


The Genetic Architecture of Congenital Heart Disease in Neonatal Intensive Care Unit Patients—The Experience of University Medical Centre, Ljubljana
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Selection
2.2. Genetic and Bioinformatic Analysis
2.2.1. CMA and Classification of Results
2.2.2. Next-Generation Sequencing and Variant Interpretation
2.3. Statistics
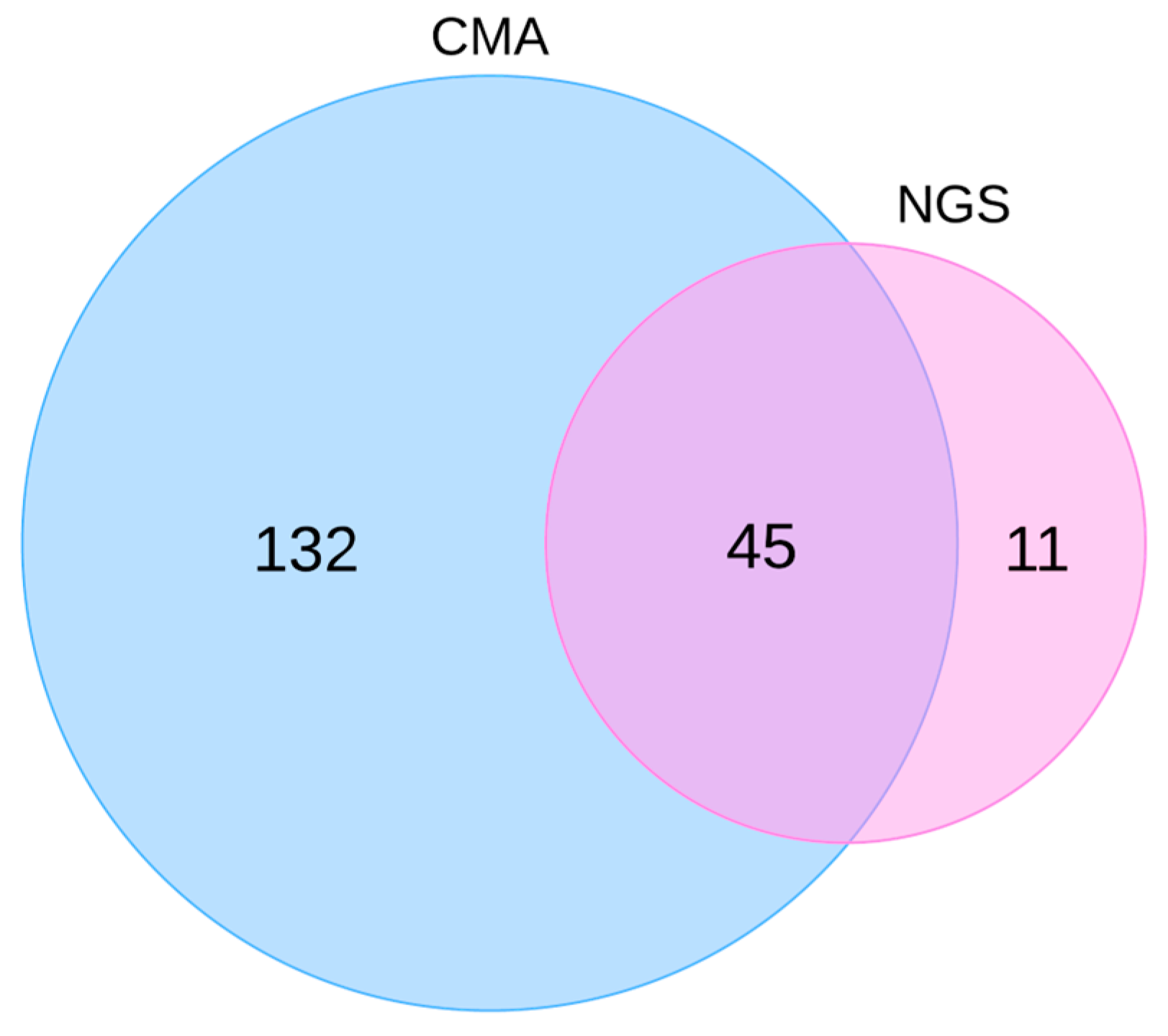
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bragança, J.; Pinto, R.; Silva, B.; Marques, N.; Leitão, H.S.; Fernandes, M.T. Charting the Path: Navigating Embryonic Development to Potentially Safeguard against Congenital Heart Defects. J. Pers. Med. 2023, 13, 1263. [Google Scholar] [CrossRef] [PubMed]
- Lalani, S.R. Other Genomic Disorders and Congenital Heart Disease. Am. J. Med. Genet. Pt C 2020, 184, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Diab, N.S.; Barish, S.; Dong, W.; Zhao, S.; Allington, G.; Yu, X.; Kahle, K.T.; Brueckner, M.; Jin, S.C. Molecular Genetics and Complex Inheritance of Congenital Heart Disease. Genes 2021, 12, 1020. [Google Scholar] [CrossRef]
- Suluba, E.; Shuwei, L.; Xia, Q.; Mwanga, A. Congenital Heart Diseases: Genetics, Non-Inherited Risk Factors, and Signaling Pathways. Egypt J. Med. Hum. Genet. 2020, 21, 11. [Google Scholar] [CrossRef]
- Fotiou, E.; Williams, S.; Martin-Geary, A.; Robertson, D.L.; Tenin, G.; Hentges, K.E.; Keavney, B. Integration of Large-Scale Genomic Data Sources With Evolutionary History Reveals Novel Genetic Loci for Congenital Heart Disease. Circ. Genom. Precis. Med. 2019, 12, e002694. [Google Scholar] [CrossRef]
- Page, D.J.; Miossec, M.J.; Williams, S.G.; Monaghan, R.M.; Fotiou, E.; Cordell, H.J.; Sutcliffe, L.; Topf, A.; Bourgey, M.; Bourque, G.; et al. Whole Exome Sequencing Reveals the Major Genetic Contributors to Nonsyndromic Tetralogy of Fallot. Circ. Res. 2019, 124, 553–563. [Google Scholar] [CrossRef]
- Shabana, N.; Shahid, S.U.; Irfan, U. Genetic Contribution to Congenital Heart Disease (CHD). Pediatr. Cardiol. 2020, 41, 12–23. [Google Scholar] [CrossRef]
- Geddes, G.C.; Basel, D.; Frommelt, P.; Kinney, A.; Earing, M. Genetic Testing Protocol Reduces Costs and Increases Rate of Genetic Diagnosis in Infants with Congenital Heart Disease. Pediatr. Cardiol. 2017, 38, 1465–1470. [Google Scholar] [CrossRef]
- Ahrens-Nicklas, R.C.; Khan, S.; Garbarini, J.; Woyciechowski, S.; D’Alessandro, L.; Zackai, E.H.; Deardorff, M.A.; Goldmuntz, E. Utility of Genetic Evaluation in Infants with Congenital Heart Defects Admitted to the Cardiac Intensive Care Unit. Am. J. Med. Genet. Pt A 2016, 170, 3090–3097. [Google Scholar] [CrossRef]
- Pierpont, M.E.; Basson, C.T.; Benson, D.W.; Gelb, B.D.; Giglia, T.M.; Goldmuntz, E.; McGee, G.; Sable, C.A.; Srivastava, D.; Webb, C.L. Genetic Basis for Congenital Heart Defects: Current Knowledge: A Scientific Statement From the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 3015–3038. [Google Scholar] [CrossRef]
- Roos-Hesselink, J.W.; Kerstjens-Frederikse, W.S.; Meijboom, F.J.; Pieper, P.G. Inheritance of Congenital Heart Disease. Neth. Heart J. 2005, 13, 88–91. [Google Scholar] [PubMed]
- Botto, L.D.; Lin, A.E.; Riehle-Colarusso, T.; Malik, S.; Correa, A. Seeking Causes: Classifying and Evaluating Congenital Heart Defects in Etiologic Studies. Birth Defects Res. 2007, 79, 714–727. [Google Scholar] [CrossRef]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T. American College of Medical Genetics Standards and Guidelines for Interpretation and Reporting of Postnatal Constitutional Copy Number Variants. Genet. Med. 2011, 13, 680–685. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast Processing of NGS Alignment Formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Desvignes, J.-P.; Bartoli, M.; Delague, V.; Krahn, M.; Miltgen, M.; Béroud, C.; Salgado, D. VarAFT: A Variant Annotation and Filtration System for Human next Generation Sequencing Data. Nucleic Acids Res. 2018, 46, W545–W553. [Google Scholar] [CrossRef]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]
- Jerves, T.; Beaton, A.; Kruszka, P. The Genetic Workup for Structural Congenital Heart Disease. Am. J. Med. Genet. Pt C 2020, 184, 178–186. [Google Scholar] [CrossRef]
- Wu, X.; Li, R.; Fu, F.; Pan, M.; Han, J.; Yang, X.; Zhang, Y.; Li, F.; Liao, C. Chromosome Microarray Analysis in the Investigation of Children with Congenital Heart Disease. BMC Pediatr. 2017, 17, 117. [Google Scholar] [CrossRef]
- Mone, F.; Stott, B.K.; Hamilton, S.; Seale, A.N.; Quinlan-Jones, E.; Allen, S.; Hurles, M.E.; McMullan, D.J.; Maher, E.R.; Kilby, M.D. The Diagnostic Yield of Prenatal Genetic Technologies in Congenital Heart Disease: A Prospective Cohort Study. Fetal Diagn. Ther. 2021, 48, 112–119. [Google Scholar] [CrossRef]
- Goldmuntz, E.; Paluru, P.; Glessner, J.; Hakonarson, H.; Biegel, J.A.; White, P.S.; Gai, X.; Shaikh, T.H. Microdeletions and Microduplications in Patients with Congenital Heart Disease and Multiple Congenital Anomalies: Copy Number Variants and Heart Defects. Congenit. Heart Dis. 2011, 6, 592–602. [Google Scholar] [CrossRef]
- Breckpot, J.; Thienpont, B.; Peeters, H.; De Ravel, T.; Singer, A.; Rayyan, M.; Allegaert, K.; Vanhole, C.; Eyskens, B.; Vermeesch, J.R.; et al. Array Comparative Genomic Hybridization as a Diagnostic Tool for Syndromic Heart Defects. J. Pediatr. 2010, 156, 810–817.e4. [Google Scholar] [CrossRef]
- Syrmou, A.; Tzetis, M.; Fryssira, H.; Kosma, K.; Oikonomakis, V.; Giannikou, K.; Makrythanasis, P.; Kitsiou-Tzeli, S.; Kanavakis, E. Array Comparative Genomic Hybridization as a Clinical Diagnostic Tool in Syndromic and Nonsyndromic Congenital Heart Disease. Pediatr. Res. 2013, 73, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Shreeve, N.; Sproule, C.; Choy, K.W.; Dong, Z.; Gajewska-Knapik, K.; Kilby, M.D.; Mone, F. Incremental Yield of Whole-genome Sequencing over Chromosomal Microarray Analysis and Exome Sequencing for Congenital Anomalies in Prenatal Period and Infancy: Systematic Review and Meta-analysis. Ultrasound Obstet. Gynecol. 2024, 63, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Hays, T.; Hernan, R.; Disco, M.; Griffin, E.L.; Goldshtrom, N.; Vargas, D.; Krishnamurthy, G.; Bomback, M.; Rehman, A.U.; Wilson, A.T.; et al. Implementation of Rapid Genome Sequencing for Critically Ill Infants with Complex Congenital Heart Disease. Circ. Genom. Precis. Med. 2023, 16, 415–420. [Google Scholar] [CrossRef]
- Li, R.; Fu, F.; Yu, Q.; Wang, D.; Jing, X.; Zhang, Y.; Li, F.; Li, F.; Han, J.; Pan, M.; et al. Prenatal Exome Sequencing in Fetuses with Congenital Heart Defects. Clin. Genet. 2020, 98, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Slavotinek, A.M.; Thompson, M.L.; Martin, L.J.; Gelb, B.D. Diagnostic Yield after Next-Generation Sequencing in Pediatric Cardiovascular Disease. Hum. Genet. Genom. Adv. 2024, 5, 100286. [Google Scholar] [CrossRef]
- D’Souza, E.E.; Findley, T.O.; Hu, R.; Khazal, Z.S.H.; Signorello, R.; Dash, C.; D’Gama, A.M.; Feldman, H.A.; Agrawal, P.B.; Wojcik, M.H.; et al. Genomic Testing and Molecular Diagnosis among Infants with Congenital Heart Disease in the Neonatal Intensive Care Unit. J. Perinatol. 2024, 44, 1196–1202. [Google Scholar] [CrossRef]
- Helm, B.M.; Ware, S.M. Clinical Decision Analysis of Genetic Evaluation and Testing in 1013 Intensive Care Unit Infants with Congenital Heart Defects Supports Universal Genetic Testing. Genes 2024, 15, 505. [Google Scholar] [CrossRef]
- Gyngell, C.; Newson, A.J.; Wilkinson, D.; Stark, Z.; Savulescu, J. Rapid Challenges: Ethics and Genomic Neonatal Intensive Care. Pediatrics 2019, 143, S14–S21. [Google Scholar] [CrossRef]
- Lantos, J.D. The Future of Newborn Genomic Testing. Children 2023, 10, 1140. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Category | N of Neonates (%) | N of Neonates CMA (%) | N of Neonates NGS (%) | |
|---|---|---|---|---|
| Isolated CHD | Simple | 29 (15) | 29 (100%) | 5 (17.2%) |
| Association | 18 (10) | 18 (100%) | 0 | |
| Complex | 20 (11) | 20 (100%) | 4 (20%) | |
| CHD with extracardiac defect | 121 (64) | 111 (91.7%) | 47 (38.8%) | |
| N | Congenital Heart Disease | Extracardiac Defects | Botto Classification | Type of Genetic Test | Results of Genetic Diagnostics | Genetic Classification | Syndrome |
|---|---|---|---|---|---|---|---|
| 1 | sASD | kidney anomaly | MCA | CMA | arr[hg38] 7q11.23(73,352,304–74,719,013)×1 | P | Williams syndrome |
| 2 | SVAS + stenosis of both pulmonary arteries | / | Isolated, association | CMA | arr[hg38] 7q11.23(74,060,601–74,079,563)×1 | P | Williams syndrome |
| 3 | mVSD, BAV | hypotonia, hypoplasia of the corpus callosum, feeding difficulties, cryptorchidism, dysmorphic facies | MCA | CMA | 46,XY, del(8)(p23.3p23.3),dup(8)(p12p23)dn | P | 8p inverted duplication/deletion syndrome |
| 4 | VSD | congenital hydronephrosis, dysmorphic facies | MCA | CMA | arr[GRCh38] 10q26.13q26.3(124,840,258–133,247,600)×1 | P | 10q26 deletion syndrome |
| 5 | VSD, ASD | dysmorphic facies | MCA | CMA | arr[GRCh38] 22q11.21(20,726,972–21,076,885)×1 | P | 22q11.2 microdeletion syndrome |
| 6 | pmVSD | coloboma of irises, hypotonia, anorectal anomaly, feeding difficulty | MCA | CMA | arr[GRCh38]11q23.3q25(1,193,69473–134,904,063)×1 | P | Jacobsen syndrome |
| 7 | TGA, ASD, PDA | LGA | MCA | CMA+NGS | arr[GRCh38] 17q12(36,792,631–37,854,407)×3, mat | P | 17q12 microduplication syndrome |
| EZH2(NM_004456.5): c.2051G>A | P | Weaver syndrome | |||||
| 8 | aortic valve stenosis, BAV, sASD | / | Isolated, complex | CMA | 47,XY,+mar.ish der(22)(pter->q11.21::p12->pter)(acro-p++,SE14/22+,CEP22+,N25+) | P | Cat eye syndrome |
| 9 | ToF, ASD | dysmorphic facies | MCA | CMA | arr[GRCh38] 1q21.1q21.2(147,147,409–143,729,392)×3 dn | P | 1q21.1 microduplication syndrome |
| 10 | VSD | hypocalcemia, dysmorphic facies | MCA | CMA | arr[GRCh38] 22q11.21(18,925,357–21,076,885)×1 dn | P | 22q11.2 microdeletion syndrome |
| 11 | pmVSD, multiple ASDs, PFO | SGA, palatoschisis, dysmorphic facies, proximal placement of thumb, pes calcaneovalgus | MCA | CMA | arr[GRCh38]18q21.31q23(57,444,618–80,244,381)×1 | P | 18q deletion syndrome |
| 12 | VSD, ASD | renal cysts | MCA | CMA | arr[GRCh38] 17p11.2(16,938,849–20,314,464)×1 | P | Smith–Magenis syndrome |
| 13 | pmVSD, truncus arteriosus, | hypothyroidism | MCA | CMA | arr[GRCh38] 22q11.21(18,930,283–21,076,885)×1 | P | 22q11.2 microdeletion syndrome |
| 14 | VSD, ASD | dysmorphic facies | MCA | CMA | arr[GRCh38] 22q11.21(18,930,283–21,076,885)×1 | P | 22q11.2 microdeletion syndrome |
| 15 | VSD, ASD | dysmorphic facies | MCA | CMA | arr[GRCh38] 1q21.1q21.2(147,147,409–148,353,946)×1 dn | P | 1q21.1 microdeletion syndrome |
| 16 | mVSD, sASD, hypoplastic aortic arch | dysmorphic facies | MCA | CMA | arr[GRCh38] 22q11.21(18,930,283–21,076,885)×1 | P | 22q11.2 microdeletion syndrome |
| 17 | ASD, PDA | dysmorphic facies | MCA | CMA | arr[GRCh38] 16p13.11(15,032,852–16,198,378)×3 | P | 16p13. 11 microdeletion syndrome |
| 18 | ASD | hypotonia, hydronephrosis | MCA | CMA | arr[GRCh38] 16p13.11(15,032,852–16,198,378)×3 | P | 16p13.11 microduplication syndrome |
| 19 | stenosis of aortic valve, BAV | dysmorphic features | MCA | CMA | arr(X)×1[0.8] | P | mosaic Turner syndrome |
| N | Congenital Heart Disease | Extracardiac Defects | Botto Classification | Type of Genetic Test | Results of Genetic Diagnostics | Genetic Classification | Syndrome |
|---|---|---|---|---|---|---|---|
| 1 | ToF | EA/TEF | MCA | CMA+NGS | CHD7(NM_017780):c.5405-8C>G | P | CHARGE syndrome |
| 2 | valvular pumonary stenosis, SVPS | dysmorphic facies, macrosomia, unilateral cryptorchidism, aplasia cutis | MCA | NGS | PTPN11(NM_002834.5):c.923A>G, p.Asn308Ser | P | Noonan syndrome |
| 3 | sASD | hypotonia, hypoplasia of the corpus callosum, dysmorphic features, palatoschisis, glossoschissis, hypermobility of joints, clinodactyly of 5th fingers | MCA | NGS | OFD1(NM_003611.3):c.1193_1196del, p.Gln398Leufs*2 | LP | Orofaciodigital syndrome I |
| 4 | AVSD | coloboma of iris, facial nerve palsy, mixed hearing loss, hypotonia dysmorphic features, feeding difficulties | MCA | NGS | CHD7(NM_017780.4):c.4353+1G>A | P | CHARGE syndrome |
| 5 | sASD, BAV, PDA | dysmorphic facies, palatoschisis, widely spaced nipples, barrel chest, hypermobility of joints, clinodactyly of 5th fingers | MCA | CMA+NGS | KMT2D(NM_003482.4):c.4364dup, p.Tyr1455* | P | Kabuki syndrome |
| 6 | sASD, cleft mitral valve with mild MVR, PDA | dysmorphic facies, chorioretinal coloboma, vocal cord paresis, feeding difficulties, hearing loss | MCA | NGS | CHD7(NM_017780.4):c.3655C>T, p.Arg1219* | P | CHARGE syndrome |
| 7 | ToF | brachycephaly, ptosis of right eyelid, coloboma of optic nerve papilla, gnatoschisis, choanal atresia, feeding difficulties, unilateral renal agenesis, dysmorphic features, hockey-stick palmar crease, partial 2–3 toe syndactyly, hypotonia, hearing loss | MCA | CMA+NGS | CHD7(NM_017780.3):c.4203_4204delT, p.His1401Glnfs*20 | P | CHARGE syndrome |
| 8 | sASD, aortic valve stenosis, BAV | AMC, dynamic upper airway obstruction, ptosis of right eyelid, cryptorchidism, bilateral congenital hip dislocation, clubfoot, fibromatosis colli | MCA | NGS | CHRNG(NM_005199.5):c.753_754del, p.Val253Alafs*44 | P | Multiple pterygium syndrome— Escobar type |
| CHRNG(NM_005199.5):c.250G>A, p.Asp84Asn | LP | ||||||
| 9 | sASD, PPS, PDA | dysmorphic facies, direct hyperbilirubinemia | MCA | NGS | JAG1(NM_000214.3):c.2122_2125del, p.Gln708Valfs*34 | P | Alagille syndrome |
| 10 | pulmonary valve stenosis, PDA, PFO | dysmorphic facies, LGA, renal cyst | MCA | NGS | PTPN11(NM_002834.5):c.922A>G, p.Asn308Asp | P | Noonan syndrome |
| 11 | pulmonary valve stenosis, BAV, bicuspid pulmonary valve, PFO | dysmorphic facies, bilateral coloboma of iris, macula and papilla, horseshoe kidney, ankyloglossia | MCA | CMA+NGS | CHD7(NM_017780.4):c.6292C>T, p.Arg2098* | P | CHARGE syndrome |
| 12 | pmVSD | hypotonia, abnormal cortical gyration, feeding difficulties, dysmorphic facies, single palmar crease | MCA | CMA+NGS | SMARCA4(NM_003072.5):c.4114C>T, p.Arg1372Cys | LP | Coffin-Siris syndrome 4 |
| 13 | left atrial isomerism | heterotaxy, polysplenia | MCA | NGS | DNAAF3(NM_001256715.2):c.73_82del, p.Leu25Lysfs*20 | LP | Ciliary dyskinesia, primary, 2 |
| N | Congenital Heart Disease | Extracardiac Defects | Botto Classification | Type of Genetic Test | Results of Genetic Diagnostics | Genetic Classification |
|---|---|---|---|---|---|---|
| 1 | ASD, PDA | Partial ACC, feeding difficulties, dysmorphic features, occipital subcutaneous vascular malformation | MCA | CMA+NGS | arr[GRCh38]15q25.2q25.3(85,149,691–85,666,309)×1 | VUS |
| 2 | CoA, hypoplastic distal aortic arch, BAV, pmVSD, ASD, PDA | hypotonia, hypocalcemia, dysmorphic facies | MCA | CMA | arr[GRCh38]9p21.2(25,713,811–26,334,159)×1 | VUS |
| 3 | ASD, pmVSD | Isolatated, association | CMA | arr[GRCh38]2q32.3(19,661,4800–196,837,193)×1dn | VUS | |
| arr[GRCh38]8p23.2(2,470,593–4,801,373)×3 mat | VUS | |||||
| 4 * | ASD, VSD | hypotonia, HCC, moderate ventriculomegaly, dysmorphic facies, hypoplasia of distal phalanx of fifth finger | MCA | CMA+NGS | arr[GRCh38]Xp22.2(14,325,346–14,757,768)×2 mat | VUS |
| 5 | CoA, HLHS | MCA | CMA | arr[GRCh38]2q24.2q24.3(163#,517,376–164,167,131)×3 pat | VUS | |
| 6 | CoA, HLHS | hypotonia | CMA | arr[GRCh38]16q24.1(85,002,354–85,508,509)×1 dn | VUS | |
| 7 | ToF | coloboma of iris, dysmorphic features | MCA | CMA+NGS | NOTCH1(NM_017617.5):c.3190G>A, p.Asp1064Asn | VUS |
| 8 | ASD | EA/TEF, annular pancreas, horseshoe kidney, extrarenal pelvis, spina bifida occulta, billiary ducts anomaly | MCA | CMA+NGS | ZNF462(NM_021224.6):c.6334C>T, p.Leu2112Phe | VUS |
| 9 | CoA | polydactyly, hypospadias, SGA | MCA | CMA+NGS | TLL1(NM_012464.5):c.283G>A(pat), p.Gly95Arg | VUS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peterlin, A.; Bertok, S.; Writzl, K.; Lovrečić, L.; Maver, A.; Peterlin, B.; Debeljak, M.; Nosan, G. The Genetic Architecture of Congenital Heart Disease in Neonatal Intensive Care Unit Patients—The Experience of University Medical Centre, Ljubljana. Life 2024, 14, 1118. https://doi.org/10.3390/life14091118
Peterlin A, Bertok S, Writzl K, Lovrečić L, Maver A, Peterlin B, Debeljak M, Nosan G. The Genetic Architecture of Congenital Heart Disease in Neonatal Intensive Care Unit Patients—The Experience of University Medical Centre, Ljubljana. Life. 2024; 14(9):1118. https://doi.org/10.3390/life14091118
Chicago/Turabian StylePeterlin, Ana, Sara Bertok, Karin Writzl, Luca Lovrečić, Aleš Maver, Borut Peterlin, Maruša Debeljak, and Gregor Nosan. 2024. "The Genetic Architecture of Congenital Heart Disease in Neonatal Intensive Care Unit Patients—The Experience of University Medical Centre, Ljubljana" Life 14, no. 9: 1118. https://doi.org/10.3390/life14091118