


Detection of Insulin in Insulin-Deficient Islets of Patients with Type 1 Diabetes


Abstract
:1. Introduction
2. Materials and Methods
3. Results
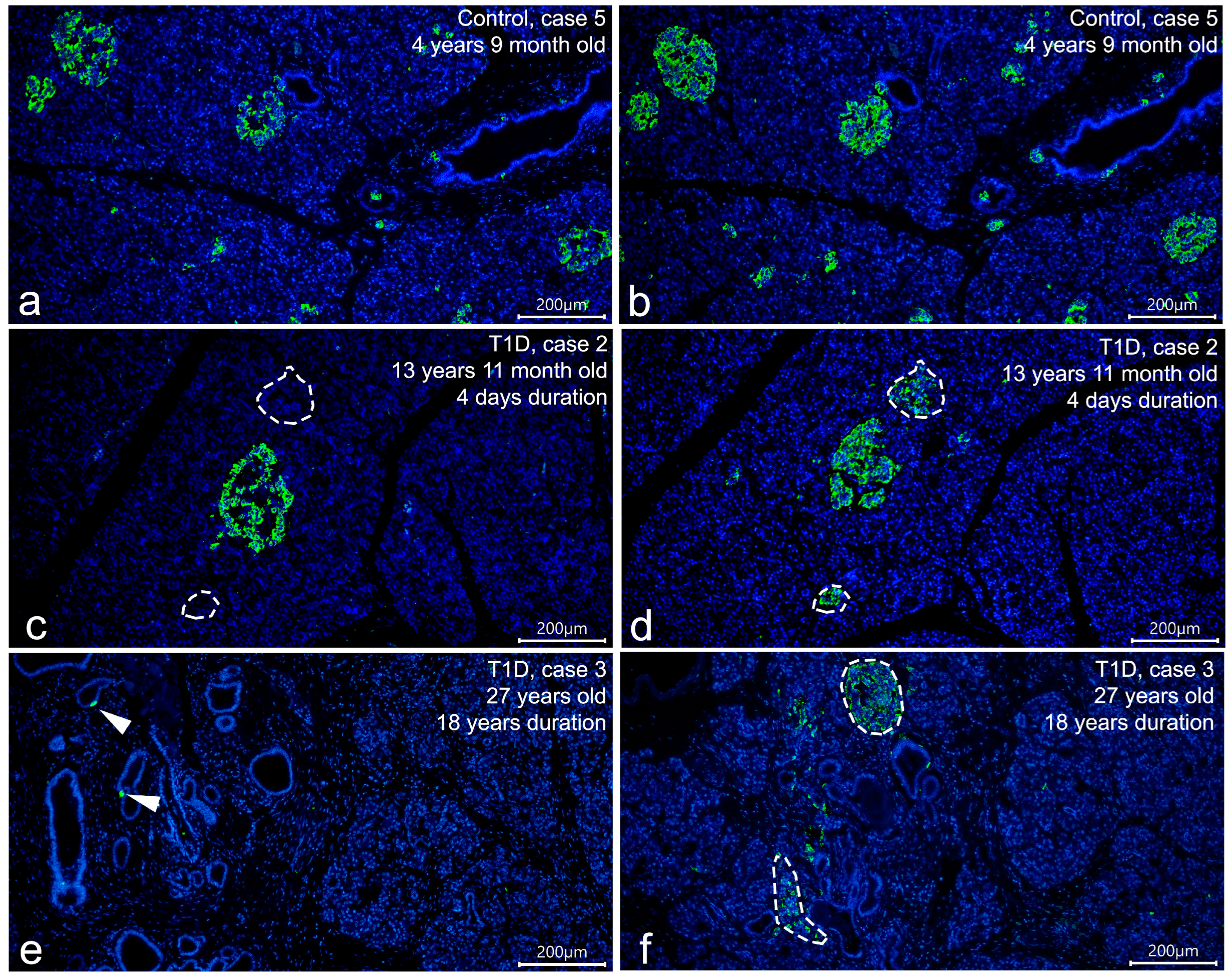
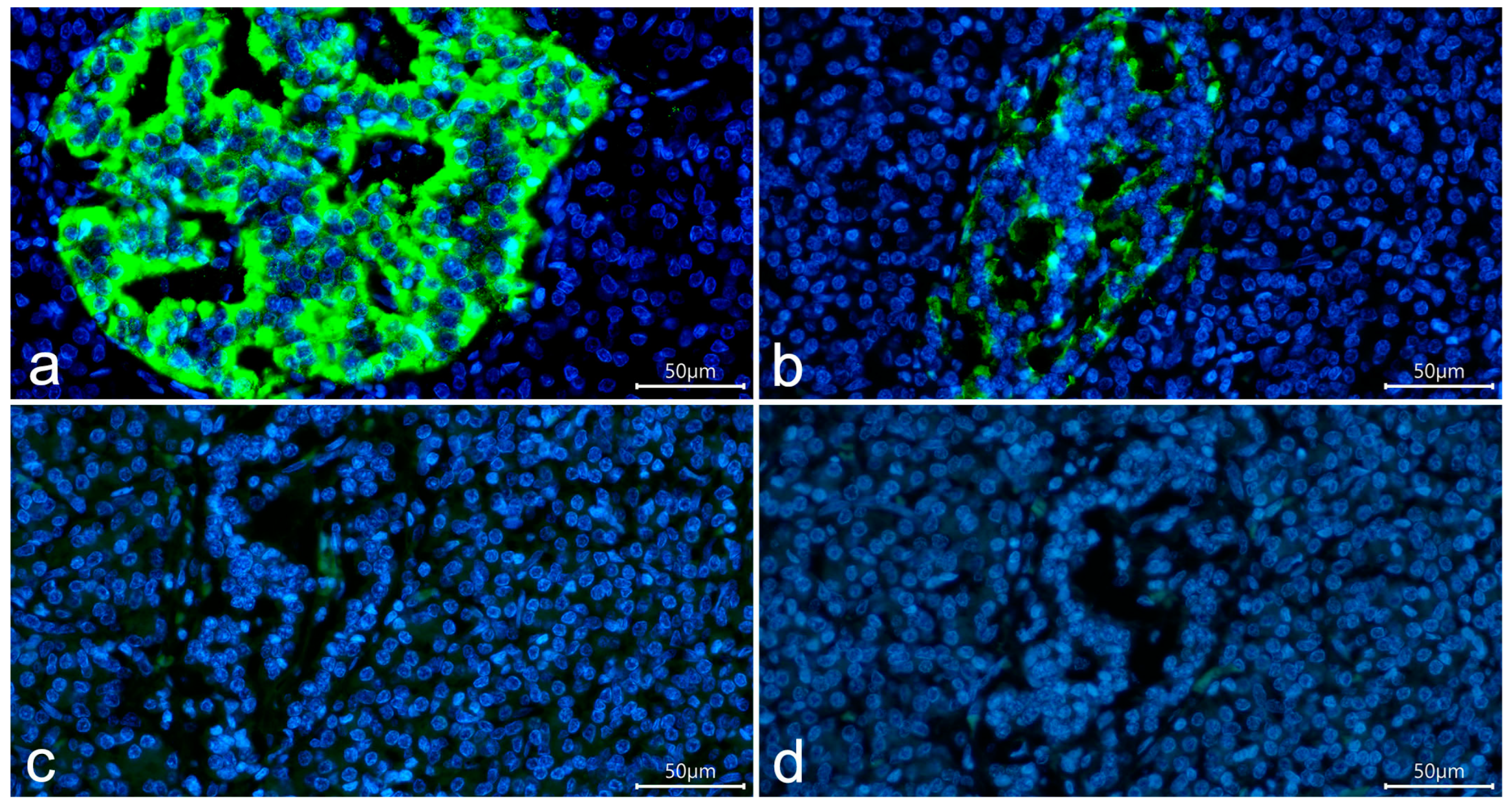
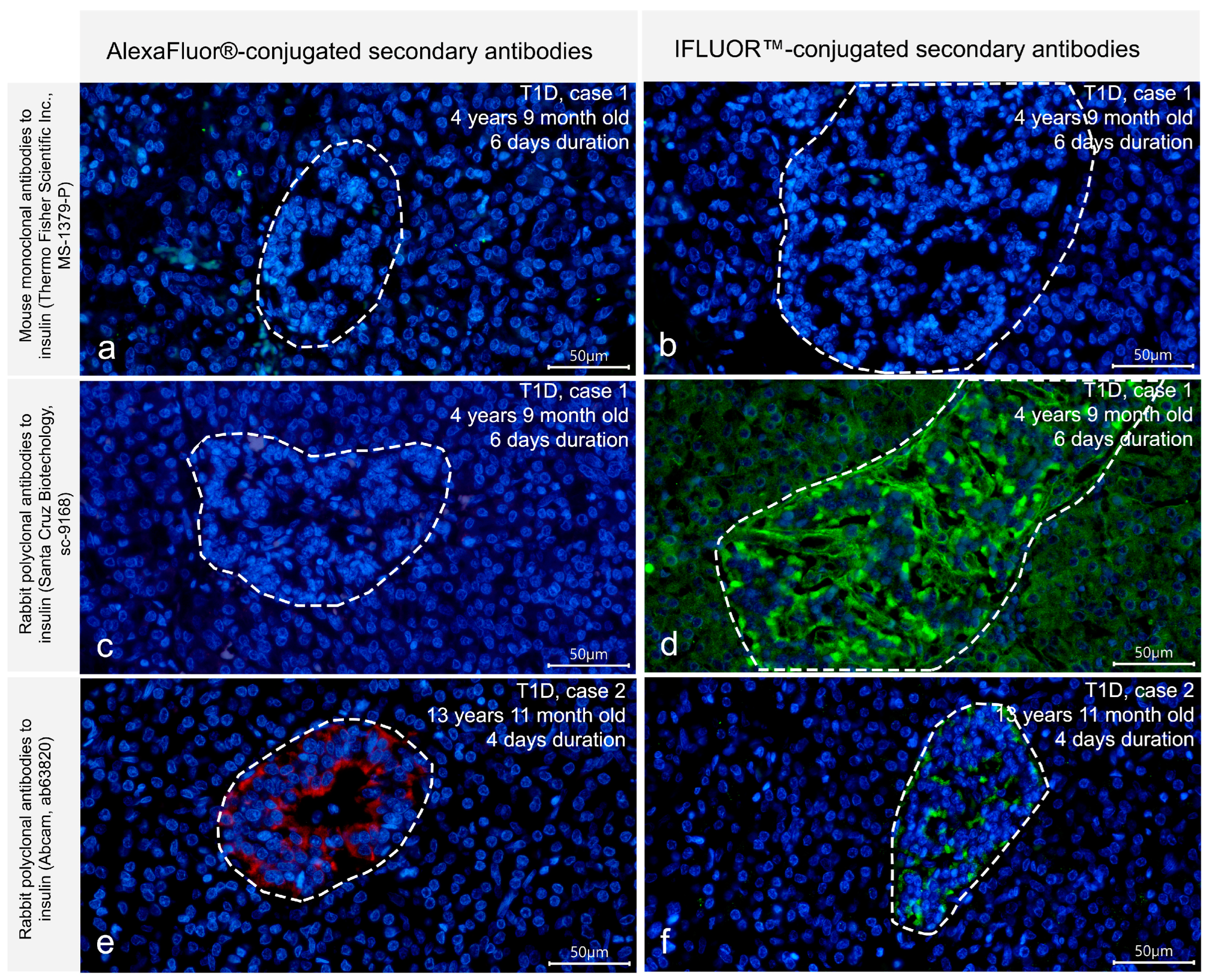
3.1. Detection of Insulin in the T1D Pancreas
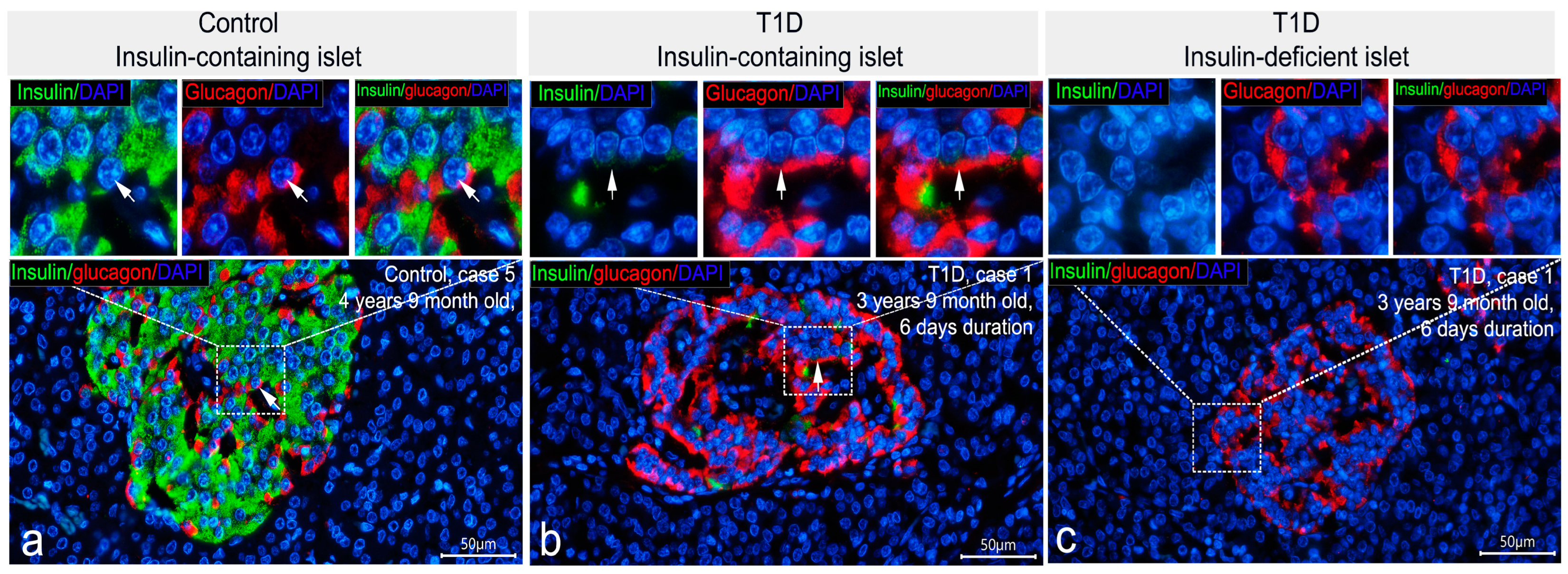
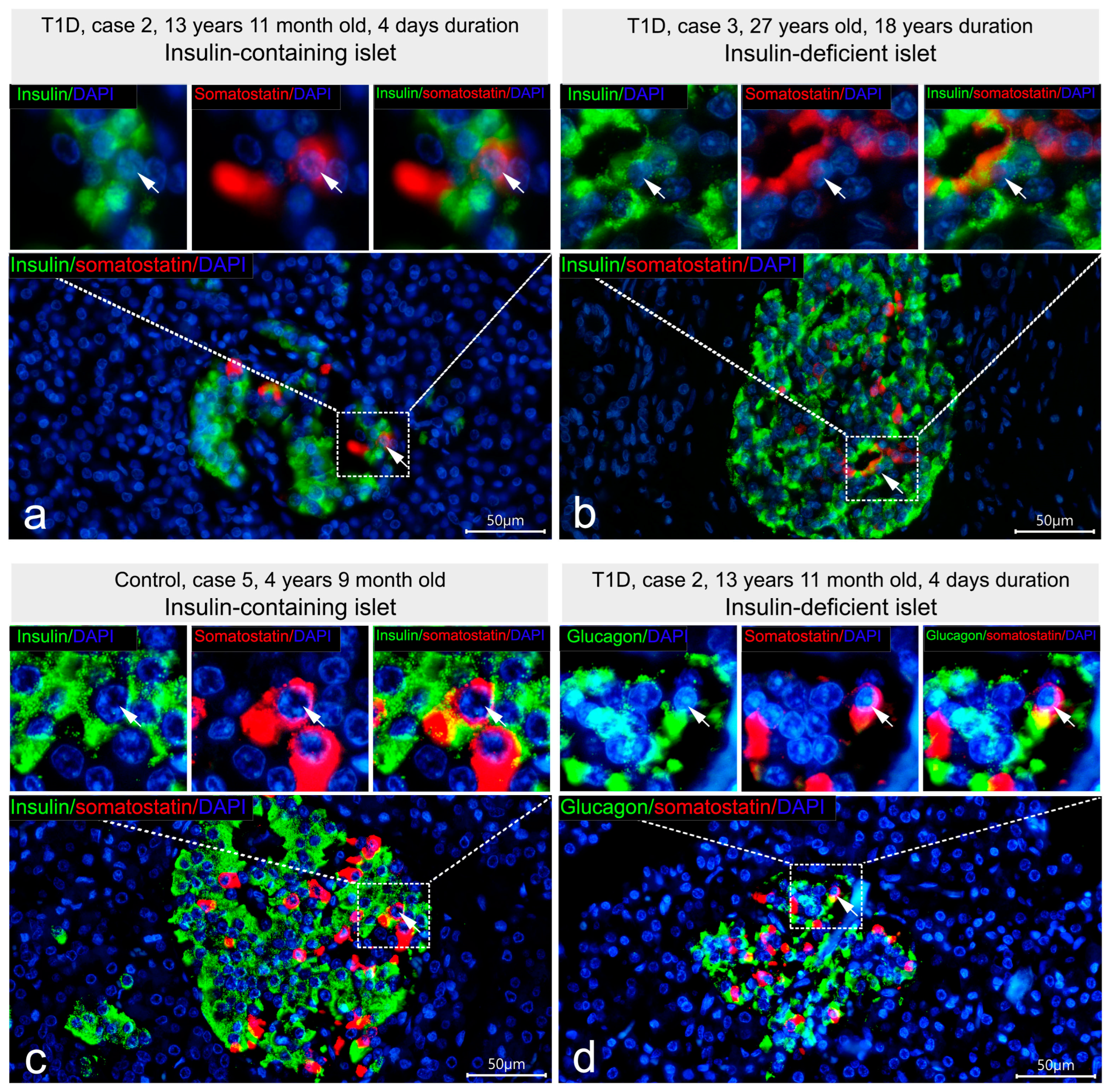
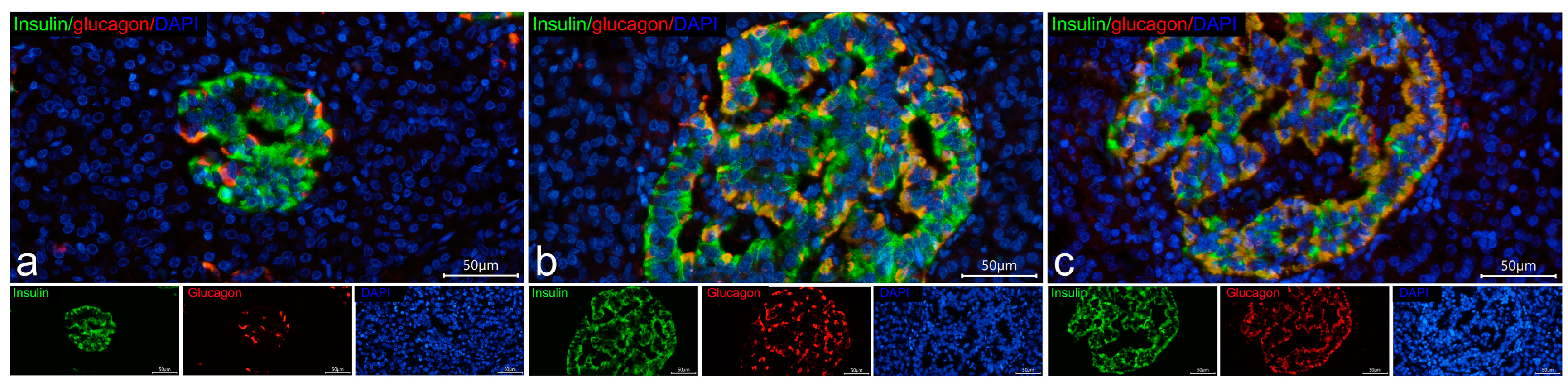
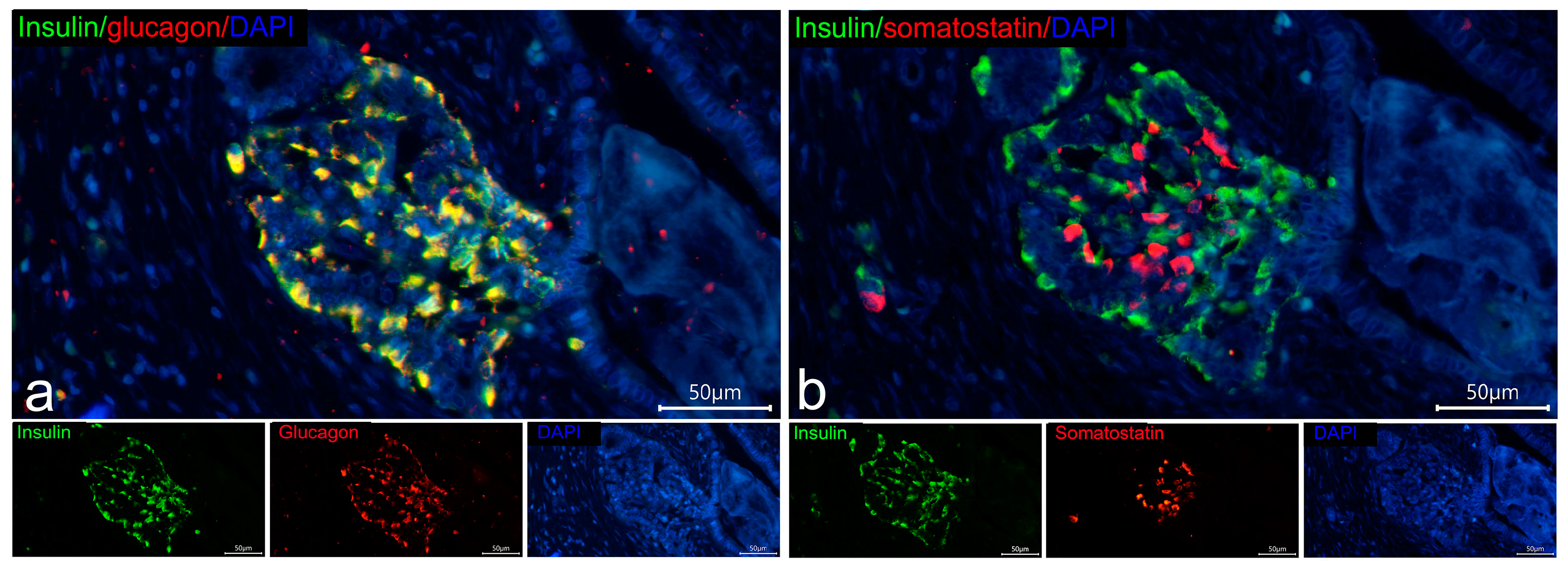
3.2. Co-Localization of Insulin with Glucagon and Somatostatin
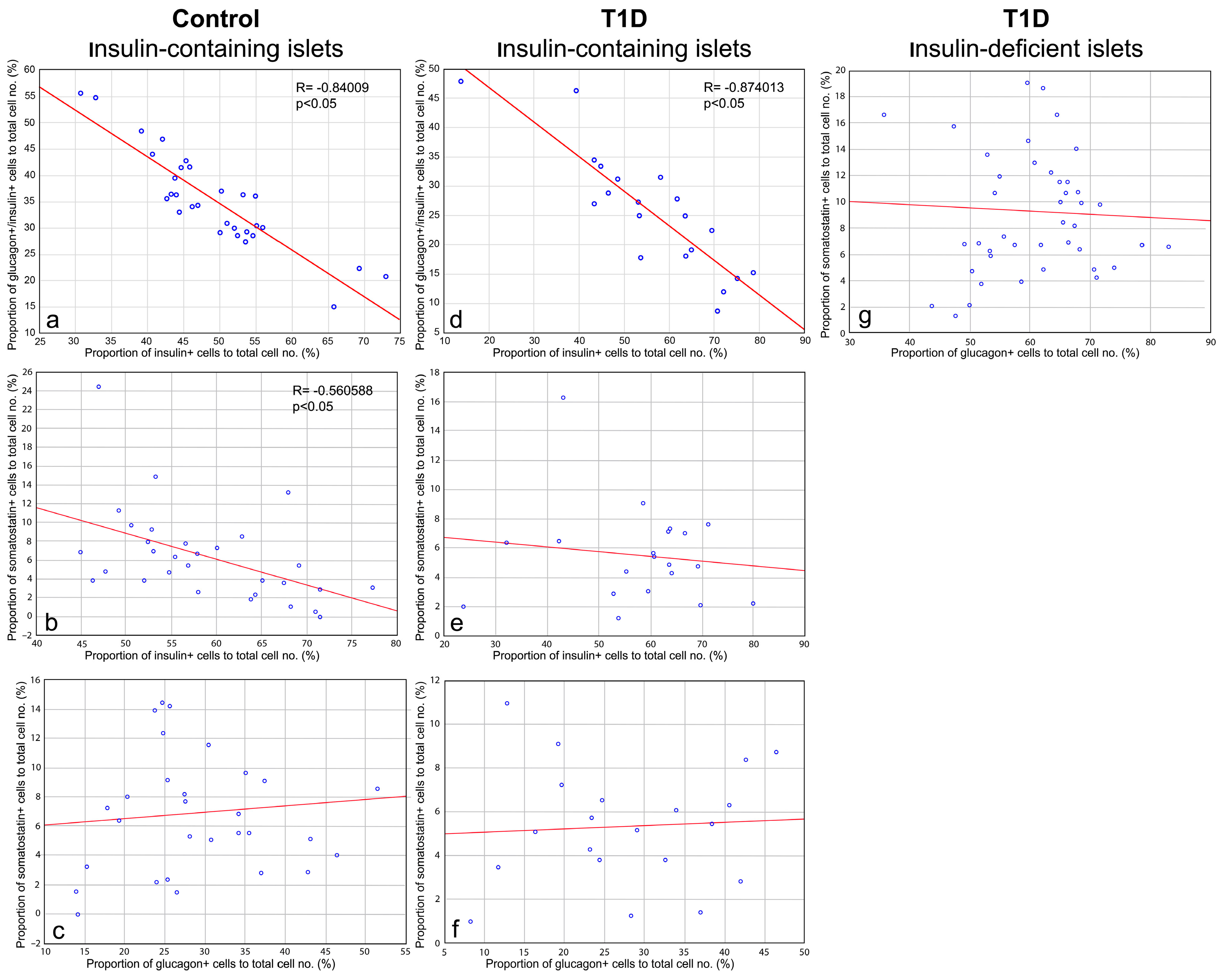
3.3. Cellular Composition of Insulin-Containing and Insulin-Deficient Islets
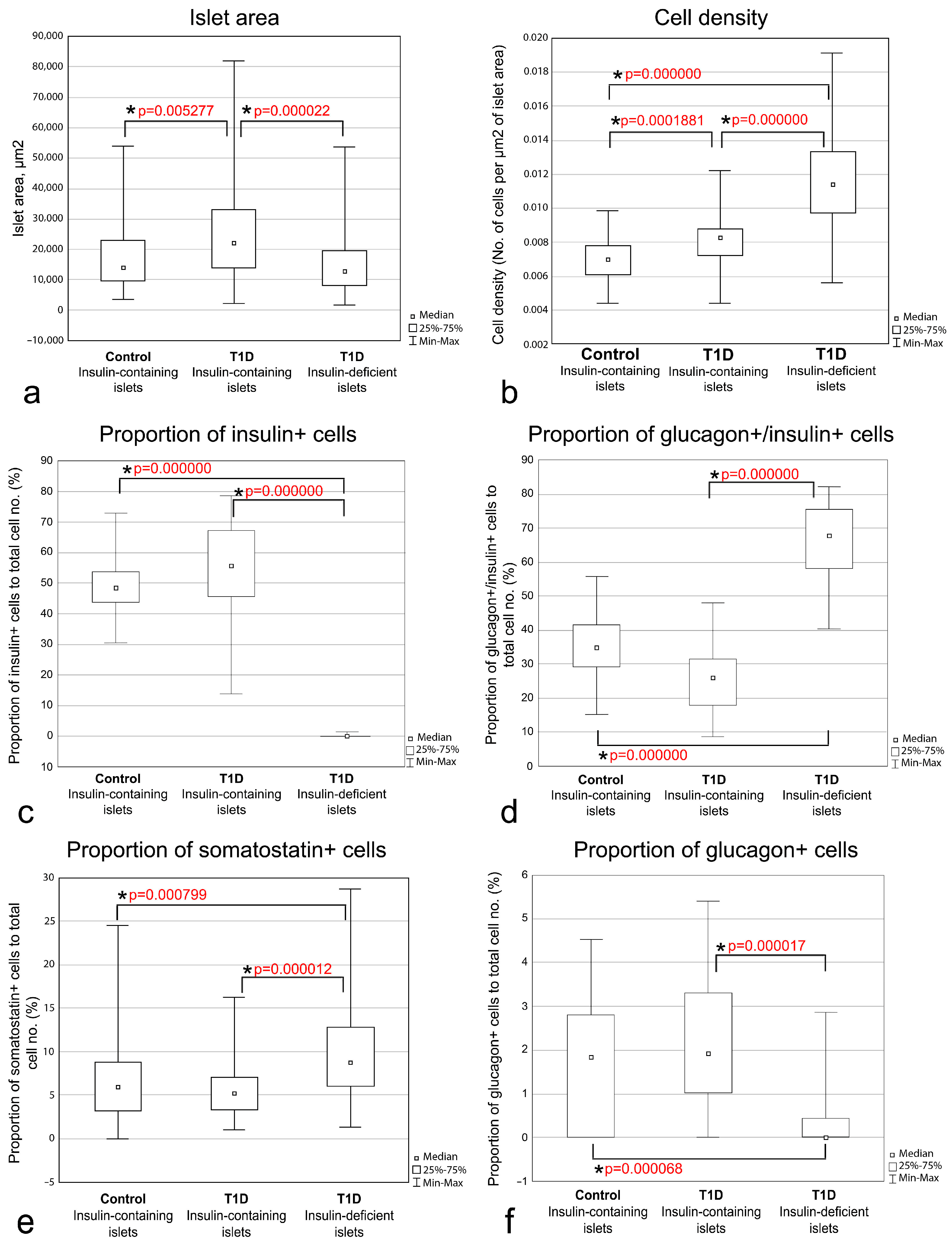
3.4. Quantitative Morphometric Analysis of Cellular Composition of Insulin-Containing and Insulin-Deficient Islets
4. Discussion
5. Conclusions
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campbell-Thompson, M.; Fu, A.; Kaddis, J.S.; Wasserfall, C.; Schatz, D.A.; Pugliese, A.; Atkinson, M.A. Insulitis and β-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes 2016, 65, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Foulis, A.K.; Farquharson, M.A.; Hardman, R. Aberrant Expression of Class II Major Histocompatibility Complex Molecules by B Cells and Hyperexpression of Class I Major Histocompatibility Complex Molecules by Insulin Containing Islets in Type 1 (Insulin-Dependent) Diabetes Mellitus. Diabetologia 1987, 30, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Foulis, A.K.; Liddle, C.N.; Farquharson, M.A.; Richmond, J.A.; Weir, R.S. The Histopathology of the Pancreas in Type I (Insulin-Dependent) Diabetes Mellitus: A 25-Year Review of Deaths in Patients under 20 Years of Age in the United Kingdom. Diabetologia 1986, 29, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Foulis, A.K.; Stewart, J.A. The Pancreas in Recent-Onset Type 1 (Insulin-Dependent) Diabetes Mellitus: Insulin Content of Islets, Insulitis and Associated Changes in the Exocrine Acinar Tissue. Diabetologia 1984, 26, 456–461. [Google Scholar] [CrossRef]
- Gepts, W. Pathologic Anatomy of the Pancreas in Juvenile Diabetes Mellitus. Diabetes 1965, 14, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Gianani, R.; Campbell-Thompson, M.; Sarkar, S.A.; Wasserfall, C.; Pugliese, A.; Solis, J.M.; Kent, S.C.; Hering, B.J.; West, E.; Steck, A.; et al. Dimorphic Histopathology of Long-Standing Childhood-Onset Diabetes. Diabetologia 2010, 53, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Klöppel, G.; Drenck, C.R.; Oberholzer, M.; Heitz, P.U. Morphometric Evidence for a Striking B-Cell Reduction at the Clinical Onset of Type 1 Diabetes. Virchows Arch. A Pathol. Anat. Histopathol. 1984, 403, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Krogvold, L.; Wiberg, A.; Edwin, B.; Buanes, T.; Jahnsen, F.L.; Hanssen, K.F.; Larsson, E.; Korsgren, O.; Skog, O.; Dahl-Jørgensen, K. Insulitis and Characterisation of Infiltrating T Cells in Surgical Pancreatic Tail Resections from Patients at Onset of Type 1 Diabetes. Diabetologia 2016, 59, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.J.; Jacobson, D.R.; Rankin, M.M.; Cox, A.R.; Kushner, J.A. β Cells Persist in T1D Pancreata Without Evidence of Ongoing β-Cell Turnover or Neogenesis. J. Clin. Endocrinol. Metab. 2017, 102, 2647–2659. [Google Scholar] [CrossRef]
- Lernmark, Å.; Stenger, D.; Baskin, D.G.; Palmer, J.P.; Li, L.; Klöppel, G.; Vathanaprida, C.; Fält, K.; Landin-Olsson, M.; Gown, A.M.; et al. Heterogeneity of Islet Pathology in Two Infants with Recent Onset Diabetes Mellitus. Virchows Arch. 1995, 425, 631–640. [Google Scholar] [CrossRef]
- Löhr, M.; Klöppel, G. Residual Insulin Positivity and Pancreatic Atrophy in Relation to Duration of Chronic Type 1 (Insulin-Dependent) Diabetes Mellitus and Microangiopathy. Diabetologia 1987, 30, 757–762. [Google Scholar] [CrossRef]
- Meier, J.J.; Bhushan, A.; Butler, A.E.; Rizza, R.A.; Butler, P.C. Sustained Beta Cell Apoptosis in Patients with Long-Standing Type 1 Diabetes: Indirect Evidence for Islet Regeneration? Diabetologia 2005, 48, 2221–2228. [Google Scholar] [CrossRef]
- Rahier, J.; Goebbels, R.M.; Henquin, J.C. Cellular Composition of the Human Diabetic Pancreas. Diabetologia 1983, 24, 366–371. [Google Scholar] [CrossRef]
- Stefan, Y.; Orci, L.; Malaisse-Lagae, F.; Perrelet, A.; Patel, Y.; Unger, R.H. Quantitation of Endocrine Cell Content in the Pancreas of Nondiabetic and Diabetic Humans. Diabetes 1982, 31, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Klinke, D.J. Extent of Beta Cell Destruction Is Important but Insufficient to Predict the Onset of Type 1 Diabetes Mellitus. PLoS ONE 2008, 3, e1374. [Google Scholar] [CrossRef] [PubMed]
- Klinke, D.J. Age-Corrected Beta Cell Mass Following Onset of Type 1 Diabetes Mellitus Correlates with Plasma C-Peptide in Humans. PLoS ONE 2011, 6, e26873. [Google Scholar] [CrossRef]
- Greenbaum, C.J.; Mandrup-Poulsen, T.; McGee, P.F.; Battelino, T.; Haastert, B.; Ludvigsson, J.; Pozzilli, P.; Lachin, J.M.; Kolb, H. Mixed-Meal Tolerance Test Versus Glucagon Stimulation Test for the Assessment of β-Cell Function in Therapeutic Trials in Type 1 Diabetes. Diabetes Care 2008, 31, 1966–1971. [Google Scholar] [CrossRef] [PubMed]
- VanBuecken, D.E.; Greenbaum, C.J. Residual C-Peptide in Type 1 Diabetes: What Do We Really Know? Pediatr. Diabetes 2014, 15, 84–90. [Google Scholar] [CrossRef]
- Sherr, J.L.; Ghazi, T.; Wurtz, A.; Rink, L.; Herold, K.C. Characterization of Residual β Cell Function in Long-standing Type 1 Diabetes. Diabetes Metab. Res. Rev. 2014, 30, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.; Lee, H.I.; Lee, M.; Song, K.; Choi, H.S.; Kwon, A.; Kim, H.-S.; Chae, H.W. Insulin Requirement and Complications Associated With Serum C-Peptide Decline in Patients With Type 1 Diabetes Mellitus During 15 Years After Diagnosis. Front. Endocrinol. 2022, 13, 869204. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lovejoy, N.F.; Faustman, D.L. Persistence of Prolonged C-Peptide Production in Type 1 Diabetes as Measured With an Ultrasensitive C-Peptide Assay. Diabetes Care 2012, 35, 465–470. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, M.H.; Dantas, J.R.; Barone, B.; Serfaty, F.M.; Kupfer, R.; Albernaz, M.; Bencke, M.R.; Zajdenverg, L.; Rodacki, M.; de Oliveira, J.E.P. Residual C-Peptide in Patients with Type 1 Diabetes and Multiethnic Backgrounds. Clinics 2013, 68, 123–126. [Google Scholar] [CrossRef]
- Uno, S.; Imagawa, A.; Kozawa, J.; Fukui, K.; Iwahashi, H.; Shimomura, I. Complete Loss of Insulin Secretion Capacity in Type 1A Diabetes Patients during Long-term Follow Up. J. Diabetes Investig. 2018, 9, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Wellens, M.J.; Vollenbrock, C.E.; Dekker, P.; Boesten, L.S.M.; Geelhoed-Duijvestijn, P.H.; de Vries-Velraeds, M.M.C.; Nefs, G.; Wolffenbuttel, B.H.R.; Aanstoot, H.-J.; van Dijk, P.R. Residual C-Peptide Secretion and Hypoglycemia Awareness in People with Type 1 Diabetes. BMJ Open Diabetes Res. Care 2021, 9, e002288. [Google Scholar] [CrossRef] [PubMed]
- Keenan, H.A.; Sun, J.K.; Levine, J.; Doria, A.; Aiello, L.P.; Eisenbarth, G.; Bonner-Weir, S.; King, G.L. Residual Insulin Production and Pancreatic β-Cell Turnover After 50 Years of Diabetes: Joslin Medalist Study. Diabetes 2010, 59, 2846–2853. [Google Scholar] [CrossRef]
- Oram, R.A.; McDonald, T.J.; Shields, B.M.; Hudson, M.M.; Shepherd, M.H.; Hammersley, S.; Pearson, E.R.; Hattersley, A.T. Most People With Long-Duration Type 1 Diabetes in a Large Population-Based Study Are Insulin Microsecretors. Diabetes Care 2015, 38, 323–328. [Google Scholar] [CrossRef]
- Oram, R.A.; Jones, A.G.; Besser, R.E.J.; Knight, B.A.; Shields, B.M.; Brown, R.J.; Hattersley, A.T.; McDonald, T.J. The Majority of Patients with Long-Duration Type 1 Diabetes Are Insulin Microsecretors and Have Functioning Beta Cells. Diabetologia 2014, 57, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Saveliev, S.; Shustov, S.; Romashevskiy, B.; Proshina, A.; Chekavskaya, V.; Appolonova, S.; Savelieva, E.; Balyasnikova, E. Clinical Evaluation of Insulin Synthesis of β-Cells Pancreas during Long-Term Diabetes Mellitus Type 1. Bull. Russ. Mil. Med. Acad. 2015, 3, 78–82. [Google Scholar]
- Lam, C.J.; Chatterjee, A.; Shen, E.; Cox, A.R.; Kushner, J.A. Low-Level Insulin Content Within Abundant Non-β Islet Endocrine Cells in Long-Standing Type 1 Diabetes. Diabetes 2019, 68, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Brereton, M.F.; Iberl, M.; Shimomura, K.; Zhang, Q.; Adriaenssens, A.E.; Proks, P.; Spiliotis, I.I.; Dace, W.; Mattis, K.K.; Ramracheya, R.; et al. Reversible Changes in Pancreatic Islet Structure and Function Produced by Elevated Blood Glucose. Nat. Commun. 2014, 5, 4639. [Google Scholar] [CrossRef] [PubMed]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of β-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef]
- Marselli, L.; Suleiman, M.; Masini, M.; Campani, D.; Bugliani, M.; Syed, F.; Martino, L.; Focosi, D.; Scatena, F.; Olimpico, F.; et al. Are We Overestimating the Loss of Beta Cells in Type 2 Diabetes? Diabetologia 2014, 57, 362–365. [Google Scholar] [CrossRef]
- Sherry, N.A.; Kushner, J.A.; Glandt, M.; Kitamura, T.; Brillantes, A.-M.B.; Herold, K.C. Effects of Autoimmunity and Immune Therapy on β-Cell Turnover in Type 1 Diabetes. Diabetes 2006, 55, 3238–3245. [Google Scholar] [CrossRef] [PubMed]
- Kitabchi, A.E. Proinsulin and C-Peptide: A Review. Metabolism 1977, 26, 547–587. [Google Scholar] [CrossRef] [PubMed]
- Madsen, O.D. Proinsulin-Specific Monoclonal Antibodies: Immunocytochemical Application as β-Cell Markers and as Probes for Conversion. Diabetes 1987, 36, 1203–1211. [Google Scholar] [CrossRef]
- Schroer, J.A.; Bender, T.; Feldmann, R.J.; Kim, K.J. Mapping Epitopes on the Insulin Molecule Using Monoclonal Antibodies. Eur. J. Immunol. 1983, 13, 693–700. [Google Scholar] [CrossRef]
- Storch, M.-J.; Licht, T.; Petersen, K.-G.; Obermeier, R.; Kerp, L. Specificity of Monoclonal Anti-Human Insulin Antibodies. Diabetes 1987, 36, 1005–1009. [Google Scholar] [CrossRef]
- Moin, A.S.M.; Dhawan, S.; Cory, M.; Butler, P.C.; Rizza, R.A.; Butler, A.E. Increased Frequency of Hormone Negative and Polyhormonal Endocrine Cells in Lean Individuals with Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 3628–3636. [Google Scholar] [CrossRef] [PubMed]
- Spijker, H.S.; Song, H.; Ellenbroek, J.H.; Roefs, M.M.; Engelse, M.A.; Bos, E.; Koster, A.J.; Rabelink, T.J.; Hansen, B.C.; Clark, A.; et al. Loss of β-Cell Identity Occurs in Type 2 Diabetes and Is Associated With Islet Amyloid Deposits. Diabetes 2015, 64, 2928–2938. [Google Scholar] [CrossRef] [PubMed]
- White, M.G.; Marshall, H.L.; Rigby, R.; Huang, G.C.; Amer, A.; Booth, T.; White, S.; Shaw, J.A.M. Expression of Mesenchymal and α-Cell Phenotypic Markers in Islet β-Cells in Recently Diagnosed Diabetes. Diabetes Care 2013, 36, 3818–3820. [Google Scholar] [CrossRef] [PubMed]
- Elgamal, R.M.; Kudtarkar, P.; Melton, R.L.; Mummey, H.M.; Benaglio, P.; Okino, M.-L.; Gaulton, K.J. An Integrated Map of Cell Type–Specific Gene Expression in Pancreatic Islets. Diabetes 2023, 72, 1719–1728. [Google Scholar] [CrossRef]
- Fasolino, M.; Schwartz, G.W.; Patil, A.R.; Mongia, A.; Golson, M.L.; Wang, Y.J.; Morgan, A.; Liu, C.; Schug, J.; Liu, J.; et al. Single-Cell Multi-Omics Analysis of Human Pancreatic Islets Reveals Novel Cellular States in Type 1 Diabetes. Nat. Metab. 2022, 4, 284–299. [Google Scholar] [CrossRef]
- Segerstolpe, Å.; Palasantza, A.; Eliasson, P.; Andersson, E.-M.; Andréasson, A.-C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Seiron, P.; Wiberg, A.; Kuric, E.; Krogvold, L.; Jahnsen, F.L.; Dahl-Jørgensen, K.; Skog, O.; Korsgren, O. Characterisation of the Endocrine Pancreas in Type 1 Diabetes: Islet Size Is Maintained but Islet Number Is Markedly Reduced. J. Pathol. Clin. Res. 2019, 5, 248–255. [Google Scholar] [CrossRef]
- Wang, Z.; York, N.W.; Nichols, C.G.; Remedi, M.S. Pancreatic β Cell Dedifferentiation in Diabetes and Redifferentiation Following Insulin Therapy. Cell Metab. 2014, 19, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Jonas, J.-C.; Sharma, A.; Hasenkamp, W.; Ilkova, H.; Patanè, G.; Laybutt, R.; Bonner-Weir, S.; Weir, G.C. Chronic Hyperglycemia Triggers Loss of Pancreatic β Cell Differentiation in an Animal Model of Diabetes. J. Biol. Chem. 1999, 274, 14112–14121. [Google Scholar] [CrossRef] [PubMed]
- Rui, J.; Deng, S.; Arazi, A.; Perdigoto, A.L.; Liu, Z.; Herold, K.C. β Cells That Resist Immunological Attack Develop during Progression of Autoimmune Diabetes in NOD Mice. Cell Metab. 2017, 25, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; McKenna, B.; Li, C.; Reichert, M.; Nguyen, J.; Singh, T.; Yang, C.; Pannikar, A.; Doliba, N.; Zhang, T.; et al. Pdx1 Maintains β Cell Identity and Function by Repressing an α Cell Program. Cell Metab. 2014, 19, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, G.D.; Bender, A.S.; Cirulli, V.; Mastracci, T.L.; Kelly, S.M.; Tsirigos, A.; Kaestner, K.H.; Sussel, L. Pancreatic β Cell Identity Requires Continual Repression of Non–β Cell Programs. J. Clin. Investig. 2016, 127, 244–259. [Google Scholar] [CrossRef]
- Jeon, J.; Correa-Medina, M.; Ricordi, C.; Edlund, H.; Diez, J.A. Endocrine Cell Clustering During Human Pancreas Development. J. Histochem. Cytochem. 2009, 57, 811–824. [Google Scholar] [CrossRef]
- Riedel, M.J.; Asadi, A.; Wang, R.; Ao, Z.; Warnock, G.L.; Kieffer, T.J. Immunohistochemical Characterisation of Cells Co-Producing Insulin and Glucagon in the Developing Human Pancreas. Diabetologia 2012, 55, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Riopel, M.; Li, J.; Fellows, G.F.; Goodyer, C.G.; Wang, R. Ultrastructural and Immunohistochemical Analysis of the 8-20 Week Human Fetal Pancreas. Islets 2014, 6, e982949. [Google Scholar] [CrossRef]
- Olaniru, O.E.; Kadolsky, U.; Kannambath, S.; Vaikkinen, H.; Fung, K.; Dhami, P.; Persaud, S.J. Single-Cell Transcriptomic and Spatial Landscapes of the Developing Human Pancreas. Cell Metab. 2023, 35, 184–199.e5. [Google Scholar] [CrossRef] [PubMed]
- Proshchina, A.; Krivova, Y.; Gurevich, L.; Otlyga, D.; Saveliev, S. 2153-P: PDX1- and Nkx6.1-Immunonegative ß Cells in the Developing Human Pancreas. Diabetes 2019, 68, 2153-P. [Google Scholar] [CrossRef]
- Fiori, J.L.; Shin, Y.-K.; Kim, W.; Krzysik-Walker, S.M.; González-Mariscal, I.; Carlson, O.D.; Sanghvi, M.; Moaddel, R.; Farhang, K.; Gadkaree, S.K.; et al. Resveratrol Prevents β-Cell Dedifferentiation in Nonhuman Primates Given a High-Fat/High-Sugar Diet. Diabetes 2013, 62, 3500–3513. [Google Scholar] [CrossRef] [PubMed]
- Nostro, M.C.; Keller, G. Generation of beta cells from human pluripotent stem cells: Potential for regenerative medicine. Semin. Cell Dev. Biol. 2012, 23, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Bruin, J.E.; Erener, S.; Vela, J.; Hu, X.; Johnson, J.D.; Kurata, H.T.; Lynn, F.C.; Piret, J.M.; Asadi, A.; Rezania, A.; et al. Characterization of polyhormonal insulin-producing cells derived in vitro from human embryonic stem cells. Stem Cell Res. 2014, 12, 194–208. [Google Scholar] [CrossRef]
- Russ, H.A.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015, 34, 1759–1772. [Google Scholar] [CrossRef] [PubMed]
- Aigha, I.I.; Abdelalim, E.M. NKX6.1 transcription factor: A crucial regulator of pancreatic β cell development, identity, and proliferation. Stem Cell Res. Ther. 2020, 11, 459. [Google Scholar] [CrossRef] [PubMed]
- Leete, P.; Oram, R.A.; McDonald, T.J.; Shields, B.M.; Ziller, C.; TIGI study team; Hattersley, A.T.; Richardson, S.J.; Morgan, N.G. Studies of insulin and proinsulin in pancreas and serum support the existence of aetiopathological endotypes of type 1 diabetes associated with age at diagnosis. Diabetologia 2020, 63, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Sims, E.K.; Bahnson, H.T.; Nyalwidhe, J.; Haataja, L.; Davis, A.K.; Speake, C.; DiMeglio, L.A.; Blum, J.; Morris, M.A.; Mirmira, R.G.; et al. T1D Exchange Residual C-peptide Study Group. Proinsulin Secretion Is a Persistent Feature of Type 1 Diabetes. Diabetes Care 2019, 42, 258–264. [Google Scholar] [CrossRef]
- Rodriguez-Calvo, T.; Chen, Y.C.; Verchere, C.B.; Haataja, L.; Arvan, P.; Leete, P.; Richardson, S.J.; Morgan, N.G.; Qian, W.J.; Pugliese, A.; et al. Altered β-Cell Prohormone Processing and Secretion in Type 1 Diabetes. Diabetes 2021, 70, 1038–1050. [Google Scholar] [CrossRef] [PubMed]
- Sims, E.K.; Syed, F.; Nyalwidhe, J.; Bahnson, H.T.; Haataja, L.; Speake, C.; Morris, M.A.; Balamurugan, A.N.; Mirmira, R.G.; Nadler, J.; et al. Abnormalities in proinsulin processing in islets from individuals with longstanding T1D. Transl. Res. 2019, 213, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Brissova, M.; Haliyur, R.; Saunders, D.; Shrestha, S.; Dai, C.; Blodgett, D.M.; Bottino, R.; Campbell-Thompson, M.; Aramandla, R.; Poffenberger, G.; et al. α Cell Function and Gene Expression Are Compromised in Type 1 Diabetes. Cell Rep. 2018, 22, 2667–2676. [Google Scholar] [CrossRef]
- Siafarikas, A.; Johnston, R.J.; Bulsara, M.K.; O’Leary, P.; Jones, T.W.; Davis, E.A. Early loss of the glucagon response to hypoglycemia in adolescents with type 1 diabetes. Diabetes Care 2012, 35, 1757–1762. [Google Scholar] [CrossRef] [PubMed]
- Zenz, S.; Mader, J.K.; Regittnig, W.; Brunner, M.; Korsatko, S.; Boulgaropoulos, B.; Magnes, C.; Raml, R.; Narath, S.H.; Eller, P.; et al. Impact of C-Peptide Status on the Response of Glucagon and Endogenous Glucose Production to Induced Hypoglycemia in T1DM. J. Clin. Endocrinol. Metab. 2018, 103, 1408–1417. [Google Scholar] [CrossRef]
- Dagogo-Jack, S. Hypoglycemia in type 1 diabetes mellitus: Pathophysiology and prevention. Treat. Endocrinol. 2004, 3, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Gibb, F.W.; McKnight, J.A.; Clarke, C.; Strachan, M.W.J. Preserved C-peptide secretion is associated with fewer low-glucose events and lower glucose variability on flash glucose monitoring in adults with type 1 diabetes. Diabetologia 2020, 63, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Gubitosi-Klug, R.A.; Braffett, B.H.; Hitt, S.; Arends, V.; Uschner, D.; Jones, K.; Diminick, L.; Karger, A.B.; Paterson, A.D.; Roshandel, D.; et al. DCCT/EDIC Research Group. Residual β cell function in long-term type 1 diabetes associates with reduced incidence of hypoglycemia. J. Clin. Investig. 2021, 13, e143011. [Google Scholar] [CrossRef]
- Rickels, M.R.; Evans-Molina, C.; Bahnson, H.T.; Ylescupidez, A.; Nadeau, K.J.; Hao, W.; Clements, M.A.; Sherr, J.L.; Pratley, R.E.; Hannon, T.S.; et al. T1D Exchange β-Cell Function Study Group. High residual C-peptide likely contributes to glycemic control in type 1 diabetes. J. Clin. Investig. 2020, 130, 1850–1862. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Sex | Age | T1D Duration | Blood Glucose Level | Insulinotherapy | Cause of Death | No. of Blocks Studied |
|---|---|---|---|---|---|---|---|
| 1 | Male | 3 years 9 month | 6 days | 23.51 nmol/L | Actrapid (20 U/D) | Diabetic coma complicated by acute pulmonary insufficiency and cardiac decompensation due to acute respiratory viral infection | 6 |
| 2 | Male | 13 years 11 month | 4 days | 33 nmol/L | Actrapid (25 U/D) | Multiple organ failure associated with diabetic coma and ketoacidosis | 7 |
| 3 | Male | 27 years | 18 years | N/A 1 | N/A | Anemia due to esophageal variceal hemorrhage | 1 |
| 4 | Male | 41 years | 26 years | N/A | N/A | Chronic renal insufficiency associated with T1D | 1 |
| 5 | Male | 4 years 9 month | Nondiabetic | ≤5.5 nmol/L | Not treated | Congenital combined immunodeficiency and bacterial sepsis complicated by anemic brain infarct, disseminated intravascular coagulation, pulmonary insufficiency and cardiac decompensation | 1 |
| 6 | Female | 8 years 11 month | Nondiabetic | ≤5.5 nmol/L | Not treated | Jungle fever complicated by disseminated intravascular coagulation, hemorrhagic brain edema, cardio-pulmonary insufficiency and cardiac decompensation | 1 |
| 7 | Female | 25 years | Nondiabetic | ≤5.5 nmol/L | Not treated | Dilated cardiomyopathy complicated by pulmonary edema and cardio-pulmonary insufficiency | 1 |
| Primary Antibodies | Company | Cat#/RRID | Dilution Ranges | Optimal Dilutions |
|---|---|---|---|---|
| Mouse monoclonal antibodies to insulin, clone K36aC10 | Sigma, St. Louis, MO, USA | I2018/RRID:AB_260137 | 1:1000–1:64,000 | 1:16,000 |
| Mouse monoclonal antibodies to insulin | Thermo Fisher Scientific Inc., Fremont, CA, USA | MS-1379-P/RRID:AB_62834 | 1:50–1:500 | 1:100 |
| Rabbit polyclonal antibodies to insulin | Santa Cruz Biotechnology, Santa Cruz, CA, USA | Sc-9168/ RRID:AB_2126540 | 1:50–1:200 | 1:100 |
| Rabbit polyclonal antibodies to insulin | Abcam, Cambridge, UK | Ab63820/RRID:AB_1925116 | 1:50–1:2000 | 1:200 |
| Rabbit polyclonal antibodies to glucagon | Thermo Fisher Scientific Inc., Regensburg, Germany | PA5-13442/RRID:AB_2107206 | 1:50–1:200 | 1:50 |
| Rabbit polyclonal antibodies to glucagon, clone K79bB10 | Sigma, St. Louis, MO, USA | G2654/RRID:AB_259852 | 1:1000–1:16,000 | 1:4000 |
| Rabbit polyclonal antibodies to somatostatin | Abcam, Cambrige, UK | Cat#ab103790/RRID:AB_10711731 | 1:50–1:400 | 1:50 |
| Parameter | Insulin-Containing Islets (Control) | Insulin-Containing Islets (T1D) | Insulin-Deficient Islets (T1D) |
|---|---|---|---|
| Islet area, µm2 | 14,233.90 (9719.22–23,035.77) (n = 90) 1 | 22,129.98 (13,898.48–32,860.76) (n = 90) | 12,735.23 (81,4840–19,678.74) (n = 90) |
| Cell density (no. of cells per µm2 of islet area) | 0.007003 (0.006128–0.007818) (n = 90) | 0.008307 (0.007224–0.008815) (n = 90) | 0.011385 (0.009767–0.013352) (n = 90) |
| Proportion of insulin+ cells to total cell no. (%) | 48.43750 (43.75000–53.65854) (n = 30) | 55.75768 (45.59944–67.14421) (n = 20) | 0.00000 (0.00000–0.00000) (n = 40) |
| Proportion of glucagon+/insulin+ cells to total cell no. (%) | 35.01359 (29.26829–41.56627) (n = 30) | 26.01351 (18.01948–31.44325) (n = 20) | 67.79580 (58.11443–75.43831) (n = 40) |
| Proportion of glucagon+ to total cell no. (%) | 1.847458 (0.000000–2.797203) (n = 30) | 1.923785 (1.020408–3.308411) (n = 20) | 0.000000 (0.000000–0.439569) (n = 40) |
| Proportion of somatostatin+ cells to total cell no. (%) | 5.96927 (3.21271–8.86587) (n = 60) | 5.28891 (3.27808–7.09256) (n = 40) | 8.81834 (6.06592–12.88623) (n = 80) |
| Proportion of insulin+/somatostatin+ cells to total cell no. (%) | 0.000000 (0.000000–0.000000) (n = 30) | 0.403661 (0.00000–0.756471) (n = 20) | 0.000000 (0.000000–0.000000) (n = 40) |
| Proportion of glucagon+/somatostatin+ cells to total cell no. (%) | 0.00000 (0.000000–0.76336) (n = 30) | 0.35357 (0.00000–0.73309) (n = 20) | 0.94340 (0.000000–0.439569) (n = 40) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krivova, Y.; Proshchina, A.; Otlyga, D.; Kharlamova, A.; Saveliev, S. Detection of Insulin in Insulin-Deficient Islets of Patients with Type 1 Diabetes. Life 2025, 15, 125. https://doi.org/10.3390/life15010125
Krivova Y, Proshchina A, Otlyga D, Kharlamova A, Saveliev S. Detection of Insulin in Insulin-Deficient Islets of Patients with Type 1 Diabetes. Life. 2025; 15(1):125. https://doi.org/10.3390/life15010125
Chicago/Turabian StyleKrivova, Yuliya, Alexandra Proshchina, Dmitry Otlyga, Anastasia Kharlamova, and Sergey Saveliev. 2025. "Detection of Insulin in Insulin-Deficient Islets of Patients with Type 1 Diabetes" Life 15, no. 1: 125. https://doi.org/10.3390/life15010125
APA StyleKrivova, Y., Proshchina, A., Otlyga, D., Kharlamova, A., & Saveliev, S. (2025). Detection of Insulin in Insulin-Deficient Islets of Patients with Type 1 Diabetes. Life, 15(1), 125. https://doi.org/10.3390/life15010125