protective effect of estradiol on isolated mitochondria. Biochim. Biophys. Acta 2010, 1802, 205–211. [Google Scholar] [CrossRef]
- Moreno, A.J.; Moreira, P.I.; Custódio, J.B.; Santos, M.S. Mechanism of inhibition of mitochondrial ATP synthase by 17β-estradiol. J. Bioenerg. Biomembr. 2013, 45, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Pandya, J.D.; Pauly, J.R.; Nukala, V.N.; Sebastian, A.H.; Day, K.M.; Korde, A.S.; Maragos, W.F.; Hall, E.D.; Sullivan, P.G. Post-Injury Administration of Mitochondrial Uncouplers Increases Tissue Sparing and Improves Behavioral Outcome following Traumatic Brain Injury in Rodents. J. Neurotrauma 2007, 24, 798–811. [Google Scholar] [CrossRef] [PubMed]
- Pandya, J.D.; Pauly, J.R.; Sullivan, P.G. The optimal dosage and window of opportunity to maintain mitochondrial homeostasis following traumatic brain injury using the uncoupler FCCP. Exp. Neurol. 2009, 218, 381–389. [Google Scholar] [CrossRef]
- Hubbard, W.B.; Harwood, C.L.; Geisler, J.G.; Vekaria, H.J.; Sullivan, P.G. Mitochondrial uncoupling prodrug improves tissue sparing, cognitive outcome, and mitochondrial bioenergetics after traumatic brain injury in male mice. J. Neurosci. Res. 2018, 96, 1677–1688. [Google Scholar] [CrossRef] [PubMed]
- Kalimon, O.J.; Vekaria, H.J.; Gerhardt, G.A.; Sullivan, P.G. Inhibition of monoamine oxidase-a increases respiration in isolated mouse cortical mitochondria. Exp. Neurol. 2023, 363, 114356. [Google Scholar] [CrossRef]
- Hubbard, W.B.; Harwood, C.L.; Prajapati, P.; Springer, J.E.; Saatman, K.E.; Sullivan, P.G. Fractionated mitochondrial magnetic separation for isolation of synaptic mitochondria from brain tissue. Sci. Rep. 2019, 9, 9656. [Google Scholar] [CrossRef]
- Brown, M.R.; Sullivan, P.G.; Dorenbos, K.A.; Modafferi, E.A.; Geddes, J.W.; Steward, O. Nitrogen disruption of synaptoneurosomes: An alternative method to isolate brain mitochondria. J. Neurosci. Methods 2004, 137, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Visavadiya, N.P.; McEwen, M.L.; Pandya, J.D.; Sullivan, P.G.; Gwag, B.J.; Springer, J.E. Antioxidant properties of Neu2000 on mitochondrial free radicals and oxidative damage. Toxicol. In Vitro Int. J. Publ. Assoc. BIBRA 2013, 27, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Visavadiya, N.P.; Patel, S.P.; VanRooyen, J.L.; Sullivan, P.G.; Rabchevsky, A.G. Cellular and subcellular oxidative stress parameters following severe spinal cord injury. Redox Biol. 2016, 8, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Vekaria, H.J.; Kalimon, O.J.; Prajapati, P.; Velmurugan, G.V.; Sullivan, P.G. An efficient and high-throughput method for the evaluation of mitochondrial dysfunction in frozen brain samples after traumatic brain injury. Front. Mol. Biosci. 2024, 11, 1378536. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Lin, C.; Zuo, Q.; Liu, Y.; Xiao, M.; Xu, X.; Li, Z.; Bao, Z.; Chen, H.; You, Y.; et al. Cardiolipin-Dependent Mitophagy Guides Outcome after Traumatic Brain Injury. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 1930–1943. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, W.B.; Velmurugan, G.V.; Sullivan, P.G. The role of mitochondrial uncoupling in the regulation of mitostasis after traumatic brain injury. Neurochem. Int. 2024, 174, 105680. [Google Scholar] [CrossRef]
- Clark, R.S.; Bayir, H.; Chu, C.T.; Alber, S.M.; Kochanek, P.M.; Watkins, S.C. Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy 2008, 4, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.S.; Kochanek, P.M.; Watkins, S.C.; Chen, M.; Dixon, C.E.; Seidberg, N.A.; Melick, J.; Loeffert, J.E.; Nathaniel, P.D.; Jin, K.L.; et al. Caspase-3 mediated neuronal death after traumatic brain injury in rats. J. Neurochem. 2000, 74, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Prins, M.L.; Samii, M.; Hovda, D.A. Cerebral metabolic response to traumatic brain injury sustained early in development: A 2-deoxy-D-glucose autoradiographic study. J. Neurotrauma 2000, 17, 649–665. [Google Scholar] [CrossRef]
- Yoshino, A.; Hovda, D.A.; Kawamata, T.; Katayama, Y.; Becker, D.P. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: Evidence of a hyper- and subsequent hypometabolic state. Brain Res. 1991, 561, 106–119. [Google Scholar] [CrossRef]
- Bergsneider, M.; Hovda, D.A.; Shalmon, E.; Kelly, D.F.; Vespa, P.M.; Martin, N.A.; Phelps, M.E.; McArthur, D.L.; Caron, M.J.; Kraus, J.F.; et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: A positron emission tomography study. J. Neurosurg. 1997, 86, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Har-Even, M.; Rubovitch, V.; Ratliff, W.A.; Richmond-Hacham, B.; Citron, B.A.; Pick, C.G. Ketogenic Diet as a potential treatment for traumatic brain injury in mice. Sci. Rep. 2021, 11, 23559. [Google Scholar] [CrossRef] [PubMed]
- Greco, T.; Glenn, T.C.; Hovda, D.A.; Prins, M.L. Ketogenic diet decreases oxidative stress and improves mitochondrial respiratory complex activity. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2016, 36, 1603–1613. [Google Scholar] [CrossRef]
- Oosthuyse, T.; Bosch, A.N. Oestrogen’s regulation of fat metabolism during exercise and gender specific effects. Curr. Opin. Pharmacol. 2012, 12, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Scafidi, S.; Jernberg, J.; Fiskum, G.; McKenna, M.C. Metabolism of Exogenous [2,4-(13)C]β-Hydroxybutyrate following Traumatic Brain Injury in 21-22-Day-Old Rats: An Ex Vivo NMR Study. Metabolites 2022, 12, 710. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Springer, J.E.; Hall, E.D.; Scheff, S.W. Mitochondrial uncoupling as a therapeutic target following neuronal injury. J. Bioenerg. Biomembr. 2004, 36, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.L.; Kulbe, J.R.; Singh, I.N.; Wang, J.A.; Hall, E.D. Synaptic Mitochondria are More Susceptible to Traumatic Brain Injury-induced Oxidative Damage and Respiratory Dysfunction than Non-synaptic Mitochondria. Neuroscience 2018, 386, 265–283. [Google Scholar] [CrossRef] [PubMed]
- Mnatsakanyan, N.; Llaguno, M.C.; Yang, Y.; Yan, Y.; Weber, J.; Sigworth, F.J.; Jonas, E.A. A mitochondrial megachannel resides in monomeric F(1)F(O) ATP synthase. Nat. Commun. 2019, 10, 5823. [Google Scholar] [CrossRef] [PubMed]
- Mnatsakanyan, N.; Park, H.A.; Wu, J.; He, X.; Llaguno, M.C.; Latta, M.; Miranda, P.; Murtishi, B.; Graham, M.; Weber, J.; et al. Mitochondrial ATP synthase c-subunit leak channel triggers cell death upon loss of its F(1) subcomplex. Cell Death Differ. 2022, 29, 1874–1887. [Google Scholar] [CrossRef]
- Mnatsakanyan, N.; Jonas, E.A. ATP synthase c-subunit ring as the channel of mitochondrial permeability transition: Regulator of metabolism in development and degeneration. J. Mol. Cell. Cardiol. 2020, 144, 109–118. [Google Scholar] [CrossRef]
- Horvat, A.; Nikezić, G.; Petrović, S.; Kanazir, D.T. Binding of estradiol to synaptosomal mitochondria: Physiological significance. Cell. Mol. Life Sci. 2001, 58, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Horvat, A.; Petrović, S.; Nedeljković, N.; Martinović, J.V.; Nikezić, G. Estradiol affect Na-dependent Ca2+ efflux from synaptosomal mitochondria. Gen. Physiol. Biophys. 2000, 19, 59–71. [Google Scholar] [PubMed]
- Glenn, T.C.; Kelly, D.F.; Boscardin, W.J.; McArthur, D.L.; Vespa, P.; Oertel, M.; Hovda, D.A.; Bergsneider, M.; Hillered, L.; Martin, N.A. Energy dysfunction as a predictor of outcome after moderate or severe head injury: Indices of oxygen, glucose, and lactate metabolism. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2003, 23, 1239–1250. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Huang, S.C.; Wu, H.M.; Yeh, E.; Glenn, T.C.; Vespa, P.M.; McArthur, D.; Phelps, M.E.; Hovda, D.A.; Bergsneider, M. Correlation of regional metabolic rates of glucose with glasgow coma scale after traumatic brain injury. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2003, 44, 1709–1716. [Google Scholar]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Svedung Wettervik, T.; Hånell, A.; Howells, T.; Enblad, P.; Lewén, A. Females Exhibit Better Cerebral Pressure Autoregulation, Less Mitochondrial Dysfunction, and Reduced Excitotoxicity after Severe Traumatic Brain Injury. J. Neurotrauma 2022, 39, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535.e1514. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Qi, G.; Vitali, F.; Shang, Y.; Raikes, A.C.; Wang, T.; Jin, Y.; Brinton, R.D.; Gu, H.; Yin, F. Loss of fatty acid degradation by astrocytic mitochondria triggers neuroinflammation and neurodegeneration. Nat. Metab. 2023, 5, 445–465. [Google Scholar] [CrossRef]
- Morant-Ferrando, B.; Jimenez-Blasco, D.; Alonso-Batan, P.; Agulla, J.; Lapresa, R.; Garcia-Rodriguez, D.; Yunta-Sanchez, S.; Lopez-Fabuel, I.; Fernandez, E.; Carmeliet, P.; et al. Fatty acid oxidation organizes mitochondrial supercomplexes to sustain astrocytic ROS and cognition. Nat. Metab. 2023, 5, 1290–1302. [Google Scholar] [CrossRef]
- Villapol, S.; Loane, D.J.; Burns, M.P. Sexual dimorphism in the inflammatory response to traumatic brain injury. Glia 2017, 65, 1423–1438. [Google Scholar] [CrossRef]
- Carter, S.L.; Rennie, C.; Tarnopolsky, M.A. Substrate utilization during endurance exercise in men and women after endurance training. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E898–E907. [Google Scholar] [CrossRef] [PubMed]
- Montero, D.; Madsen, K.; Meinild-Lundby, A.K.; Edin, F.; Lundby, C. Sexual dimorphism of substrate utilization: Differences in skeletal muscle mitochondrial volume density and function. Exp. Physiol. 2018, 103, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.; Mayorov, V.I.; Dikalov, S. Metabolic Syndrome and β-Oxidation of Long-Chain Fatty Acids in the Brain, Heart, and Kidney Mitochondria. Int. J. Mol. Sci. 2022, 23, 4047. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalimon, O.J.; Vekaria, H.J.; Prajapati, P.; Short, S.L.; Hubbard, W.B.; Sullivan, P.G. The Uncoupling Effect of 17β-Estradiol Underlies the Resilience of Female-Derived Mitochondria to Damage after Experimental TBI. Life 2024, 14, 961. https://doi.org/10.3390/life14080961
Kalimon OJ, Vekaria HJ, Prajapati P, Short SL, Hubbard WB, Sullivan PG. The Uncoupling Effect of 17β-Estradiol Underlies the Resilience of Female-Derived Mitochondria to Damage after Experimental TBI. Life. 2024; 14(8):961. https://doi.org/10.3390/life14080961
Chicago/Turabian StyleKalimon, Olivia J., Hemendra J. Vekaria, Paresh Prajapati, Sydney L. Short, W. Brad Hubbard, and Patrick G. Sullivan. 2024. "The Uncoupling Effect of 17β-Estradiol Underlies the Resilience of Female-Derived Mitochondria to Damage after Experimental TBI" Life 14, no. 8: 961. https://doi.org/10.3390/life14080961