
Investigating the Effects of Chelidonic Acid on Oxidative Stress-Induced Premature Cellular Senescence in Human Skin Fibroblast Cells


Abstract
1. Introduction
2. Materials and Methods
2.1. Cells Culture and Treatment
2.2. Developing a Stress-Induced Premature Senescence (SIPS) Model
2.2.1. Doubling Time
2.2.2. Staining Cells for SA-βgal
2.2.3. Determining the Sublethal Dose of H2O2 as an Oxidative Stress Agent
2.2.4. Determining the Sublethal Dose of CA
2.3. Free Radical Measurements
2.3.1. Staining Cells for SA-βgal
2.3.2. Cytotoxicity Testing: AO/DAPI Double Staining
4′,6-Diamino-2-Phenylindole (DAPI) Staining
Acridine-Orange Staining (AO)
2.3.3. Genotoxicity Testing: Comet Assay
2.3.4. H2O2 Measurements
2.3.5. Malondialdehyde Levels
2.4. Determination of Antioxidant Activity
2.4.1. Superoxide Dismutase Activity
2.4.2. Glutathione Peroxidase (GSH-Px) Activity
2.5. Molecular Docking Experiments
2.6. Gen Expression and Protein Analysis
2.6.1. Real-Time PCR Analysis
Total RNA Isolation and First-Strand cDNA Synthesis
Quantitative, Real-Time PCR
2.6.2. Western Blot Analysis
2.7. Statistical Analysis
3. Results
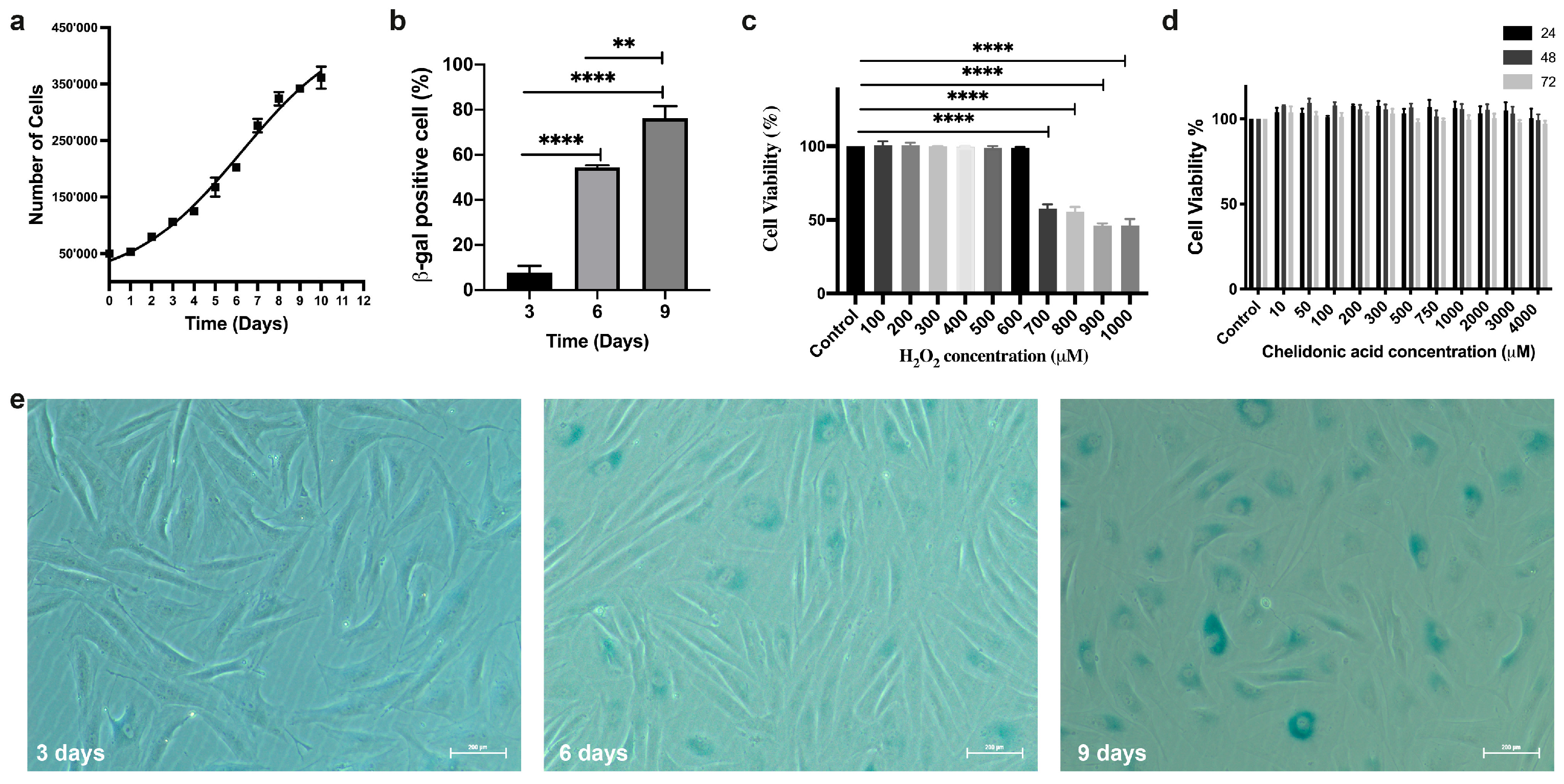
3.1. Developing a Stress-Induced Premature Senescence (SIPS) Model
3.1.1. Doubling Time
3.1.2. Staining Cells for SA-βgal
3.1.3. Determining the Sublethal Dose of H2O2 as an Oxidative Stress Agent
3.1.4. Determining the Sublethal Dose of CA
3.2. Free Radical Measurements
3.2.1. Staining Cells for SA-βgal
3.2.2. DAPI-Acridine Orange (AO) Staining
3.2.3. Comet Assay
3.2.4. H2O2 Measurements
3.2.5. MDA Levels
3.3. Antioxidant Activity
3.4. Molecular Docking
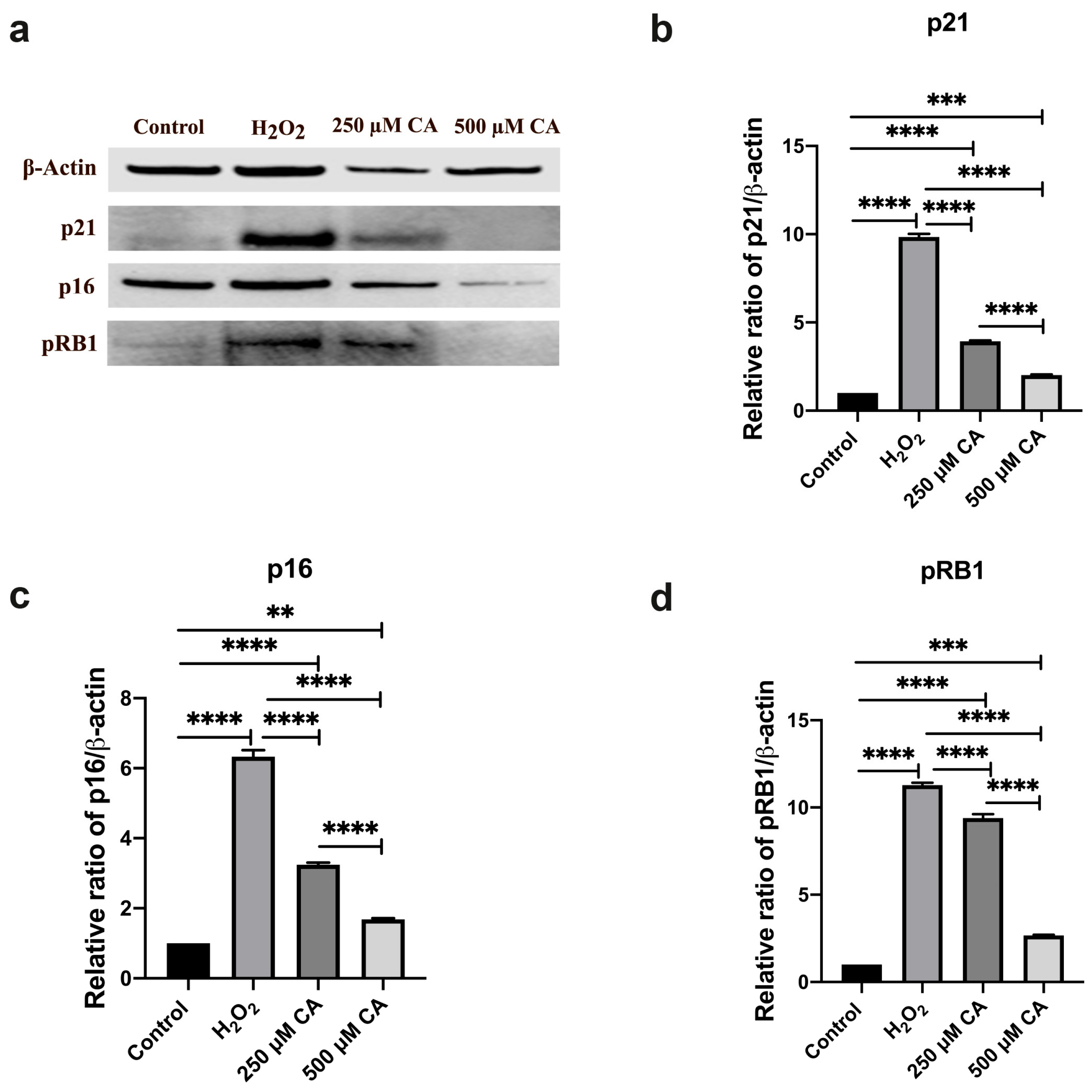
3.5. Gene Expression and Protein Analysis Results
3.5.1. RT PCR Results
Evaluation of Total RNA Isolation Results
Selection of Candidate Reference Genes and Evaluation of Amplification Specificity, PCR Efficiency, and Gene Expression Levels
3.5.2. Protein Analysis Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bellu, E.; Cruciani, S.; Garroni, G.; Balzano, F.; Satta, R.; Montesu, M.A.; Fadda, A.; Mulas, M.; Sarais, G.; Bandiera, P. Natural compounds and PCL nanofibers: A novel tool to counteract stem cell senescence. Cells 2021, 10, 1415. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular senescence: Aging, cancer, and injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Zhu, Y.; Van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Clinical strategies and animal models for developing senolytic agents. Exp. Gerontol. 2015, 68, 19–25. [Google Scholar] [CrossRef]
- He, S.; Sharpless, N.E. Senescence in health and disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef]
- Jeyapalan, J.C.; Sedivy, J.M. Cellular senescence and organismal aging. Mech. Ageing Dev. 2008, 129, 467–474. [Google Scholar] [CrossRef]
- Tchkonia, T.; Morbeck, D.E.; Von Zglinicki, T.; Van Deursen, J.; Lustgarten, J.; Scrable, H.; Khosla, S.; Jensen, M.D.; Kirkland, J.L. Fat tissue, aging, and cellular senescence. Aging Cell 2010, 9, 667–684. [Google Scholar] [CrossRef]
- Weyemi, U.; Lagente-Chevallier, O.; Boufraqech, M.; Prenois, F.; Courtin, F.; Caillou, B.; Talbot, M.; Dardalhon, M.; Al Ghuzlan, A.; Bidart, J.-M. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene 2012, 31, 1117–1129. [Google Scholar] [CrossRef]
- Sitte, N.; Merker, K.; von Zglinicki, T.; Grune, T.; Davies, K.J. Protein oxidation and degradation during cellular senescence of human BJ fibroblasts: Part I—Effects of proliferative senescence. FASEB J. 2000, 14, 2495–2502. [Google Scholar] [CrossRef]
- Bian, Y.; Wei, J.; Zhao, C.; Li, G. Natural polyphenols targeting senescence: A novel prevention and therapy strategy for cancer. Int. J. Mol. Sci. 2020, 21, 684. [Google Scholar] [CrossRef]
- Buranasudja, V.; Rani, D.; Malla, A.; Kobtrakul, K.; Vimolmangkang, S. Insights into antioxidant activities and anti-skin-aging potential of callus extract from Centella asiatica (L.). Sci. Rep. 2021, 11, 13459. [Google Scholar] [CrossRef]
- Han, X.; Zhang, T.; Zhang, X.; Zhang, R.; Lao, K.; Mi, Y.; Gou, X. AMPK alleviates oxidative stress-induced premature senescence via inhibition of NF-κB/STAT3 axis-mediated positive feedback loop. Mech. Ageing Dev. 2020, 191, 111347. [Google Scholar] [CrossRef] [PubMed]
- Ok, C.Y.; Park, S.; Jang, H.-O.; Takata, T.; Lee, O.-H.; Bae, M.-K.; Bae, S.-K. FK866 protects human dental pulp cells against oxidative stress-induced cellular senescence. Antioxidants 2021, 10, 271. [Google Scholar] [CrossRef] [PubMed]
- Mbara, K.C.; Devnarain, N.; Owira, P.M. Potential Role of Polyphenolic Flavonoids as Senotherapeutic Agents in Degenerative Diseases and Geroprotection. Pharm. Med. 2022, 36, 331–352. [Google Scholar] [CrossRef]
- Gurău, F.; Baldoni, S.; Prattichizzo, F.; Espinosa, E.; Amenta, F.; Procopio, A.D.; Albertini, M.C.; Bonafè, M.; Olivieri, F. Anti-senescence compounds: A potential nutraceutical approach to healthy aging. Ageing Res. Rev. 2018, 46, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; di Fagagna, A. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Wang, E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl 2 is involved. Cancer Res. 1995, 55, 2284–2292. [Google Scholar]
- Khalil, R.; Diab-Assaf, M.; Lemaitre, J.-M. Emerging Therapeutic Approaches to Target the Dark Side of Senescent Cells: New Hopes to Treat Aging as a Disease and to Delay Age-Related Pathologies. Cells 2023, 12, 915. [Google Scholar] [CrossRef]
- Malavolta, M.; Bracci, M.; Santarelli, L.; Sayeed, M.A.; Pierpaoli, E.; Giacconi, R.; Costarelli, L.; Piacenza, F.; Basso, A.; Cardelli, M. Inducers of senescence, toxic compounds, and senolytics: The multiple faces of Nrf2-activating phytochemicals in cancer adjuvant therapy. Mediat. Inflamm. 2018, 2018, 4159013. [Google Scholar] [CrossRef]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.-P.; Patil, C.K.; Rodier, F.; Krtolica, A.; Beausejour, C.M.; Parrinello, S.; Hodgson, J.G.; Chin, K.; Desprez, P.-Y.; Campisi, J. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS ONE 2010, 5, e9188. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Kucheryavenko, O.; Wordsworth, J.; von Zglinicki, T. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech. Ageing Dev. 2018, 170, 30–36. [Google Scholar] [CrossRef]
- Lagoumtzi, S.M.; Chondrogianni, N. Senolytics and senomorphics: Natural and synthetic therapeutics in the treatment of aging and chronic diseases. Free Radic. Biol. Med. 2021, 171, 169–190. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef]
- Gasek, N.S.; Kuchel, G.A.; Kirkland, J.L.; Xu, M. Strategies for targeting senescent cells in human disease. Nat. Aging 2021, 1, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; He, S.; Wang, H.; Li, J.; Liu, Y.; Liu, S. Targeting Cellular Senescence in Aging and Age-Related Diseases: Challenges, Considerations, and the Emerging Role of Senolytic and Senomorphic Therapies. Aging Dis 2024, 16, 1. [Google Scholar] [CrossRef]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting cellular senescence with senotherapeutics: Senolytics and senomorphics. FEBS J. 2023, 290, 1362–1383. [Google Scholar] [CrossRef]
- Prašnikar, E.; Borišek, J.; Perdih, A. Senescent cells as promising targets to tackle age-related diseases. Ageing Res. Rev. 2021, 66, 101251. [Google Scholar] [CrossRef]
- Liao, K.-M.; Chen, C.-J.; Luo, W.-J.; Hsu, C.-W.; Yu, S.-L.; Yang, P.-C.; Su, K.-Y. Senomorphic effect of diphenyleneiodonium through AMPK/MFF/DRP1 mediated mitochondrial fission. Biomed. Pharmacother. 2023, 162, 114616. [Google Scholar] [CrossRef]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef] [PubMed]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef]
- Singh, D.K.; Gulati, K.; Ray, A. Effects of chelidonic acid, a secondary plant metabolite, on mast cell degranulation and adaptive immunity in rats. Int. Immunopharmacol. 2016, 40, 229–234. [Google Scholar] [CrossRef]
- Shin, H.-J.; Kim, H.-L.; Kim, S.-J.; Chung, W.-S.; Kim, S.-S.; Um, J.-Y. Inhibitory effects of chelidonic acid on IL-6 production by blocking NF-κB and caspase-1 in HMC-1 cells. Immunopharmacol. Immunotoxicol. 2011, 33, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Sharma, A.; Wadje, B.N.; Bharate, S.B. Benzopyrone, a privileged scaffold in drug discovery: An overview of FDA-approved drugs and clinical candidates. Med. Res. Rev. 2024, 44, 2035–2077. [Google Scholar] [CrossRef]
- Banik, B.K. Green Approaches in Medicinal Chemistry for Sustainable Drug Design; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Borah, B.; Dwivedi, K.D.; Chowhan, L.R. Review on synthesis and medicinal application of dihydropyrano [3, 2-b] pyrans and spiro-pyrano [3, 2-b] pyrans by employing the reactivity of 5-hydroxy-2-(hydroxymethyl)-4 H-pyran-4-one. Polycycl. Aromat. Compd. 2022, 42, 5893–5937. [Google Scholar] [CrossRef]
- Zielińska, S.; Jezierska-Domaradzka, A.; Wójciak-Kosior, M.; Sowa, I.; Junka, A.; Matkowski, A.M. Greater Celandine’s ups and downs− 21 centuries of medicinal uses of Chelidonium majus from the viewpoint of today’s pharmacology. Front. Pharmacol. 2018, 9, 299. [Google Scholar] [CrossRef]
- Liu, H.; Zheng, Y.-F.; Li, C.-Y.; Zheng, Y.-Y.; Wang, D.-Q.; Wu, Z.; Huang, L.; Wang, Y.-G.; Li, P.-B.; Peng, W. Discovery of anti-inflammatory ingredients in chinese herbal formula kouyanqing granule based on relevance analysis between chemical characters and biological effects. Sci. Rep. 2015, 5, 18080. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.L.; Bosisio, E. Pharmacological activities ofchelidonium majusl. (papaveraceae). Pharmacol. Res. 1996, 33, 127–134. [Google Scholar] [CrossRef]
- Carraher, C.E., Jr.; Ayoub, M.; Roner, M.R.; Moric, A.; Trang, N.T. Synthesis, structural characterization, and ability to inhibit the growth of pancreatic cancer by organotin polymers containing chelidonic acid. J. Chin. Adv. Mater. Soc. 2013, 1, 65–73. [Google Scholar] [CrossRef]
- Porter, T.G.; Martin, D.L. Chelidonic acid and other conformationally restricted substrate analogues as inhibitors of rat brain glutamate decarboxylase. Biochem. Pharmacol. 1985, 34, 4145–4150. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.-A.; Kim, H.-M.; Jeong, H.-J. Beneficial effects of chelidonic acid on a model of allergic rhinitis. Int. Immunopharmacol. 2011, 11, 39–45. [Google Scholar] [CrossRef]
- Kim, S.-J.; Kim, D.-S.; Lee, S.-H.; Ahn, E.-M.; Kee, J.-Y.; Hong, S.-H. Chelidonic acid ameliorates atopic dermatitis symptoms through suppression the inflammatory mediators in in vivo and in vitro. Appl. Biol. Chem. 2023, 66, 12. [Google Scholar] [CrossRef]
- Gerasymchuk, M.; Robinson, G.I.; Kovalchuk, O.; Kovalchuk, I. Modeling of the senescence-associated phenotype in human skin fibroblasts. Int. J. Mol. Sci. 2022, 23, 7124. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Kapuscinski, J. DAPI: A DNA-specific fluorescent probe. Biotech. Histochem. 1995, 70, 220–233. [Google Scholar] [CrossRef]
- Thomé, M.P.; Filippi-Chiela, E.C.; Villodre, E.S.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.B.; Lenz, G. Ratiometric analysis of Acridine Orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef]
- Gyori, B.M.; Venkatachalam, G.; Thiagarajan, P.; Hsu, D.; Clement, M.-V. OpenComet: An automated tool for comet assay image analysis. Redox Biol. 2014, 2, 457–465. [Google Scholar] [CrossRef]
- Marnett, L.J. Lipid peroxidation—DNA damage by malondialdehyde. Mutat. Res./Fundam. Mol. Mech. Mutagen. 1999, 424, 83–95. [Google Scholar] [CrossRef]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Sun, Y.; Oberley, L.W.; Li, Y. A simple method for clinical assay of superoxide dismutase. Clin. Chem. 1988, 34, 497–500. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Biovia, D.S. Discovery Studio Modeling Environment; Release San Diego: San Diego, CA, USA, 2017; Volume 936, pp. 240–249. [Google Scholar]
- DeLano, W.L. The PyMOL Molecular Graphics System, version 1.8; Schrödinger, LLC: San Carlos, CA, USA, 2002. [Google Scholar]
- Wong, M.L.; Medrano, J.F. Real-time PCR for mRNA quantitation. Biotechniques 2005, 39, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hnasko, T.S.; Hnasko, R.M. The western blot. In ELISA: Methods and Protocols; Humana Press: New York, NY, USA, 2015; pp. 87–96. [Google Scholar]
- Sanders, Y.Y.; Liu, H.; Zhang, X.; Hecker, L.; Bernard, K.; Desai, L.; Liu, G.; Thannickal, V.J. Histone modifications in senescence-associated resistance to apoptosis by oxidative stress. Redox Biol. 2013, 1, 8–16. [Google Scholar] [CrossRef]
- Hao, C.; Zhao, F.; Song, H.; Guo, J.; Li, X.; Jiang, X.; Huan, R.; Song, S.; Zhang, Q.; Wang, R. Structure-based design of 6-chloro-4-aminoquinazoline-2-carboxamide derivatives as potent and selective p21-activated kinase 4 (PAK4) inhibitors. J. Med. Chem. 2018, 61, 265–285. [Google Scholar] [CrossRef]
- Ikuta, M.; Kamata, K.; Fukasawa, K.; Honma, T.; Machida, T.; Hirai, H.; Suzuki-Takahashi, I.; Hayama, T.; Nishimura, S. Crystallographic approach to identification of cyclin-dependent kinase 4 (CDK4)-specific inhibitors by using CDK4 mimic CDK2 protein. J. Biol. Chem. 2001, 276, 27548–27554. [Google Scholar] [CrossRef]
- Emamzadah, S.; Tropia, L.; Vincenti, I.; Falquet, B.; Halazonetis, T.D. Reversal of the DNA-binding-induced loop L1 conformational switch in an engineered human p53 protein. J. Mol. Biol. 2014, 426, 936–944. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Cho, Y. Structural similarity between the pocket region of retinoblastoma tumour suppressor and the cyclin-box. Nat. Struct. Biol. 1997, 4, 390–395. [Google Scholar] [CrossRef]
- Fukutomi, T.; Takagi, K.; Mizushima, T.; Ohuchi, N.; Yamamoto, M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol. Cell. Biol. 2014, 34, 832–846. [Google Scholar] [CrossRef]
- Muzaffer, U.; Paul, V.; Prasad, N.R. Molecular docking of selected phytoconstituents with signaling molecules of Ultraviolet-B induced oxidative damage. In Silico Pharmacol. 2017, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Toms, A.V.; Deshpande, A.; McNally, R.; Jeong, Y.; Rogers, J.M.; Kim, C.U.; Gruner, S.M.; Ficarro, S.B.; Marto, J.A.; Sattler, M. Structure of a pseudokinase-domain switch that controls oncogenic activation of Jak kinases. Nat. Struct. Mol. Biol. 2013, 20, 1221–1223. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, N.J.; Murphy, J.M.; Liau, N.P.; Varghese, L.N.; Laktyushin, A.; Whitlock, E.L.; Lucet, I.S.; Nicola, N.A.; Babon, J.J. SOCS3 binds specific receptor–JAK complexes to control cytokine signaling by direct kinase inhibition. Nat. Struct. Mol. Biol. 2013, 20, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Sok, P.; Gross, F.; Albert, K.; Kobori, E.; Póti, Á.L.; Gógl, G.; Bento, I.; Kuang, E.; Taylor, S.S. A non-catalytic herpesviral protein reconfigures ERK-RSK signaling by targeting kinase docking systems in the host. Nat. Commun. 2022, 13, 472. [Google Scholar] [CrossRef]
- Kinoshita, T.; Sugiyama, H.; Mori, Y.; Takahashi, N.; Tomonaga, A. Identification of allosteric ERK2 inhibitors through in silico biased screening and competitive binding assay. Bioorganic Med. Chem. Lett. 2016, 26, 955–958. [Google Scholar] [CrossRef]
- Zhou, C.; Zhang, C.; Zhu, H.; Liu, Z.; Su, H.; Zhang, X.; Chen, T.; Zhong, Y.; Hu, H.; Xiong, M. Allosteric regulation of Hsp90α’s activity by small molecules targeting the middle domain of the chaperone. Iscience 2020, 23, 100857. [Google Scholar] [CrossRef]
- Boura, E.; Rezabkova, L.; Brynda, J.; Obsilova, V.; Obsil, T. Structure of the human FOXO4-DBD–DNA complex at 1.9 Å resolution reveals new details of FOXO binding to the DNA. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 1351–1357. [Google Scholar] [CrossRef]
- Fleige, S.; Pfaffl, M.W. RNA integrity and the effect on the real-time qRT-PCR performance. Mol. Asp. Med. 2006, 27, 126–139. [Google Scholar] [CrossRef]
- Kim, D.-S.; Kim, S.-J.; Kim, M.-C.; Jeon, Y.-D.; Um, J.-Y.; Hong, S.-H. The therapeutic effect of chelidonic acid on ulcerative colitis. Biol. Pharm. Bull. 2012, 35, 666–671. [Google Scholar] [CrossRef]
- Khairnar, S.I.; Kulkarni, Y.A.; Singh, K. Mitigation of cisplatin-induced nephrotoxicity by chelidonic acid in Wistar rats. J. Trace Elem. Med. Biol. 2024, 81, 127321. [Google Scholar] [CrossRef] [PubMed]
- Bielak-Zmijewska, A.; Wnuk, M.; Przybylska, D.; Grabowska, W.; Lewinska, A.; Alster, O.; Korwek, Z.; Cmoch, A.; Myszka, A.; Pikula, S. A comparison of replicative senescence and doxorubicin-induced premature senescence of vascular smooth muscle cells isolated from human aorta. Biogerontology 2014, 15, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef] [PubMed]
- Macieira-Coelho, A. Growth inhibition of human fibroblasts in vitro. In Inhibitors of Cell Growth; Springer: Berlin/Heidelberg, Germany, 1998; pp. 249–270. [Google Scholar]
- Valieva, Y.; Ivanova, E.; Fayzullin, A.; Kurkov, A.; Igrunkova, A. Senescence-associated β-galactosidase detection in pathology. Diagnostics 2022, 12, 2309. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Campisi, J.; Dimri, G.P. Methods to detect biomarkers of cellular senescence: The senescence-associated β-galactosidase assay. In Biological Aging: Methods and Protocols; Humana Press: Clifton, NJ, USA, 2007; pp. 21–31. [Google Scholar]
- Hoogstraten, C.A.; Smeitink, J.A.; Russel, F.G.; Schirris, T.J. Dissecting drug-induced cytotoxicity and metabolic dysfunction in conditionally immortalized human proximal tubule cells. Front. Toxicol. 2022, 4, 842396. [Google Scholar] [CrossRef]
- Zhu, X.; Yue, H.; Guo, X.; Yang, J.; Liu, J.; Liu, J.; Wang, R.; Zhu, W. The preconditioning of berberine suppresses hydrogen peroxide-induced premature senescence via regulation of sirtuin 1. Oxidative Med. Cell. Longev. 2017, 2017, 2391820. [Google Scholar] [CrossRef]
- Miquel, J. An update of the oxidation-inflammation theory of aging: The involvement of the immune system in oxi-inflamm-aging. Curr. Pharm. Des. 2009, 15, 3003–3026. [Google Scholar]
- Kandlur, A.; Satyamoorthy, K.; Gangadharan, G. Oxidative stress in cognitive and epigenetic aging: A retrospective glance. Front. Mol. Neurosci. 2020, 13, 41. [Google Scholar] [CrossRef]
- Kumar, R.; Sharma, A.; Kumari, A.; Gulati, A.; Padwad, Y.; Sharma, R. Epigallocatechin gallate suppresses premature senescence of preadipocytes by inhibition of PI3K/Akt/mTOR pathway and induces senescent cell death by regulation of Bax/Bcl-2 pathway. Biogerontology 2019, 20, 171–189. [Google Scholar] [CrossRef]
- Kirkland, J.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Yang, J.; Wu, X.; Zhang, G.; Li, T.; Wang, X.E.; Zhang, H.; Wang, C.C.; Liu, G.H.; Wang, L. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell 2018, 17, e12765. [Google Scholar] [CrossRef]
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κ B activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef]
- de Godoy, M.C.X.; Macedo, J.A.; Gambero, A. Researching New Drug Combinations with Senolytic Activity Using Senescent Human Lung Fibroblasts MRC-5 Cell Line. Pharmaceuticals 2024, 17, 70. [Google Scholar] [CrossRef] [PubMed]
- Birar, V.C.; Sheerin, A.N.; Ostler, E.L.; Faragher, R.G. Novel resveratrol derivatives have diverse effects on the survival, proliferation and senescence of primary human fibroblasts. Biogerontology 2020, 21, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.G.; Feig, Y.S. The use of DAPI for identifying and counting aquatic microflora 1. Limnol. Oceanogr. 1980, 25, 943–948. [Google Scholar] [CrossRef]
- Swanson, E.C.; Manning, B.; Zhang, H.; Lawrence, J.B. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J. Cell Biol. 2013, 203, 929–942. [Google Scholar] [CrossRef]
- Hayashi, M.; Sofuni, T.; Ishidate, M., Jr. An application of acridine orange fluorescent staining to the micronucleus test. Mutat. Res. Lett. 1983, 120, 241–247. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G. Cellular senescence: Defining a path forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Kurz, T.; Eaton, J.W.; Brunk, U.T. Redox activity within the lysosomal compartment: Implications for aging and apoptosis. Antioxid. Redox Signal. 2010, 13, 511–523. [Google Scholar] [CrossRef]
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-associated β-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113, 3613–3622. [Google Scholar] [CrossRef]
- Copp, M.E.; Chubinskaya, S.; Bracey, D.N.; Shine, J.; Sessions, G.; Loeser, R.F.; Diekman, B.O. Comet assay for quantification of the increased DNA damage burden in primary human chondrocytes with aging and osteoarthritis. Aging Cell 2022, 21, e13698. [Google Scholar] [CrossRef]
- Morlière, P.; Santus, R. Pro-oxidant role of superoxide dismutase in ultraviolet-A-induced lipid peroxidation in cultured normal human skin fibroblasts. Eur. J. Biochem. 1998, 256, 184–189. [Google Scholar] [CrossRef]
- Panieri, E.; Gogvadze, V.; Norberg, E.; Venkatesh, R.; Orrenius, S.; Zhivotovsky, B. Reactive oxygen species generated in different compartments induce cell death, survival, or senescence. Free Radic. Biol. Med. 2013, 57, 176–187. [Google Scholar] [CrossRef]
- Zhang, Y.; Unnikrishnan, A.; Deepa, S.S.; Liu, Y.; Li, Y.; Ikeno, Y.; Sosnowska, D.; Van Remmen, H.; Richardson, A. A new role for oxidative stress in aging: The accelerated aging phenotype in Sod1−/− mice is correlated to increased cellular senescence. Redox Biol. 2017, 11, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Pole, A.; Dimri, M.; Dimri, G.P. Oxidative stress, cellular senescence and ageing. AIMS Mol. Sci. 2016, 3, 300–324. [Google Scholar] [CrossRef]
- Xiao, Y.-Z.; Yang, M.; Xiao, Y.; Guo, Q.; Huang, Y.; Li, C.-J.; Cai, D.; Luo, X.-H. Reducing hypothalamic stem cell senescence protects against aging-associated physiological decline. Cell Metab. 2020, 31, 534–548.e5. [Google Scholar] [CrossRef]
- Aviv, G.; Gal-Mor, O. Real-time reverse transcription PCR as a tool to study virulence gene regulation in bacterial pathogens. In Host-Pathogen Interactions: Methods and Protocols; Humana Press: New York, NY, USA, 2018; pp. 23–32. [Google Scholar]
- Cohn, R.L.; Gasek, N.S.; Kuchel, G.A.; Xu, M. The heterogeneity of cellular senescence: Insights at the single-cell level. Trends Cell Biol. 2023, 33, 9–17. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef]
- Wong, H.; Riabowol, K. Differential CDK-inhibitor gene expression in aging human diploid fibroblasts. Exp. Gerontol. 1996, 31, 311–325. [Google Scholar] [CrossRef]
- Stein, G.H.; Dulić, V. Molecular mechanisms for the senescent cell cycle arrest. J. Investig. Dermatol. Symp. Proc. 1998, 3, 14–18. [Google Scholar] [PubMed]
- Campisi, J.; Robert, L. Cell senescence: Role in aging and age-related diseases. Aging 2014, 39, 45–61. [Google Scholar]
- Kamal, N.S.M.; Safuan, S.; Shamsuddin, S.; Foroozandeh, P. Aging of the cells: Insight into cellular senescence and detection Methods. Eur. J. Cell Biol. 2020, 99, 151108. [Google Scholar]
- Whittaker, S.R.; Mallinger, A.; Workman, P.; Clarke, P.A. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol. Ther. 2017, 173, 83–105. [Google Scholar] [CrossRef]
- Newton, H.B. Handbook of Brain Tumor Chemotherapy, Molecular Therapeutics, and Immunotherapy; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Olsen, J.V.; Mann, M. Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol. Cell. Proteom. 2013, 12, 3444–3452. [Google Scholar] [CrossRef]
- Ziegler, S.J.; Mallinson, S.J.; John, P.C.S.; Bomble, Y.J. Advances in integrative structural biology: Towards understanding protein complexes in their cellular context. Comput. Struct. Biotechnol. J. 2021, 19, 214–225. [Google Scholar] [CrossRef]
- Zhang, M.; Ying, W. NAD+ deficiency is a common central pathological factor of a number of diseases and aging: Mechanisms and therapeutic implications. Antioxid. Redox Signal. 2019, 30, 890–905. [Google Scholar] [CrossRef]
- Hwang, J.-w.; Yao, H.; Caito, S.; Sundar, I.K.; Rahman, I. Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic. Biol. Med. 2013, 61, 95–110. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Auer, B.; Stingl, L.; Berghammer, H.; Haidacher, D.; Schweiger, M.; Wagner, E.F. Mice lacking ADPRT and poly (ADP-ribosyl) ation develop normally but are susceptible to skin disease. Genes Dev. 1995, 9, 509–520. [Google Scholar] [CrossRef]
- Xue, Z.; Demple, B. Knockout and inhibition of Ape1: Roles of Ape1 in base excision DNA repair and modulation of gene expression. Antioxidants 2022, 11, 1817. [Google Scholar] [CrossRef] [PubMed]
- Rios-Covian, D.; Butcher, L.D.; Ablack, A.L.; den Hartog, G.; Matsubara, M.T.; Ly, H.; Oates, A.W.; Xu, G.; Fisch, K.M.; Ahrens, E.T. A Novel Hypomorphic Apex1 Mouse Model Implicates Apurinic/Apyrimidinic Endonuclease 1 in Oxidative DNA Damage Repair in Gastric Epithelial Cells. Antioxid. Redox Signal. 2023, 38, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, W.; Su, Z.-Y.; Kong, A.-N.T. The complexity of the Nrf2 pathway: Beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef]
- Hiebert, P.; Wietecha, M.S.; Cangkrama, M.; Haertel, E.; Mavrogonatou, E.; Stumpe, M.; Steenbock, H.; Grossi, S.; Beer, H.-D.; Angel, P. Nrf2-mediated fibroblast reprogramming drives cellular senescence by targeting the matrisome. Dev. Cell 2018, 46, 145–161.e10. [Google Scholar] [CrossRef]
- Yu, C.; Xiao, J.-H. The Keap1-Nrf2 system: A mediator between oxidative stress and aging. Oxidative Med. Cell. Longev. 2021, 2021, 6635460. [Google Scholar] [CrossRef]
- Xu, W.; Luo, Y.; Yin, J.; Huang, M.; Luo, F. Targeting AMPK signaling by polyphenols: A novel strategy for tackling aging. Food Funct. 2023, 14, 56–73. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, C.; Zhou, N.; Wang, X.; Su, D.; Qi, Y. Metformin alleviates oxidative stress-induced senescence of human lens epithelial cells via AMPK activation and autophagic flux restoration. J. Cell. Mol. Med. 2021, 25, 8376–8389. [Google Scholar] [CrossRef]
- Liang, C.; Curry, B.J.; Brown, P.L.; Zemel, M.B. Leucine modulates mitochondrial biogenesis and SIRT1-AMPK signaling in C2C12 myotubes. J. Nutr. Metab. 2014, 2014, 239750. [Google Scholar] [CrossRef]
- Cai, M.; Ding, C.; Cao, X.; Wang, F.; Zhang, C.; Xian, Y. Label-free fluorometric assay for cytochrome c in apoptotic cells based on near infrared Ag2S quantum dots. Anal. Chim. Acta 2019, 1056, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond—Mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef] [PubMed]
- Mavrogonatou, E.; Kletsas, D. Plant-Derived Senotherapeutics for the Prevention and Treatment of Intervertebral Disc Degeneration and Aging. Metabolites 2024, 14, 146. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The clinical potential of senolytic drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Source | Catalog |
|---|---|---|
| Anti-p16 polyclonal antibody | Proteintech | 10883-1-AP |
| Anti-p21 polyclonal antibody | Proteintech | 10355-1-AP |
| Anti-pRB1 monoclonal antibody | Proteintech | 67521-1-16 |
| Anti- β-Actin Rabbit mAb | ABclonal | AC026 |
| HRP-conjugated Goat anti-Rabbit IgG (H + L) | ABclonal | AS014 |
| HRP-conjugated Goat anti- Mouse IgG (H + L) | ABclonal | AS071 |
| Number | Complexes | PDB Code | Docking Scores (kcal/mol) | Types of Interactions | Interacting Residues |
|---|---|---|---|---|---|
| 1 | P21-Chelidonic acid | 5XVG [63] | −12.68 | Conventional H-bonding | LEU 475. TYR 492, VAL 476, LYS 473 |
| 2 | P16-Chelidonic acid | 1GIH [64] | −11.99 | Conventional H-bonding, π Anion, π Sigma | ARG 126, VAL 164, THR 165, THR 14, ASP 127, ARG 169, LYS 129 |
| 3 | P53-Chelidonic acid | 4MZI [65] | −10.13 | Conventional H-bonding, π Alkyl | LYS 132, ARG 273, PRO 250, ARG 248, SER 240 |
| 4 | PRB1-Chelidonic acid | 1AD6 [66] | −6.81 | Van der waals, Conventional and Carbon H-bonding, π Alkyl | ASN 390, SER 499, ARG 445, ARG 500, |
| 5 | NRF2-Chelidonic acid | 3WN7 [67] | −7.71 | Conventional and Carbon H-bonding | Val 463, ILE 416, ARG 415, VAL 465, GLY 511, GLY 509 |
| 6 | NF-κB-Chelidonic acid | 1SVC [68] | −7.89 | Van der waals, Conventional and Carbon H-bonding, π Alkyl | ARG 57, SER 243, ASN 250, LYS 244 |
| 7 | JAK1-Chelidonic acid | 4L00 [69] | −11.31 | Van der waals, Conventional and Carbon H-bonding, π Alkyl | ARG 577, ARG 643 |
| 8 | JAK2-Chelidonic acid | 4GL9 [70] | −7.17 | Conventional and Carbon H-bonding, π Cation, π Sigma | LYS 1009, GLU 1006, LYS 1005, LYS 1030 |
| 9 | AMPK-Chelidonic acid | 7OPM [71] | −7.72 | Conventional and Carbon H-bonding | GLU 186, LEU 184, LYS 203 |
| 10 | STAT3-Chelidonic acid | 5AX3 [72] | −6.94 | Van der waals, Conventional H-bonding | LYS 221, THR 181, LYS 142 |
| 11 | HSP90-Chelidonic acid | 6KSQ [73] | −7.68 | Van der waals, Conventional and Carbon H-bonding, π Alkyl | THR 425, SER 476, GLU 461, THR 425, ALA 428, LYS 478, ARG 464 |
| 12 | FOXO4-Chelidonic acid | 3L2C [74] | −7.09 | Conventional H-bonding | Glu 125, ARG 429 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turkoglu, B.; Mansuroglu, B. Investigating the Effects of Chelidonic Acid on Oxidative Stress-Induced Premature Cellular Senescence in Human Skin Fibroblast Cells. Life 2024, 14, 1070. https://doi.org/10.3390/life14091070
Turkoglu B, Mansuroglu B. Investigating the Effects of Chelidonic Acid on Oxidative Stress-Induced Premature Cellular Senescence in Human Skin Fibroblast Cells. Life. 2024; 14(9):1070. https://doi.org/10.3390/life14091070
Chicago/Turabian StyleTurkoglu, Burcu, and Banu Mansuroglu. 2024. "Investigating the Effects of Chelidonic Acid on Oxidative Stress-Induced Premature Cellular Senescence in Human Skin Fibroblast Cells" Life 14, no. 9: 1070. https://doi.org/10.3390/life14091070
APA StyleTurkoglu, B., & Mansuroglu, B. (2024). Investigating the Effects of Chelidonic Acid on Oxidative Stress-Induced Premature Cellular Senescence in Human Skin Fibroblast Cells. Life, 14(9), 1070. https://doi.org/10.3390/life14091070