
Cataloging the Genetic Response: Unveiling Drought-Responsive Gene Expression in Oil Tea Camellia (Camellia oleifera Abel.) through Transcriptomics
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Site
2.2. Plant Materials, Drought Treatment, and Experimental Design
2.3. Measurements and Analysis
2.3.1. Soil Volumetric Moisture Content Measurement
2.3.2. RWC Measurement
2.3.3. Antioxidant Enzyme Activities Measurement
2.3.4. RNA Isolation and Qualification
2.3.5. Library Preparation for Transcriptome Sequencing
2.3.6. Cleaning and Mapping of Sequenced Reads
2.3.7. Differential Expression Analysis
2.3.8. GO and KEGG Pathway Enrichment Analysis
2.3.9. Quantitative Real-Time PCR Analysis
2.4. Statistical Analysis
3. Results
3.1. Phenotypic Observation and Physiological Analysis
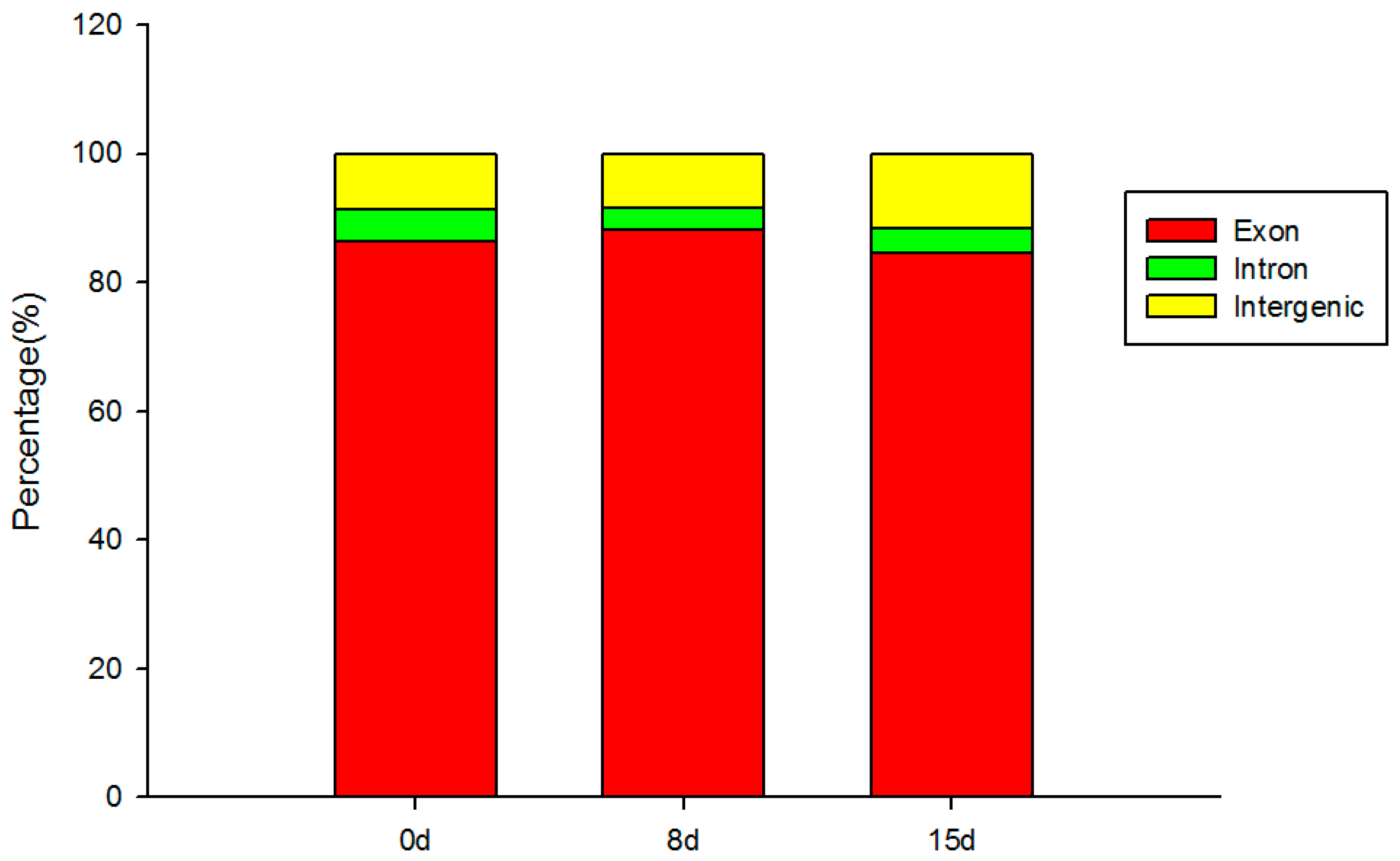
3.2. Transcriptome Sequencing and Assembly
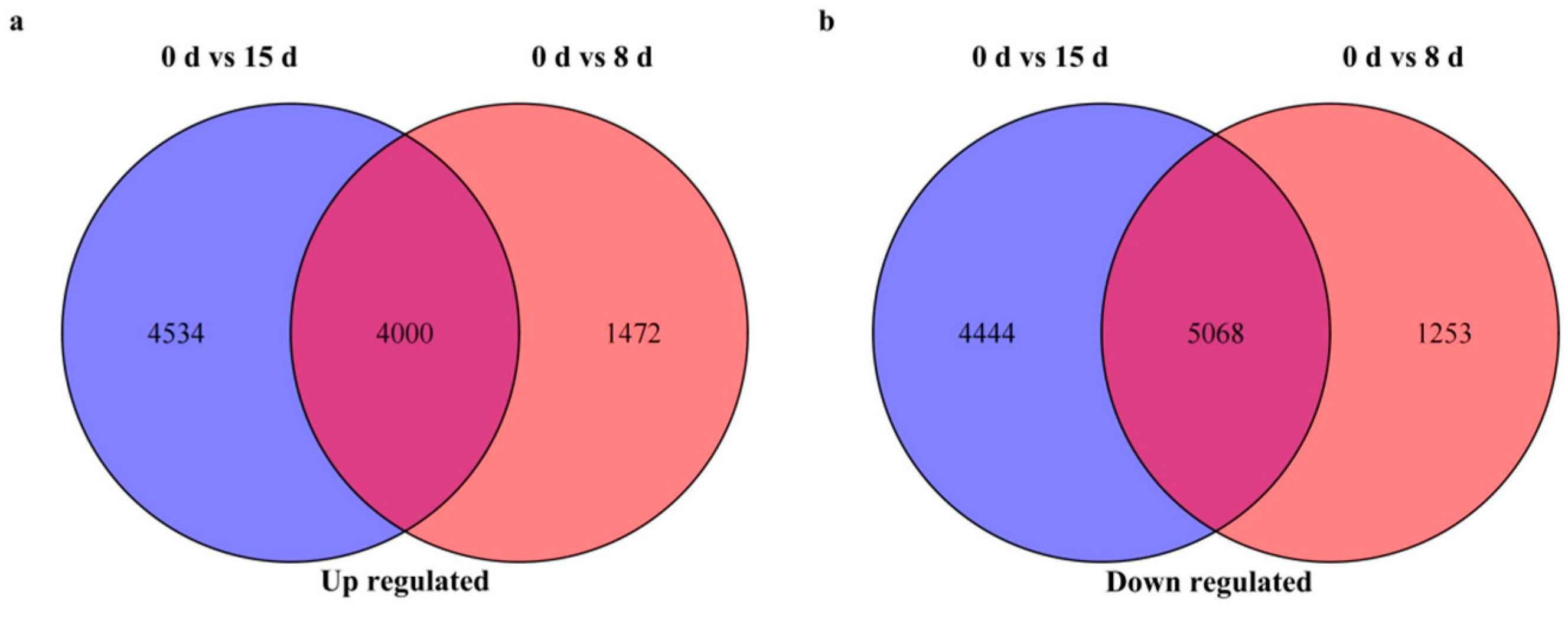
3.3. Identification and Analysis of DEGs
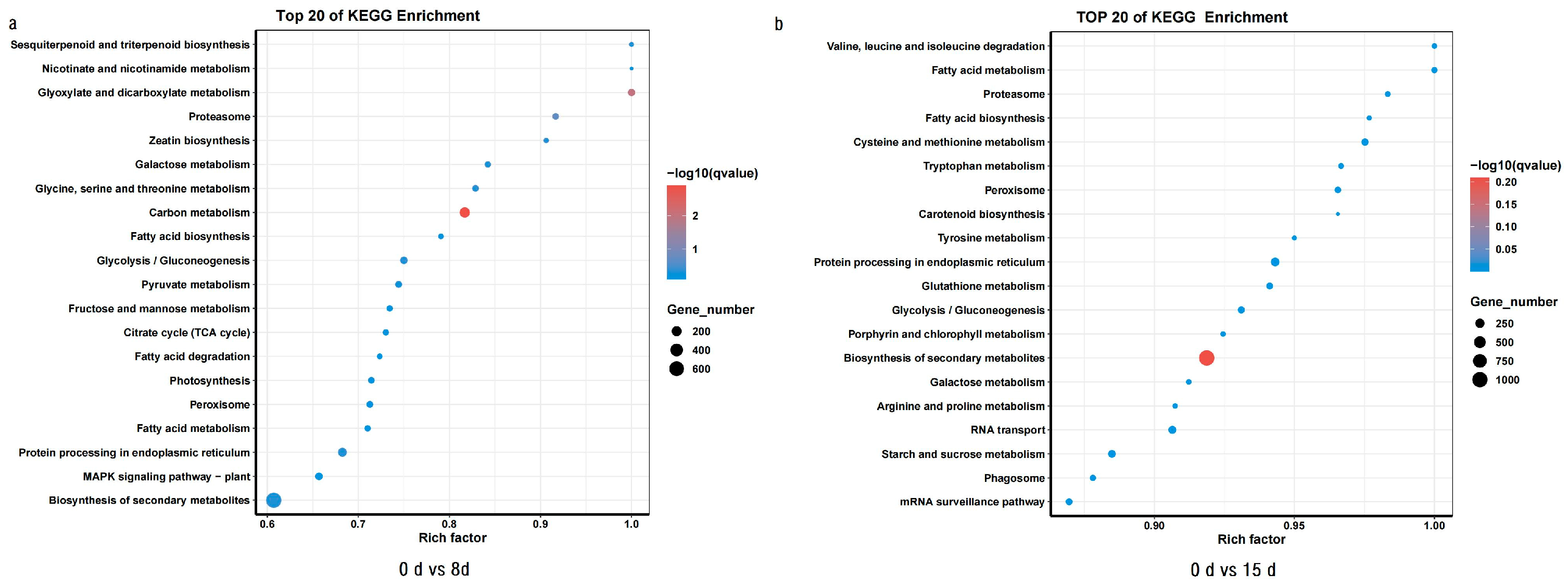
3.4. GO and KEGG Enrichment Analysis of DEGs
3.5. Verification of Selected DEGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bashir, S.S.; Hussain, A.; Hussain, S.J.; Wani, O.A.; Zahid Nabi, S.; Dar, N.A.; Mansoor, S. Plant drought stress tolerance: Understanding its physiological, biochemical and molecular mechanisms. Biotechnol. Biotechnol. Equip. 2021, 35, 1912–1925. [Google Scholar] [CrossRef]
- Razi, K.; Muneer, S. Drought stress-induced physiological mechanisms, signaling pathways and molecular response of chloroplasts in common vegetable crops. Crit. Rev. Biotechnol. 2021, 41, 669–691. [Google Scholar] [CrossRef]
- Sinha, R.; Fritschi, F.B.; Zandalinas, S.I.; Mittler, R. The impact of stress combination on reproductive processes in crops. Plant Sci. 2021, 311, 111007. [Google Scholar] [CrossRef]
- Suzuki, N.; Rivero, R.M.; Shulaev, V.; Blumwald, E.; Mittler, R. Abiotic and biotic stress combinations. New Phytol. 2014, 203, 32–43. [Google Scholar] [CrossRef]
- Sharma, A.; Choudhary, P.; Chakdar, H.; Shukla, P. Molecular insights and omics-based understanding of plant-microbe interactions under drought stress. World J. Microbiol. Biotechnol. 2023, 40, 42. [Google Scholar] [CrossRef]
- Osakabe, Y.; Osakabe, K.; Shinozaki, K.; Tran, L.S. Response of plants to water stress. Front. Plant. Sci. 2014, 5, 76566. [Google Scholar] [CrossRef]
- Chaves, M.M.; Flexas, J.; Pinheiro, C. Photosynthesis under drought and salt stress: Regulation mechanisms from whole plant to cell. Ann. Bot. 2009, 103, 551–560. [Google Scholar] [CrossRef]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef]
- Krasensky, J.; Jonak, C. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 2012, 63, 1593–1608. [Google Scholar] [CrossRef]
- Mardis, E.R. Next-generation DNA sequencing methods. Annu. Rev. Genom. Hum. Genet. 2008, 9, 387–402. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Imadi, S.R.; Kazi, A.G.; Ahanger, M.A.; Gucel, S.; Ahmad, P. Plant transcriptomics and responses to environmental stress: An overview. J. Genet. 2015, 94, 525–537. [Google Scholar] [CrossRef]
- Sun, L.F.; Liu, L.B.; Wang, Y.T.; Feng, Y.F.; Yang, W.; Wang, D.; Gao, S.R.; Miao, X.F.; Sun, W.T. Integration of Metabolomics and Transcriptomics for Investigating the Tolerance of Foxtail Millet (Setaria italica) to Atrazine Stress. Front. Plant Sci. 2022, 3, 890550. [Google Scholar] [CrossRef]
- Ding, X.P.; Li, X.K.; Xiong, L.Z. Insight into differential responses of upland and paddy rice to drought stress by comparative expression profiling analysis. Int. J. Mol. Sci. 2013, 14, 5214–5238. [Google Scholar] [CrossRef]
- Dong, B.; Wu, B.; Hong, W.H.; Li, X.P.; Li, Z.; Xue, L.; Huang, Y.F. Transcriptome analysis of the tea oil camellia (Camellia oleifera) reveals candidate drought stress genes. PLoS ONE 2017, 12, e0181835. [Google Scholar] [CrossRef]
- Yoshida, T.; Mogami, J.; Yamaguchi-Shinozaki, K. ABA-dependent and ABA-independent signaling in response to osmotic stress in plants. Curr. Opin. Plant Biol. 2014, 21, 133–139. [Google Scholar] [CrossRef]
- Manna, M.; Thakur, T.; Chirom, O.; Mandlik, R.; Deshmukh, R.; Salvi, P. Transcription factors as key molecular target to strengthen the drought stress tolerance in plants. Physiol. Plant 2020, 172, 847–868. [Google Scholar] [CrossRef]
- Guo, Q.Q.; Li, X.; Niu, L.; Jameson, P.E.; Zhou, W.B. Transcription-associated metabolomic adjustments in maize occur during combined drought and cold stress. Plant Physiol. 2021, 186, 677–695. [Google Scholar] [CrossRef]
- Wang, X.Y.; Song, S.; Wang, X.; Liu, J.; Dong, S.K. Transcriptomic and Metabolomic Analysis of Seedling-Stage Soybean Responses to PEG-Simulated Drought Stress. Int. J. Mol. Sci. 2022, 23, 6869. [Google Scholar] [CrossRef]
- Wu, L.L.; Li, J.A.; Li, Z.; Zhang, F.H.; Tan, X.F. Transcriptomic analyses of Camellia oleifera ‘Huaxin’leaf reveal candidate genes related to long-term cold stress. Int. J. Mol. Sci. 2020, 21, 846. [Google Scholar] [CrossRef]
- Li, G.H.; Ma, L.; Yan, Z.P.; Zhu, Q.H.; Cai, J.T.; Wang, S.Y.; Yuan, Y.; Chen, Y.Z.; Deng, S.W. Extraction of oils and phytochemicals from Camellia oleifera seeds: Trends, challenges, and innovations. Processes 2022, 10, 1489. [Google Scholar] [CrossRef]
- Chen, J.M.; Yang, X.Q.; Huang, X.M.; Duan, S.H.; Long, C.; Chen, J.K.; Rong, J. Leaf transcriptome analysis of a subtropical evergreen broadleaf plant, wild oil-tea camellia (Camellia oleifera), revealing candidate genes for cold acclimation. BMC Genom. 2017, 18, 211. [Google Scholar] [CrossRef]
- He, Z.L.; Liu, C.X.; Zhang, Z.; Wang, R.; Chen, Y.Z. Integration of mRNA and miRNA analysis reveals the differentially regulatory network in two different Camellia oleifera cultivars under drought stress. Front. Plant Sci. 2022, 13, 1001357. [Google Scholar] [CrossRef]
- Ye, H.L.; Folz, J.; Li, C.; Zhang, Y.; Hou, Z.X.; Zhang, L.Y.; Su, S.C. Response of metabolic and lipid synthesis gene expression changes in Camellia oleifera to mulched ecological mat under drought conditions. Sci. Total. Environ. 2021, 795, 148856. [Google Scholar] [CrossRef]
- He, Z.L.; Cui, K.P.; Wang, R.; Xu, T.; Zhang, Z.; Wang, X.N.; Chen, Y.Z.; Zhu, Y.H. Multi-omics joint analysis reveals how Streptomyces albidoflavus OsiLf-2 assists Camellia oleifera to resist drought stress and improve fruit quality. Front. Microbiol. 2023, 14, 1152632. [Google Scholar] [CrossRef]
- He, X.S.; Xu, L.C.; Pan, C.; Gong, C.; Wang, Y.J.; Liu, X.L.; Yu, Y.C. Drought resistance of Camellia oleifera under drought stress: Changes in physiology and growth characteristics. PLoS ONE 2020, 15, e0235795. [Google Scholar] [CrossRef]
- Guo, P.R.; Wu, L.L.; Wang, Y.; Liu, D.; Li, J.A. Effects of Drought Stress on the Morphological Structure and Flower Organ Physiological Characteristics of Camellia oleifera Flower Buds. Plants 2023, 12, 2585. [Google Scholar] [CrossRef]
- Chen, J.J.; Han, X.J.; Ye, S.C.; Liu, L.X.; Yang, B.B.; Cao, Y.Q.; Zhuo, R.Y.; Yao, X.H. Integration of small RNA, degradome, and transcriptome sequencing data illustrates the mechanism of low phosphorus adaptation in Camellia oleifera. Front. Plant. Sci. 2022, 13, 932926. [Google Scholar] [CrossRef]
- Long, W.; Yao, X.H.; Wang, K.L.; Sheng, Y.; Lv, L.Y. De novo transcriptome assembly of the cotyledon of Camellia oleifera for discovery of genes regulating seed germination. BMC. Plant. Biol. 2022, 22, 265. [Google Scholar] [CrossRef]
- Gong, W.F.; Song, Q.L.; Ji, K.; Gong, S.F.; Wang, L.K.; Chen, L.; Zhang, J.; Yuan, D.Y. Full-length transcriptome from Camellia oleifera seed provides insight into the transcript variants involved in oil biosynthesis. J. Agric. Food. Chem. 2020, 68, 14670–14683. [Google Scholar] [CrossRef]
- Sheng, Y.; Yao, X.H.; Liu, L.X.; Yu, C.L.; Wang, K.X.; Wang, K.L.; Chang, J.; Chen, J.J.; Cao, Y.Q. Transcriptomic time-course sequencing: Insights into the cell wall macromolecule-mediated fruit dehiscence during ripening in Camellia oleifera. Plants 2023, 12, 3314. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, R.F.; Yang, Y.; Liang, G.C.; Zhang, H.; Deng, X.M.; Xi, R.C. Sugar metabolism and transcriptome analysis reveal key sugar transporters during Camellia oleifera fruit development. Int. J. Mol. Sci. 2022, 23, 822. [Google Scholar] [CrossRef]
- Zhao, N.N.; Cui, S.L.; Li, X.K.; Liu, B.K.; Deng, H.T.; Liu, Y.R.; Hou, M.Y.; Yang, X.L.; Mu, G.J.; Liu, L.F. Transcriptome and co-expression network analyses reveal differential gene expression and pathways in response to severe drought stress in peanut (Arachis hypogaea L.). Front. Genet. 2021, 12, 672884. [Google Scholar] [CrossRef]
- Zhu, K.K.; Wei, L.; Ma, W.J.; Hu, X.L.; Zhao, J.; Tan, P.P.; Liu, H.; Feng, G.; Fan, P.H.; Peng, F.R. Transcriptome analysis of pecan (Carya illinoinensis) differentially expressed genes in response to drought stress. Forests 2023, 14, 608. [Google Scholar] [CrossRef]
- Bartlett, M.K.; Scoffoni, C.; Sack, L. The determinants of leaf turgor loss point and prediction of drought tolerance of species and biomes: A global meta-analysis. Ecol. Lett. 2012, 15, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Castro-Díez, P.; Navarro, J. Water relations of seedlings of three Quercus species: Variations across and within species grown in contrasting light and water regimes. Tree Physiol. 2007, 27, 1011–1018. [Google Scholar] [CrossRef]
- Chen, J.J.; Sun, Y.; Kopp, K.; Oki, L.; Jones, S.B.; Hipps, L. Effects of Water Availability on Leaf Trichome Density and Plant Growth and Development of Shepherdia ×utahensis. Front. Plant Sci. 2022, 13, 855858. [Google Scholar] [CrossRef]
- Medyouni, I.; Zouaoui, R.; Rubio, E.; Serino, S.; Ahmed, H.B.; Bertin, N. Effects of water deficit on leaves and fruit quality during the development period in tomato plant. Food Sci. Nutr. 2021, 9, 1949–1960. [Google Scholar] [CrossRef]
- Qiu, R.; Han, G.; Li, S.W.; Tian, F.; Ma, X.; Gong, W. Soil moisture dominates the variation of gross primary productivity during hot drought in drylands. Sci. Total Environ. 2023, 899, 165686. [Google Scholar] [CrossRef]
- Zheng, C.; Wang, S.Q.; Chen, J.H.; Xiang, N.; Sun, L.G.; Chen, B.; Fu, Z.; Zhu, K.; He, X.L. Divergent impacts of VPD and SWC on ecosystem carbon-water coupling under different dryness conditions. Sci. Total Environ. 2023, 905, 167007. [Google Scholar] [CrossRef]
- Chen, G.Q.; Ren, L.; Zhang, J.; Reed, B.M.; Zhang, D.; Shen, X.H. Cryopreservation affects ROS-induced oxidative stress and antioxidant response in Arabidopsis seedlings. Cryobiology 2015, 70, 38–47. [Google Scholar] [CrossRef]
- Shoaib, M.; Hussain, S.; Cheng, X.R.; Cui, Y.L.; Liu, H.; Chen, Q.; Ma, M.G.; Gu, Y.F.; Zhao, K.; Xiang, Q.J.; et al. Synergistic anti-oxidative effects of Pongamia pinnata against nickel mediated by Rhizobium pisi and Ochrobacterium pseudogrignonense. Ecotoxicol. Environ. Saf. 2021, 217, 112244. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, H.Y.; Yang, X.N.; Zhang, J.J.; Zhou, H.; Wang, C.; Yu, Y.; Lu, X.Y.; Chen, Y.Z.; Tian, Y. Transcriptomic analysis of Camellia oleifera in response to drought stress using high throughput RNA-seq. Russ. J. Plant Physiol. 2017, 64, 728–737. [Google Scholar] [CrossRef]
- Patanè, C.; Cosentino, S.L.; Romano, D.; Toscano, S. Relative Water Content, Proline, and Antioxidant Enzymes in Leaves of Long Shelf-Life Tomatoes under Drought Stress and Rewatering. Plants 2022, 11, 3045. [Google Scholar] [CrossRef]
- Meher; Shivakrishna, P.; Ashok Reddy, K.; Manohar Rao, D. Effect of PEG-6000 imposed drought stress on RNA content, relative water content (RWC), and chlorophyll content in peanut leaves and roots. Saudi J. Biol. Sci. 2017, 25, 285–289. [Google Scholar] [CrossRef]
- Wang, B.M.; Liu, C.; Zhang, D.F.; He, C.M.; Zhang, J.R.; Li, Z.X. Effects of maize organ-specific drought stress response on yields from transcriptome analysis. BMC Plant Biol. 2019, 19, 335. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.M.; Shukla, V.; Merewitz, E.B. Transcriptome analysis of creeping bentgrass exposed to drought stress and polyamine treatment. PLoS ONE 2017, 12, e0175848. [Google Scholar] [CrossRef]
- Zhu, J.K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Todaka, D.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Recent advances in the dissection of drought-stress regulatory networks and strategies for development of drought-tolerant transgenic rice plants. Front. Plant Sci. 2015, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Yang, M.; Zhao, C.F.; Wang, Y.F.; Zhang, R.H. Physiological and proteomic analyses revealed the response mechanisms of two different drought-resistant maize varieties. BMC Plant Biol. 2021, 21, 513. [Google Scholar] [CrossRef]
- Xiao, F.; Zhao, Y.; Wang, X.R.; Jian, X.Y.; Yang, Y. Physiological responses to drought stress of three pine species and comparative transcriptome analysis of Pinus yunnanensis var. pygmaea. BMC Genom. 2024, 25, 281. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Yamaguchi, K.; Shinozaki, K. The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Front Plant. Sci. 2014, 5, 170. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R. ROS are good. Trends. Plant. Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Hasanuzzaman, M.; Bhuyan, M.; Parvin, K. Reactive oxygen species and antioxidant defense in plants under abiotic stress: Revisiting the crucial role of a universal defense regulator. Antioxidants 2021, 10, 805. [Google Scholar] [CrossRef] [PubMed]
- Flexas, J.; Bota, J.; Loreto, F. Mesophyll conductance to CO2: Current knowledge and future prospects. Plant. Biol. 2016, 18, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Narayan, S.; Sharma, R.K.; Kumar, V.; Sanyal, I.; Shirke, P.A. Alterations in plant anatomy and higher lignin synthesis provides drought tolerance in cluster bean [Cyamopsis tetragonoloba (L.) Taub.]. Plant Physiol. Biochem. 2023, 201, 107905. [Google Scholar] [CrossRef] [PubMed]
- Hamel, L.; Nicole, M.; Sritubtim, S.; Morency, M.; Ellis, M.; Ehlting, J.; Beaudoin, N.; Barbazuk, B.; Klessig, D.; Lee, J.; et al. Ancient signals: Comparative genomics of plant MAPK and MAPKK gene families. Trends. Plant. Sci. 2014, 19, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Dekomah, S.D.; Wang, Y.; Qin, T.; Xu, D.; Sun, C.; Yao, P.; Liu, Y.; Bi, Z.; Bai, J. Identification and expression analysis of calcium-dependent protein kinases gene family in potato under drought stress. Front. Genet. 2022, 13, 874397. [Google Scholar] [CrossRef]
- Díaz, M.; Velasco, B.; González, P.; Martínez, M.; Díaz, I. C1A cysteine protease–cystatin interactions in leaf senescence. J. Exp. Bot. 2016, 67, 639–652. [Google Scholar] [CrossRef]
- Moloi, S.J.; Ngara, R. The roles of plant proteases and protease inhibitors in drought response: A review. Front. Plant Sci. 2023, 14, 1165845. [Google Scholar] [CrossRef]
- Ma, C.; Wang, H.; Macnish, A.J.; Estrada, A.C.; Lin, J.; Chang, Y.H.; Reid, M.S.; Jiang, C.Z. Transcriptomic analysis reveals numerous diverse protein kinases and transcription factors involved in desiccation tolerance in the resurrection plant Myrothamnus flabellifolia. Hortic. Res. 2015, 2, 15034. [Google Scholar] [CrossRef] [PubMed]
- D’Ippólito, S.; Rey, M.F.; Feingold, S.E.; Guevara, M.G. Role of proteases in the response of plants to drought. Plant Physiol. Bio. 2021, 168, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Dubos, C.; Dodd, I.; Holroyd, G.; Hetherington, A.; Campbell, M. AtMYB61, an R2R3-MYB transcription factor controlling stomatal aperture in Arabidopsis thaliana. Curr. Biol. 2008, 18, 1231–1236. [Google Scholar] [CrossRef] [PubMed]
- Kuki, Y.; Ohno, R.; Yoshida, K.; Takumi, S. Heterologous expression of wheat WRKY transcription factor genes transcriptionally activated in hybrid necrosis strains alters abiotic and biotic stress tolerance in transgenic Arabidopsis. Plant Physiol. Biochem. 2020, 150, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.K.; Jung, H.; Jang, G.; Jeong, J.S.; Kim, Y.S.; Ha, S.H.; Choi Do, Y.; Kim, J.K. Overexpression of the OsERF71 transcription factor alters rice root structure and drought resistance. Plant Physiol. 2016, 172, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.F.; Li, C.L.; Zhao, X.R.; Li, M.F.; Zhao, H.X.; Guo, J.Y.; Cai, Y.; Chen, H.; Wu, Q. Overexpression of a tartary buckwheat gene, FtbHLH3, enhances drought/oxidative stress tolerance in transgenic Arabidopsis. Front. Plant Sci. 2017, 8, 625. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wang, C.P.; Han, X.; Tang, S.; Liu, S.; Xia, X.L.; Yin, W.L. A novel bHLH transcription factor PebHLH35 from Populus euphratica confers drought tolerance through regulating stomatal development, photosynthesis and growth in Arabidopsis. Biochem. Biophys. Res. Commun. 2014, 450, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Hsieh, V.; Liao, P.C.; Cheng, W.H.; Liu, L.Y.; Yang, Y.W.; Lai, M.H.; Chang, M.C. The function of OsbHLH068 is partially redundant with its homolog, AtbHLH112, in the regulation of the salt stress response but has opposite functions to control flowering in Arabidopsis. Plant Mol. Biol. 2017, 94, 531–548. [Google Scholar] [CrossRef]
- Liu, Y.J.; Ji, X.Y.; Nie, X.G.; Qu, M.; Zheng, L.; Tan, Z.L.; Zhao, H.M.; Huo, L.; Liu, S.N.; Zhang, B.; et al. Arabidopsis AtbHLH112 regulates the expression of genes involved in abiotic stress tolerance by binding to their E-box and GCG-box motifs. New. Phytol. 2015, 207, 692–709. [Google Scholar] [CrossRef]
- Chen, D.D.; Chai, S.C.; McIntyre, C.L.; Xue, G.P. Overexpression of a predominantly root-expressed NAC transcription factor in wheat roots enhances root length, biomass and drought tolerance. Plant. Cell. Rep. 2018, 37, 225–237. [Google Scholar] [CrossRef]
- Mao, H.D.; Li, S.M.; Chen, B.; Jian, C.; Mei, F.M.; Zhang, Y.F.; Li, F.F.; Chen, N.; Li, T.; Du, L.Y.; et al. Variation in cis-regulation of a NAC transcription factor contributes to drought tolerance in wheat. Mol. Plant. 2022, 15, 276–292. [Google Scholar] [CrossRef]
- Shi, H.; Hou, J.; Li, D. Comparative transcriptome and coexpression network analysis reveals key pathways and hub candidate genes associated with sunflower (Helianthus annuus L.) drought tolerance. BMC. Plant Biol. 2024, 24, 224. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.W.; Huang, D.Z.; Zhao, X.J.; Zhang, M. Evaluation of drought resistance and transcriptome analysis for the identification of drought-responsive genes in Iris germanica. Sci. Rep. 2021, 11, 16308. [Google Scholar] [CrossRef]
- Hou, X.B.; Kong, Y.; Teng, Z.; Yang, C.F.; Li, Y.F.; Zhu, Z.J. Integrating genes and metabolites: Unraveling mango’s drought resilience mechanisms. BMC. Plant. Biol. 2024, 24, 208. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.L.; Zhang, Z.N.; Gu, Y.H.; Li, W.J.; Wang, W.Y.; Yuan, X.K.; Zhang, Y.X.; Yuan, M.; Du, J.D.; Zhao, Q. Genome-wide identification of the soybean cytokinin oxidase/dehydrogenase gene family and its diverse roles in response to multiple abiotic stress. Front. Plant. Sci. 2023, 14, 1163219. [Google Scholar] [CrossRef]
- Sheoran, S.; Thakur, V.; Narwal, S.; Turan, R.; Mamrutha, H.M.; Singh, V.; Tiwari, V.; Sharma, I. Differential activity and expression profile of antioxidant enzymes and physiological changes in wheat (Triticum aestivum L.) under drought. Appl. Biochem. Biotechnol. 2015, 177, 1282–1298. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.C.; Luo, W.; Ge, M.M.; Guan, Y.J.; Tang, Y.; Chen, W.M.; Lv, J.Y. A group I WRKY gene, TaWRKY133, negatively regulates drought resistance in transgenic plants. Int. J. Mol. Sci. 2022, 23, 12026. [Google Scholar] [CrossRef]
- Marino, D.; Froidure, S.; Canonne, J.; Ben, S.; Khafif, M.; Pouzet, C.; Jauneau, A.; Roby, D.; Rivas, S. Arabidopsis ubiquitin ligase MIEL1 mediates degradation of the transcription factor MYB30 weakening plant defence. Nat. Commun. 2013, 4, 1476. [Google Scholar] [CrossRef]
- Sharma, E.; Bhatnagar, A.; Bhaskar, A.; Majee, S.M.; Kieffer, M.; Kepinski, S.; Khurana, P.; Khurana, J.P. Stress-induced F-Box protein-coding gene OsFBX257 modulates drought stress adaptations and ABA responses in rice. Plant Cell Environ. 2023, 46, 1207–1231. [Google Scholar] [CrossRef]
- An, J.; Li, Q.X.; Yang, J.J.; Zhang, G.Q.; Zhao, Z.X.; Wu, Y.; Wang, Y.; Wang, W. Wheat F-box protein TaFBA1 positively regulates plant drought tolerance but negatively regulates stomatal closure. Front. Plant Sci. 2019, 10, 1242. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.C.; Hao, Y.P.; Ma, X.Y.; Shi, Y.Q.; Dang, Y.M.; Dong, Z.Y.; Zhao, Y.Y.; Zhao, T.L.; Zhu, S.J.; Zhang, Z.Y.; et al. Genome-wide analysis and identification of stress-responsive genes of the CCCH zinc finger family in Capsicum annuum L. Front. Plant Sci. 2023, 14, 1189038. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, R.; Watanabe, Y.; Fujita, Y.; Le, D.T.; Kojima, M.; Werner, T. Analysis of cytokinin mutants and regulation of cytokinin metabolic genes reveals important regulatory roles of cytokinins in drought, salt and abscisic acid responses, and abscisic acid biosynthesis. Plant Cell 2011, 23, 2169–2183. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.Y.; Chen, H.W.; Ng, C.Y.; Lin, C.Y.; Tseng, T.H.; Li, W.H. Down-regulation of cytokinin oxidase 2 expression increases tiller number and improves rice yield. Rice 2015, 8, 36. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | POD (U g–1 min–1 FM) | CAT (U g–1 min–1 FM) | RWC (%) | Soil Volumetric Moisture Content (%) |
|---|---|---|---|---|
| 0 d | 32.31 ± 1.49 b | 26.09 ± 0.66 b | 60.90 ± 0.42 a | 34.57 ± 1.10 a |
| 8 d | 35.00 ± 1.32 b | 26.19 ± 1.05 b | 55.16 ± 0.15 b | 13.95 ± 0.80 b |
| 15 d | 40.65 ± 1.35 a | 66.20 ± 0.43 a | 43.44 ± 0.45 c | 9.34 ± 0.87 c |
| Pathway | Pathway ID | 0 d vs. 8 d | 0 d vs. 15 d |
|---|---|---|---|
| Proteasome | ath03050 | 55 | 59 |
| Glycolysis/Gluconeogenesis | ath00010 | 87 | 108 |
| Galactose metabolism | ath00052 | 48 | 52 |
| Biosynthesis of secondary metabolites | ath01110 | 672 | 1017 |
| Protein processing in endoplasmic reticulum | ath04141 | 144 | 199 |
| Fatty acid biosynthesis | ath00061 | 34 | 42 |
| Peroxisome | ath04146 | 62 | 84 |
| Fatty acid metabolism | ath01212 | 49 | 69 |
| Selected Genes | Relative Expression | ||
|---|---|---|---|
| 0 d | 8 d | 15 d | |
| LG01G01976 (MAPK) | 0.91 | 1.92 | 1.01 |
| LG12G00734 (Calcium-dependent protein kinase) | 0.82 | 1.16 | 0.88 |
| LG07G00954 (Serine/threonine-protein kinase) | 1.14 | 1.43 | 1.30 |
| LG02G03733 (Cysteine protease) | 1.16 | 2.32 | 1.37 |
| LG08G01571 (NAC) | 1.07 | 1.18 | 1.28 |
| LG04G00063 (ERF) | 0.95 | 1.36 | 0.98 |
| LG03G00646 (WRKY) | 1.02 | 1.17 | 1.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Xu, Y.; Liu, C.; Chen, L.; Zhang, Y.; He, Z.; Wang, R.; Xun, C.; Ma, Y.; Yuan, X.; et al. Cataloging the Genetic Response: Unveiling Drought-Responsive Gene Expression in Oil Tea Camellia (Camellia oleifera Abel.) through Transcriptomics. Life 2024, 14, 989. https://doi.org/10.3390/life14080989
Zhang Z, Xu Y, Liu C, Chen L, Zhang Y, He Z, Wang R, Xun C, Ma Y, Yuan X, et al. Cataloging the Genetic Response: Unveiling Drought-Responsive Gene Expression in Oil Tea Camellia (Camellia oleifera Abel.) through Transcriptomics. Life. 2024; 14(8):989. https://doi.org/10.3390/life14080989
Chicago/Turabian StyleZhang, Zhen, Yanming Xu, Caixia Liu, Longsheng Chen, Ying Zhang, Zhilong He, Rui Wang, Chengfeng Xun, Yushen Ma, Xiaokang Yuan, and et al. 2024. "Cataloging the Genetic Response: Unveiling Drought-Responsive Gene Expression in Oil Tea Camellia (Camellia oleifera Abel.) through Transcriptomics" Life 14, no. 8: 989. https://doi.org/10.3390/life14080989
APA StyleZhang, Z., Xu, Y., Liu, C., Chen, L., Zhang, Y., He, Z., Wang, R., Xun, C., Ma, Y., Yuan, X., Wang, X., Chen, Y., & Yang, X. (2024). Cataloging the Genetic Response: Unveiling Drought-Responsive Gene Expression in Oil Tea Camellia (Camellia oleifera Abel.) through Transcriptomics. Life, 14(8), 989. https://doi.org/10.3390/life14080989