

Management of Pulmonary Arterial Hypertension: Current Strategies and Future Prospects
 ,
, 
Abstract
1. Introduction
- Group 1 PH—pulmonary arterial hypertension (PAH);
- Group 2 PH—PH due to Left Heart Disease;
- Group 3 PH—PH due to lung diseases and/or hypoxia;
- Group 4 PH—PH due to pulmonary artery obstructions;
- Group 5 PH—PH due to multifactorial mechanisms.
2. Endothelin Receptor Antagonists (ERAs)
3. Nitric Oxide-Cyclic Guanosine Monophosphate (cGMP) Stimulators
3.1. Phosphodiesterase-5 Inhibitors (PDE-5i)
3.2. Soluble Guanylate Cyclase (sGC) Stimulator
3.3. Prostacyclin Analogues and Receptor Agonists
3.4. Fixed-Dose Combination Drug (Macitentan/Tadalafil)
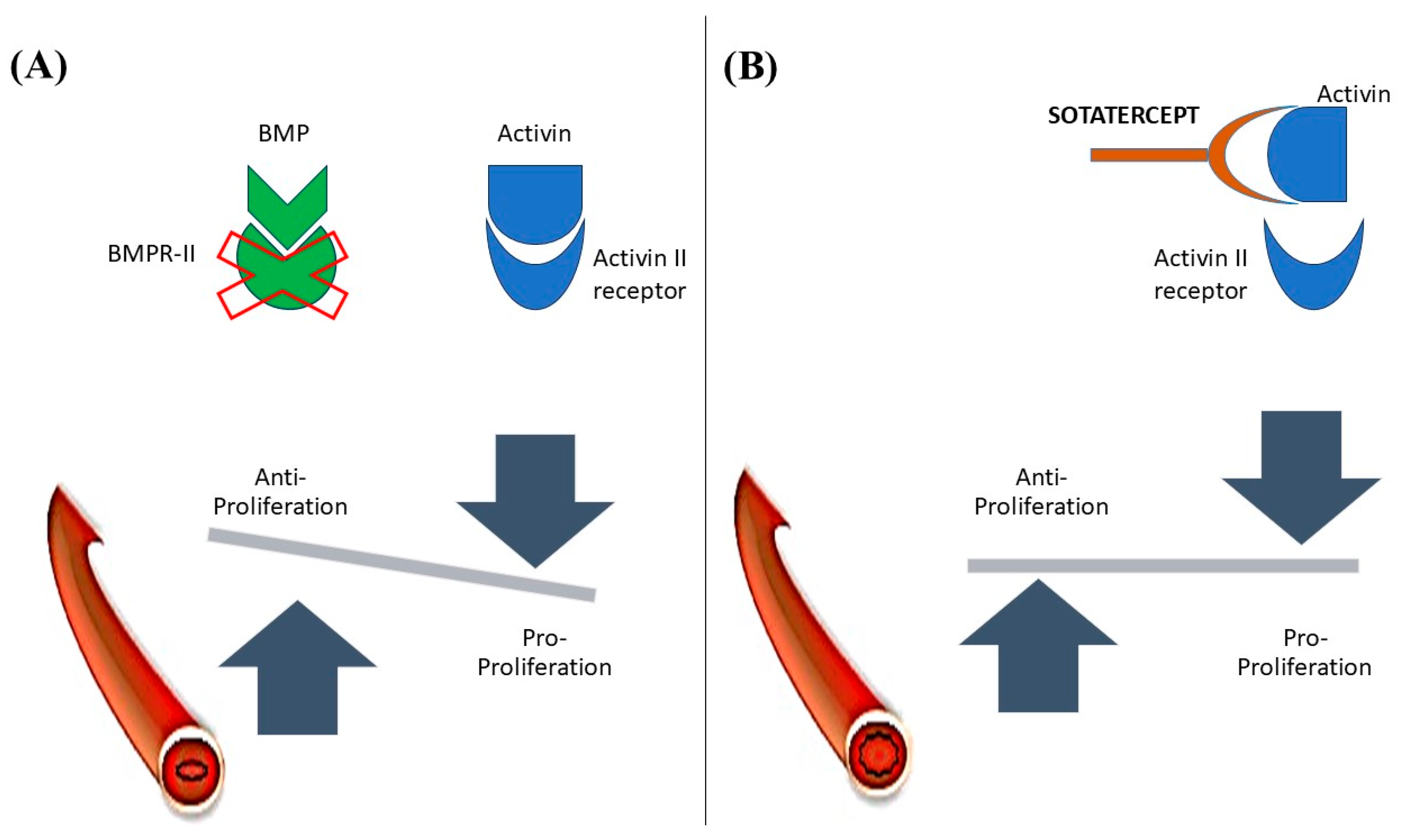
4. Activin Signaling Inhibitor
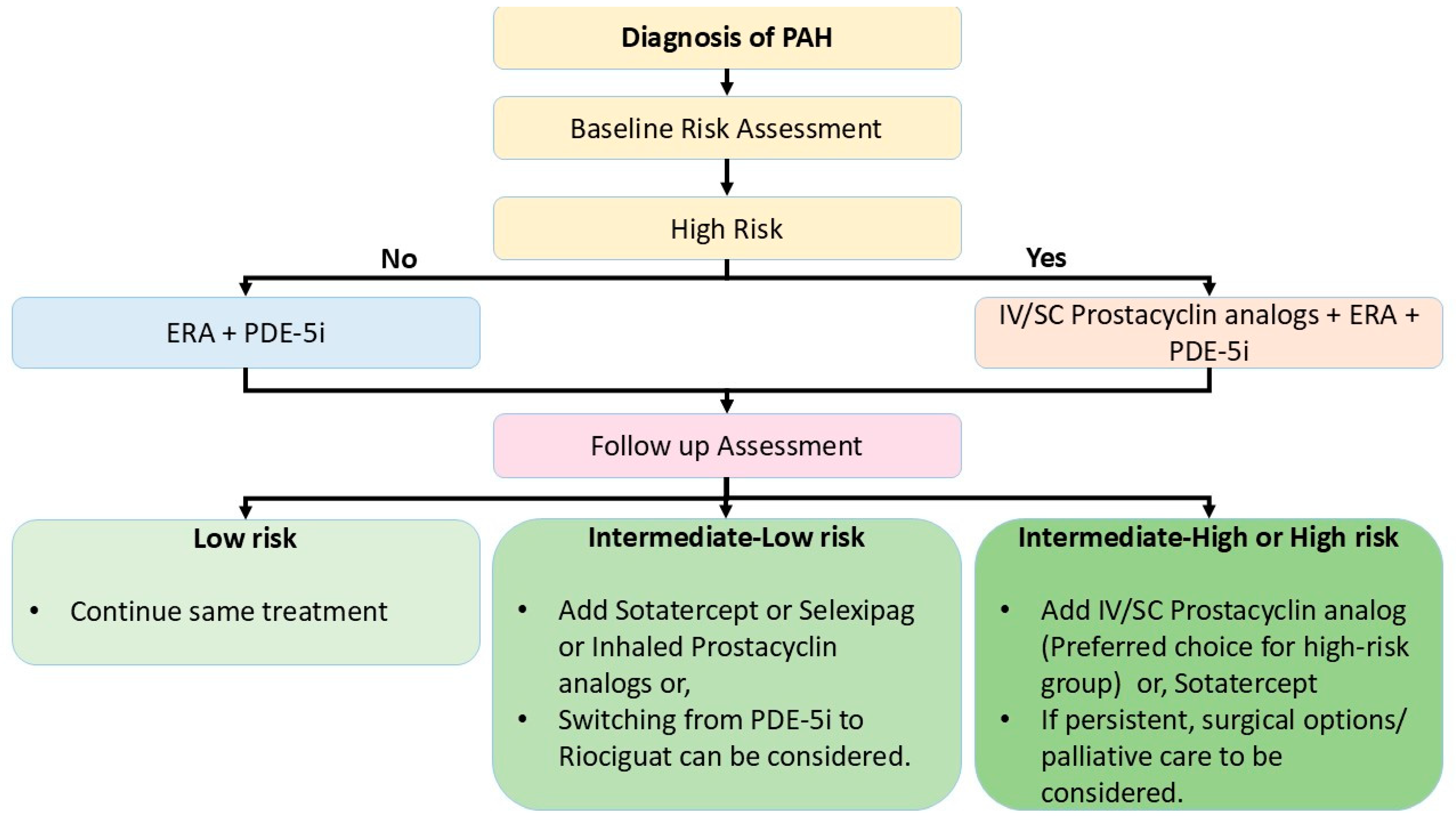
5. Treatment Algorithm
6. Surgical Strategies in Severe Pulmonary Arterial Hypertension (PAH)
6.1. Risk Assessment and Decision-Making for Treatment
Surgical Strategies
- A.
- Right to Left Shunting
- I.
- Atrial septostomy is a palliative intervention used in patients with severe pulmonary arterial hypertension (PAH) who have right ventricular (RV) failure refractory to medical therapy. It involves creating a right-to-left shunt at the atrial level to decompress the right heart, reduce right atrial pressure, and increase systemic cardiac output. This results in improved oxygen delivery despite systemic desaturation. Hemodynamic stability during the procedure is critical for success. Risks include systemic desaturation, paradoxical embolism, and worsening left ventricular filling in the setting of reduced LV compliance [81,82,83,84].
- II.
- Potts shunt, a surgical alternative, is considered for pediatric or selected adult patients with supra-systemic PAH and right ventricular failure. The procedure involves creating a left pulmonary artery-to-descending aorta anastomosis and establishing a right-to-left shunt that offloads the RV while preserving systemic oxygenation better than atrial septostomy. Originally described in congenital heart disease, the Potts shunt has gained interest in PAH, particularly in children with idiopathic or heritable PAH. Outcomes have shown hemodynamic improvement and symptom relief in select patients; however, careful patient selection is crucial to avoid excessive systemic desaturation [81,85,86].
- B.
- Pulmonary artery denervation (PADN)
- C.
- Lung Transplantation
- D.
- Balloon pulmonary angioplasty (BPA)
- E.
- Pulmonary Thromboendarterectomy (PTE)
6.2. Decision-Making for Surgical Treatment [1,81,82]
6.3. Emerging Surgical Strategies
6.3.1. Hybrid Approaches
6.3.2. Mechanical Support Devices
6.3.3. Precision Medicine in Surgical PAH Management
6.4. Palliative Care: An Overlooked Extra Panel of Support for PAH Therapy
7. What Is on the Horizon?
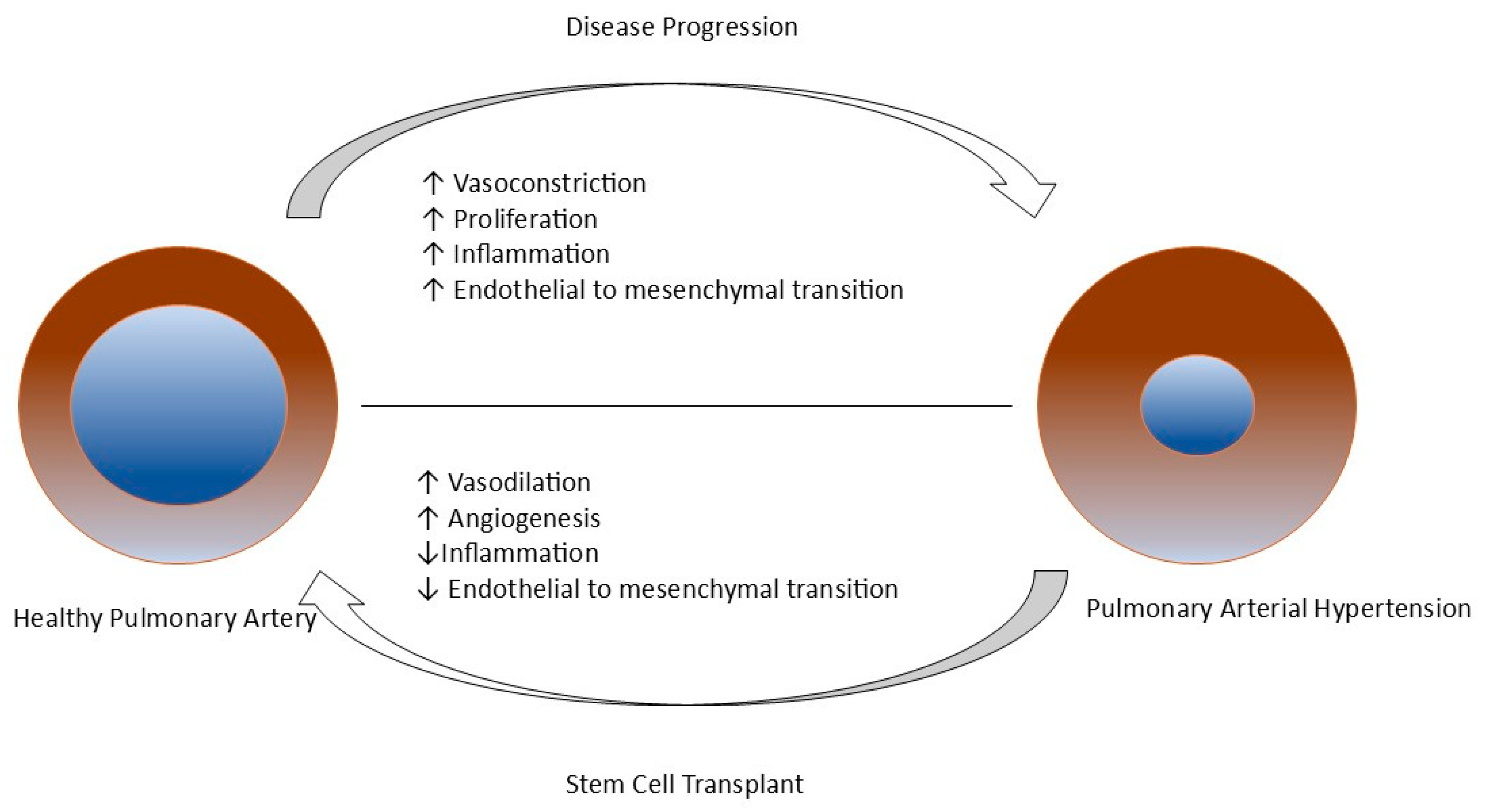
7.1. Regenerative Medicine: A Potential Curative Approach for a Patient with PAH
7.2. Stem Cell Therapy
8. Gene Therapy
9. Epigenetic Medicines
10. Personalized and AI-Driven Treatment Approaches in PAH
11. Conclusions
12. Materials and Methodology
12.1. Objective of the Review
12.2. Literature Search Strategy
12.3. Inclusion and Exclusion Criteria
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. 2023, 61, 2200879. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Wijeratne, D.T.; Lajkosz, K.; Brogly, S.B.; Lougheed, M.D.; Jiang, L.; Housin, A.; Barber, D.; Johnson, A.; Doliszny, K.M.; Archer, S.L. Increasing Incidence and Prevalence of World Health Organization Groups 1 to 4 Pulmonary Hypertension: A Population-Based Cohort Study in Ontario, Canada. Circ. Cardiovasc. Qual. Outcomes 2018, 11, e003973. [Google Scholar] [CrossRef] [PubMed]
- D’Alonzo, G.E.; Barst, R.J.; Ayres, S.M.; Bergofsky, E.H.; Brundage, B.H.; Detre, K.M.; Fishman, A.P.; Goldring, R.M.; Groves, B.M.; Kernis, J.T. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann. Intern. Med. 1991, 115, 343–349. [Google Scholar] [CrossRef]
- McGoon, M.D.; Miller, D.P. REVEAL: A contemporary US pulmonary arterial hypertension registry. Eur. Respir. Rev. 2012, 21, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Balsa, A.; Adão, R.; Brás-Silva, C. Therapeutic Approaches in Pulmonary Arterial Hypertension with Beneficial Effects on Right Ventricular Function—Preclinical Studies. Int. J. Mol. Sci. 2023, 24, 15539. [Google Scholar] [CrossRef]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.-F.; Chabot, F.; et al. Pulmonary arterial hypertension in France: Results from a national registry. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030. [Google Scholar] [CrossRef]
- Pietra, G.G.; Capron, F.; Stewart, S.; Leone, O.; Humbert, M.; Robbins, I.M.; Reid, L.M.; Tuder, R.M. Pathologic assessment of vasculopathies in pulmonary hypertension. J. Am. Coll. Cardiol. 2004, 43 (Suppl. S12), 25S–32S. [Google Scholar] [CrossRef]
- Lüscher, T.F.; Barton, M. Endothelins and Endothelin Receptor Antagonists: Therapeutic Considerations for a Novel Class of Cardiovascular Drugs. Circulation 2000, 102, 2434–2440. [Google Scholar] [CrossRef]
- Hynynen, M.M.; Khalil, R.A. The vascular endothelin system in hypertension—Recent patents and discoveries. Recent. Pat. Cardiovasc. Drug Discov. 2006, 1, 95–108. [Google Scholar] [CrossRef]
- Clozel, M.; Breu, V.; Gray, G.A.; Kalina, B.; Löffler, B.M.; Burri, K.; Cassal, J.M.; Hirth, G.; Müller, M.; Neidhart, W. Pharmacological characterization of bosentan, a new potent orally active nonpeptide endothelin receptor antagonist. J. Pharmacol. Exp. Ther. 1994, 270, 228–235. [Google Scholar] [CrossRef]
- Kingman, M.; Ruggiero, R.; Torres, F. Ambrisentan, an endothelin receptor type A-selective endothelin receptor antagonist, for the treatment of pulmonary arterial hypertension. Expert. Opin. Pharmacother. 2009, 10, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Bolli, M.H.; Boss, C.; Binkert, C.; Buchmann, S.; Bur, D.; Hess, P.; Iglarz, M.; Meyer, S.; Rein, J.; Rey, M.; et al. The Discovery of N.-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N.′-propylsulfamide (Macitentan), an Orally Active, Potent Dual Endothelin Receptor Antagonist. J. Med. Chem. 2012, 55, 7849–7861. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J.; Badesch, D.B.; Barst, R.J.; Galiè, N.; Black, C.M.; Keogh, A.; Pulido, T.; Frost, A.; Roux, S.; Leconte, I.; et al. Bosentan Therapy for Pulmonary Arterial Hypertension. N. Engl. J. Med. 2002, 346, 896–903. [Google Scholar] [CrossRef]
- Rivera-Lebron, B.N.; Risbano, M.G. Ambrisentan: A review of its use in pulmonary arterial hypertension. Ther. Adv. Respir. Dis. 2017, 11, 233–244. [Google Scholar] [CrossRef]
- Galiè, N.; Olschewski, H.; Oudiz, R.J.; Torres, F.; Frost, A.; Ghofrani, H.A.; Badesch, D.B.; McGoon, M.D.; McLaughlin, V.V.; Roecker, E.B.; et al. Ambrisentan for the treatment of pulmonary arterial hypertension: Results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008, 117, 3010–3019. [Google Scholar] [CrossRef]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galiè, N.; Ghofrani, H.-A.; Jansa, P.; Jing, Z.-C.; Le Brun, F.-O.; Mehta, S.; et al. Macitentan and Morbidity and Mortality in Pulmonary Arterial Hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Wang, N.; Gu, Z.-C.; Wei, A.-H.; Cheng, A.-N.; Fang, S.-S.; Du, H.-L.; Wang, L.-Z.; Zhang, G.-Q. A network meta-analysis for safety of endothelin receptor antagonists in pulmonary arterial hypertension. Cardiovasc. Diagn. Ther. 2019, 9, 239–249. [Google Scholar] [CrossRef]
- Galiè, N.; Channick, R.N.; Frantz, R.P.; Grünig, E.; Jing, Z.C.; Moiseeva, O.; Preston, I.R.; Pulido, T.; Safdar, Z.; Tamura, Y.; et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801889. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837, 837a–837d. [Google Scholar] [CrossRef]
- Wharton, J.; Strange, J.W.; Møller, G.M.O.; Growcott, E.J.; Ren, X.; Franklyn, A.P.; Phillips, S.C.; Wilkins, M.R. Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. Am. J. Respir. Crit. Care Med. 2005, 172, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A.; et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J.; Badesch, D.B.; Fleming, T.R.; Galiè, N.; Simonneau, G.; Ghofrani, H.A.; Oakes, M.; Layton, G.; Serdarevic-Pehar, M.; McLaughlin, V.V.; et al. Long-term treatment with sildenafil citrate in pulmonary arterial hypertension: The SUPER-2 study. Chest 2011, 140, 1274–1283. [Google Scholar] [CrossRef]
- Galiè, N.; Brundage, B.H.; Ghofrani, H.A.; Oudiz, R.J.; Simonneau, G.; Safdar, Z.; Shapiro, S.; White, R.J.; Chan, M.; Beardsworth, A.; et al. Tadalafil Therapy for Pulmonary Arterial Hypertension. Circulation 2009, 119, 2894–2903. [Google Scholar] [CrossRef]
- Wrishko, R.E.; Dingemanse, J.; Yu, A.; Darstein, C.; Phillips, D.L.; Mitchell, M.I. Pharmacokinetic interaction between tadalafil and bosentan in healthy male subjects. J. Clin. Pharmacol. 2008, 48, 610–618. [Google Scholar] [CrossRef]
- Oudiz, R.J.; Brundage, B.H.; Galiè, N.; Ghofrani, H.A.; Simonneau, G.; Botros, F.T.; Chan, M.; Beardsworth, A.; Barst, R.J. Tadalafil for the Treatment of Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 2012, 60, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Barnes, H.; Brown, Z.; Burns, A.; Williams, T. Phosphodiesterase 5 inhibitors for pulmonary hypertension. Cochrane Database Syst. Rev. 2019, 1, CD012621. [Google Scholar] [CrossRef]
- Ghofrani, H.A.; Hoeper, M.M.; Halank, M.; Meyer, F.J.; Staehler, G.; Behr, J.; Ewert, R.; Weimann, G.; Grimminger, F. Riociguat for chronic thromboembolic pulmonary hypertension and pulmonary arterial hypertension: A phase II study. Eur. Respir. J. 2010, 36, 792–799. [Google Scholar] [CrossRef]
- Ghofrani, H.-A.; Galiè, N.; Grimminger, F.; Grünig, E.; Humbert, M.; Jing, Z.-C.; Keogh, A.M.; Langleben, D.; Kilama, M.O.; Fritsch, A.; et al. Riociguat for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef]
- Rubin, L.J.; Galiè, N.; Grimminger, F.; Grünig, E.; Humbert, M.; Jing, Z.-C.; Keogh, A.; Langleben, D.; Fritsch, A.; Menezes, F.; et al. Riociguat for the treatment of pulmonary arterial hypertension: A long-term extension study (PATENT-2). Eur. Respir. J. 2015, 45, 1303–1313. [Google Scholar] [CrossRef]
- Ghofrani, H.-A.; Grimminger, F.; Grünig, E.; Huang, Y.; Jansa, P.; Jing, Z.-C.; Kilpatrick, D.; Langleben, D.; Rosenkranz, S.; Menezes, F.; et al. Predictors of long-term outcomes in patients treated with riociguat for pulmonary arterial hypertension: Data from the PATENT-2 open-label, randomised, long-term extension trial. Lancet Respir. Med. 2016, 4, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Majed, B.H.; Khalil, R.A. Molecular Mechanisms Regulating the Vascular Prostacyclin Pathways and Their Adaptation during Pregnancy and in the Newborn. Garland CJ, editor. Pharmacol. Rev. 2012, 64, 540–582. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Cool, C.D.; Geraci, M.W.; Wang, J.; Abman, S.H.; Wright, L.; Badesch, D.; Voelkel, N.F. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999, 159, 1925–1932. [Google Scholar] [CrossRef]
- Gomberg-Maitland, M.; Olschewski, H. Prostacyclin therapies for the treatment of pulmonary arterial hypertension. Eur. Respir. J. 2008, 31, 891–901. [Google Scholar] [CrossRef]
- Rubin, L.J. Treatment of Primary Pulmonary Hypertension with Continuous Intravenous Prostacyclin (Epoprostenol): Results of a Randomized Trial. Ann. Intern. Med. 1990, 112, 485. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J.; Rubin, L.J.; Long, W.A.; McGoon, M.D.; Rich, S.; Badesch, D.B.; Groves, B.M.; Tapson, V.F.; Bourge, R.C.; Brundage, B.H.; et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N. Engl. J. Med. 1996, 334, 296–301. [Google Scholar] [CrossRef]
- McLaughlin, V.V.; Shillington, A.; Rich, S. Survival in primary pulmonary hypertension: The impact of epoprostenol therapy. Circulation 2002, 106, 1477–1482. [Google Scholar] [CrossRef]
- Sitbon, O.; Humbert, M.; Nunes, H.; Parent, F.; Garcia, G.; Hervé, P.; Rainisio, M.; Simonneau, G. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: Prognostic factors and survival. J. Am. Coll. Cardiol. 2002, 40, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Barst, R.J.; Galie, N.; Naeije, R.; Rich, S.; Bourge, R.C.; Keogh, A.; Oudiz, R.; Frost, A.; Blackburn, S.D.; et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: A double-blind, randomized, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 2002, 165, 800–804. [Google Scholar] [CrossRef]
- Lang, I.; Gomez-Sanchez, M.; Kneussl, M.; Naeije, R.; Escribano, P.; Skoro-Sajer, N.; Vachiery, J.-L. Efficacy of long-term subcutaneous treprostinil sodium therapy in pulmonary hypertension. Chest 2006, 129, 1636–1643. [Google Scholar] [CrossRef]
- Tapson, V.F.; Gomberg-Maitland, M.; McLaughlin, V.V.; Benza, R.L.; Widlitz, A.C.; Krichman, A.; Barst, R.J. Safety and efficacy of IV treprostinil for pulmonary arterial hypertension: A prospective, multicenter, open-label, 12-week trial. Chest 2006, 129, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Gomberg-Maitland, M.; Tapson, V.F.; Benza, R.L.; McLaughlin, V.V.; Krichman, A.; Widlitz, A.C.; Barst, R.J. Transition from intravenous epoprostenol to intravenous treprostinil in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2005, 172, 1586–1589. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J.; Galie, N.; Naeije, R.; Simonneau, G.; Jeffs, R.; Arneson, C.; Rubin, L.J. Long-term outcome in pulmonary arterial hypertension patients treated with subcutaneous treprostinil. Eur. Respir. J. 2006, 28, 1195–1203. [Google Scholar] [CrossRef]
- McLaughlin, V.V.; Benza, R.L.; Rubin, L.J.; Channick, R.N.; Voswinckel, R.; Tapson, V.F.; Robbins, I.M.; Olschewski, H.; Rubenfire, M.; Seeger, W. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: A randomized controlled clinical trial. J. Am. Coll. Cardiol. 2010, 55, 1915–1922. [Google Scholar] [CrossRef]
- Spikes, L.A.; Bajwa, A.A.; Burger, C.D.; Desai, S.V.; Eggert, M.S.; El-Kersh, K.A.; Fisher, M.R.; Johri, S.; Joly, J.M.; Mehta, J.; et al. BREEZE: Open-label clinical study to evaluate the safety and tolerability of treprostinil inhalation powder as Tyvaso DPITM in patients with pulmonary arterial hypertension. Pulm. Circ. 2022, 12, e12063. [Google Scholar] [CrossRef]
- West, N.; Smoot, K.; Patzlaff, N.; Miceli, M.; Waxman, A. Plain language summary of the INCREASE study: Inhaled treprostinil (Tyvaso) for the treatment of pulmonary hypertension due to interstitial lung disease. Future Cardiol. 2023, 19, 229–239. [Google Scholar] [CrossRef]
- Arslan, A.; Smith, J.; Qureshi, M.R.; Uysal, A.; Patel, K.K.; Herazo-Maya, J.D.; Bandyopadhyay, D. Evolution of pulmonary hypertension in interstitial lung disease: A journey through past, present, and future. Front. Med. 2023, 10, 1306032. [Google Scholar] [CrossRef]
- Olschewski, H.; Simonneau, G.; Galiè, N.; Higenbottam, T.; Naeije, R.; Rubin, L.J.; Nikkho, S.; Speich, R.; Hoeper, M.M.; Behr, J.; et al. Inhaled Iloprost for Severe Pulmonary Hypertension. N. Engl. J. Med. 2002, 347, 322–329. [Google Scholar] [CrossRef]
- McLaughlin, V.V.; Oudiz, R.J.; Frost, A.; Tapson, V.F.; Murali, S.; Channick, R.N.; Badesch, D.B.; Barst, R.J.; Hsu, H.H.; Rubin, L.J. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2006, 174, 1257–1263. [Google Scholar] [CrossRef]
- Simonneau, G.; Torbicki, A.; Hoeper, M.M.; Delcroix, M.; Karlócai, K.; Galiè, N.; Degano, B.; Bonderman, D.; Kurzyna, M.; Efficace, M.; et al. Selexipag: An oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur. Respir. J. 2012, 40, 874–880. [Google Scholar] [CrossRef]
- Sitbon, O.; Channick, R.; Chin, K.M.; Frey, A.; Gaine, S.; Galiè, N.; Ghofrani, H.-A.; Hoeper, M.M.; Lang, I.M.; Preiss, R.; et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 2522–2533. [Google Scholar] [CrossRef]
- Galiè, N.; Barberà, J.A.; Frost, A.E.; Ghofrani, H.-A.; Hoeper, M.M.; McLaughlin, V.V.; Peacock, A.J.; Simonneau, G.; Vachiery, J.-L.; Grünig, E.; et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Grünig, E.; Jansa, P.; Fan, F.; Hauser, J.A.; Pannaux, M.; Morganti, A.; Rofael, H.; Chin, K.M. Randomized Trial of Macitentan/Tadalafil Single-Tablet Combination Therapy for Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 2024, 83, 473–484. [Google Scholar] [CrossRef]
- You, R.; Qian, X.; Tang, W.; Xie, T.; Zeng, F.; Chen, J.; Zhang, Y.; Liu, J. Cost Effectiveness of Bosentan for Pulmonary Arterial Hypertension: A Systematic Review. Can. Respir. J. 2018, 2018, 1015239. [Google Scholar] [CrossRef]
- Coyle, K.; Coyle, D.; Blouin, J.; Lee, K.; Jabr, M.F.; Tran, K.; Mielniczuk, L.; Swiston, J.; Innes, M. Cost Effectiveness of First-Line Oral Therapies for Pulmonary Arterial Hypertension: A Modelling Study. Pharmacoeconomics 2016, 34, 509–520. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Jowett, S.; Barton, P.; Malottki, K.; Hyde, C.; Gibbs, J.S.R.; Pepke-Zaba, J.; Fry-Smith, A.; Roberts, J.; Moore, D. Clinical and cost-effectiveness of epoprostenol, iloprost, bosentan, sitaxentan and sildenafil for pulmonary arterial hypertension within their licensed indications: A systematic review and economic evaluation. Health Technol. Assess. 2009, 13, 1–320. [Google Scholar] [CrossRef] [PubMed]
- Einarson, T.R.; Granton, J.T.; Vicente, C.; Walker, J.; Engel, G.; Iskedjian, M. Cost-effectiveness of treprostinil versus epoprostenol in patients with pulmonary arterial hypertension: A Canadian analysis. Can. Respir. J. 2005, 12, 419–425. [Google Scholar] [CrossRef]
- Dong, W.; Zhang, Z.; Chu, M.; Gu, P.; Hu, M.; Liu, L.; Huang, J.; Zhang, R. Cost-effectiveness analysis of selexipag for the combined treatment of pulmonary arterial hypertension. Front. Pharmacol. 2023, 14, 1122866. [Google Scholar] [CrossRef] [PubMed]
- Simbaqueba, E.; Gomez, L.M.; Huerfano, C.; Tamayo, C.; Palomino, R.A. Effectiveness and safety of macitentan in the treatment of pulmonary arterial hypertension. Value Health 2016, 19, A83. [Google Scholar] [CrossRef]
- Pharmacoeconomic Review Report: Riociguat (Adempas) [Internet]. Ottawa (ON): Canadian Agency for Drugs and Technologies in Health; 2015 December Summary. Available online: https://www.ncbi.nlm.nih.gov/books/NBK540269/ (accessed on 25 January 2025).
- Massagué, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef]
- Yndestad, A.; Larsen, K.-O.; Oie, E.; Ueland, T.; Smith, C.; Halvorsen, B.; Sjaastad, I.; Skjønsberg, O.H.; Pedersen, T.M.; Anfinsen, O.-G.; et al. Elevated levels of activin A in clinical and experimental pulmonary hypertension. J. Appl. Physiol. 2009, 106, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Nakaoka, T.; Gonda, K.; Ogita, T.; Otawara-Hamamoto, Y.; Okabe, F.; Kira, Y.; Harii, K.; Miyazono, K.; Takuwa, Y.; Fujita, T. Inhibition of rat vascular smooth muscle proliferation in vitro and in vivo by bone morphogenetic protein-2. J. Clin. Investig. 1997, 100, 2824–2832. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, C.; Stewart, S.; Upton, P.D.; Machado, R.; Thomson, J.R.; Trembath, R.C.; Morrell, N.W. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002, 105, 1672–1678. [Google Scholar] [CrossRef] [PubMed]
- Yung, L.-M.; Yang, P.; Joshi, S.; Augur, Z.M.; Kim, S.S.J.; Bocobo, G.A.; Dinter, T.; Troncone, L.; Chen, P.-S.; McNeil, M.E.; et al. ACTRIIA-Fc rebalances activin/GDF versus BMP signaling in pulmonary hypertension. Sci. Transl. Med. 2020, 12, eaaz5660. [Google Scholar] [CrossRef]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.; Escribano Subias, P.; Feldman, J.; et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 384, 1204–1215. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Badesch, D.B.; Ghofrani, H.A.; Gibbs, J.S.R.; Gomberg-Maitland, M.; McLaughlin, V.V.; Preston, I.R.; Souza, R.; Waxman, A.B.; Grünig, E.; et al. Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2023, 388, 1478–1490. [Google Scholar] [CrossRef]
- Nasrollahizadeh, A.; Soleimani, H.; Nasrollahizadeh, A.; Hashemi, S.M.; Hosseini, K. Navigating the Sotatercept landscape: A meta-analysis of clinical outcomes. Clin. Cardiol. 2024, 47, e24173. [Google Scholar] [CrossRef]
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, M.D. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef]
- Tapson, V.F.; Jing, Z.-C.; Xu, K.-F.; Pan, L.; Feldman, J.; Kiely, D.G.; Kotlyar, E.; McSwain, C.S.; Laliberte, K.; Arneson, C.; et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): A randomized controlled trial. Chest 2013, 144, 952–958. [Google Scholar] [CrossRef]
- Ruaro, B.; Salton, F.; Baratella, E.; Confalonieri, P.; Geri, P.; Pozzan, R.; Torregiani, C.; Bulla, R.; Confalonieri, M.; Matucci-Cerinic, M.; et al. An Overview of Different Techniques for Improving the Treatment of Pulmonary Hypertension Secondary in Systemic Sclerosis Patients. Diagnostics 2022, 12, 616. [Google Scholar] [CrossRef]
- Rossi, R.; Talarico, M.; Schepis, F.; Coppi, F.; Sgura, F.A.; Monopoli, D.E.; Minici, R.; Boriani, G. Effects of sildenafil on right ventricle remodelling in Portopulmonary hypertension. Pulm. Pharmacol. Ther. 2021, 70, 102071. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, W.A.; Leaderer, D.; Rowan, C.A.; Mituniewicz, J.D.; Rosenzweig, E.B. Ambrisentan for pulmonary arterial hypertension due to congenital heart disease. Am. J. Cardiol. 2011, 107, 1381–1385. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.J.C.; Piloto, B.; Castro, M.; Gavilanes Oleas, F.; Alves, J.L.; Lopes Prada, L.F.; Jardim, C.; Souza, R. Survival of patients with schistosomiasis-associated pulmonary arterial hypertension in the modern management era. Eur. Respir. J. 2018, 51, 1800307. [Google Scholar] [CrossRef]
- Kylhammar, D.; Kjellström, B.; Hjalmarsson, C.; Jansson, K.; Nisell, M.; Söderberg, S.; Wikström, G.; Rådegran, G. A comprehensive risk stratification at early follow-up determines prognosis in pulmonary arterial hypertension. Eur. Heart J. 2018, 39, 4175–4181. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Kramer, T.; Pan, Z.; Eichstaedt, C.A.; Spiesshoefer, J.; Benjamin, N.; Olsson, K.M.; Meyer, K.; Vizza, C.D.; Vonk-Noordegraaf, A.; et al. Mortality in pulmonary arterial hypertension: Prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur. Respir. J. 2017, 50, 1700740. [Google Scholar] [CrossRef]
- Boucly, A.; Weatherald, J.; Savale, L.; Jaïs, X.; Cottin, V.; Prevot, G.; Picard, F.; de Groote, P.; Jevnikar, M.; Bergot, E.; et al. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur. Respir. J. 2017, 50, 1700889. [Google Scholar] [CrossRef]
- Benza, R.L.; Gomberg-Maitland, M.; Elliott, C.G.; Farber, H.W.; Foreman, A.J.; Frost, A.E.; McGoon, M.D.; Pasta, D.J.; Selej, M.; Burger, C.D.; et al. Predicting Survival in Patients With Pulmonary Arterial Hypertension: The REVEAL Risk Score Calculator 2.0 and Comparison With ESC/ERS-Based Risk Assessment Strategies. Chest 2019, 156, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Benza, R.L.; Kanwar, M.K.; Raina, A.; Scott, J.V.; Zhao, C.L.; Selej, M.; Elliott, C.G.; Farber, H.W. Development and Validation of an Abridged Version of the REVEAL 2.0 Risk Score Calculator, REVEAL Lite 2, for Use in Patients with Pulmonary Arterial Hypertension. Chest 2021, 159, 337–346. [Google Scholar] [CrossRef]
- Chin, K.M.; Gaine, S.P.; Gerges, C.; Jing, Z.-C.; Mathai, S.C.; Tamura, Y.; McLaughlin, V.V.; Sitbon, O. Treatment algorithm for pulmonary arterial hypertension. Eur. Respir. J. 2024, 64, 2401325. [Google Scholar] [CrossRef]
- Stącel, T.; Latos, M.; Urlik, M.; Nęcki, M.; Antończyk, R.; Hrapkowicz, T.; Kurzyna, M.; Ochman, M. Interventional and Surgical Treatments for Pulmonary Arterial Hypertension. J. Clin. Med. 2021, 10, 3326. [Google Scholar] [CrossRef]
- Keogh, A.M.; Mayer, E.; Benza, R.L.; Corris, P.; Dartevelle, P.G.; Frost, A.E.; Kim, N.H.; Lang, I.M.; Pepke-Zaba, J.; Sandoval, J. Interventional and surgical modalities of treatment in pulmonary hypertension. J. Am. Coll. Cardiol. 2009, 54 (Suppl. 1), S67–S77. [Google Scholar] [CrossRef]
- Zebadua, R.; Zayas, N.; Lopez, J.; Zorrilla, L.; Pozas, M.; Villalobos, M.; Sandoval, J.; Pulido, T.; Hdz., N.G.Z. Survival of patients diagnosed with Pulmonary Hypertension undergoing Atrial Septostomy. In 1301—Pulmonary Hypertension; European Respiratory Society: Lausanne, Switzerland, 2022; p. 4186. [Google Scholar]
- Barst, R.J.; Ivy, D.D.; Foreman, A.J.; McGoon, M.D.; Rosenzweig, E.B. Four- and seven-year outcomes of patients with congenital heart disease-associated pulmonary arterial hypertension (from the REVEAL Registry). Am. J. Cardiol. 2014, 113, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Baruteau, A.-E.; Serraf, A.; Lévy, M.; Petit, J.; Bonnet, D.; Jais, X.; Vouhé, P.; Simonneau, G.; Belli, E.; Humbert, M. Potts Shunt in Children With Idiopathic Pulmonary Arterial Hypertension: Long-Term Results. Ann. Thorac. Surg. 2012, 94, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Blanc, J.; Vouhé, P.; Bonnet, D. Potts Shunt in Patients with Pulmonary Hypertension. N. Engl. J. Med. 2004, 350, 623. [Google Scholar] [CrossRef]
- Chen, S.-L.; Zhang, F.-F.; Xu, J.; Xie, D.-J.; Zhou, L.; Nguyen, T.; Stone, G.W. Pulmonary artery denervation to treat pulmonary arterial hypertension: The single-center, prospective, first-in-man PADN-1 study (first-in-man pulmonary artery denervation for treatment of pulmonary artery hypertension). J. Am. Coll. Cardiol. 2013, 62, 1092–1100. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, Y.; Zhang, C.; Yang, Z.; Kan, J.; Gu, H.; Fan, F.; Gu, H.; Wang, Q.; Xie, D.; et al. Pulmonary Artery Denervation for Pulmonary Arterial Hypertension: A Sham-Controlled Randomized PADN-CFDA Trial. JACC Cardiovasc. Interv. 2022, 15, 2412–2423. [Google Scholar] [CrossRef]
- Salazar, A.M.; Al-Asad, K.S.; Prasad, R.M.; Panama, G.; Banga, S.; Wilcox, M. Pulmonary Artery Denervation as a New Therapeutic Option for Pulmonary Hypertension: A Systematic Review and Meta-Analysis. Curr. Probl. Cardiol. 2023, 48, 101776. [Google Scholar] [CrossRef]
- Default. International Thoracic Organ Transplant (TTX) Registry. Available online: https://www.ishlt.org/registries/international-thoracic-organ-transplant-(ttx)-registry (accessed on 21 February 2025).
- Christie, J.D.; Edwards, L.B.; Kucheryavaya, A.Y.; Aurora, P.; Dobbels, F.; Kirk, R.; Rahmel, A.O.; Stehlik, J.; Hertz, M.I. The Registry of the International Society for Heart and Lung Transplantation: Twenty-seventh official adult lung and heart-lung transplant report—2010. J. Heart Lung Transplant. 2010, 29, 1104–1118. [Google Scholar] [CrossRef]
- Long, J.; Russo, M.J.; Muller, C.; Vigneswaran, W.T. Surgical treatment of pulmonary hypertension: Lung transplantation. Pulm. Circ. 2011, 1, 327–333. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Ogawa, A.; Munemasa, M.; Mikouchi, H.; Ito, H.; Matsubara, H. Refined balloon pulmonary angioplasty for inoperable patients with chronic thromboembolic pulmonary hypertension. Circ. Cardiovasc. Interv. 2012, 5, 748–755. [Google Scholar] [CrossRef]
- Ogawa, A.; Satoh, T.; Fukuda, T.; Sugimura, K.; Fukumoto, Y.; Emoto, N.; Yamada, N.; Yao, A.; Ando, M.; Ogino, H.; et al. Balloon Pulmonary Angioplasty for Chronic Thromboembolic Pulmonary Hypertension: Results of a Multicenter Registry. Circ. Cardiovasc. Qual. Outcomes 2017, 10, e004029. [Google Scholar] [CrossRef]
- Madani, M.M. Pulmonary endarterectomy for chronic thromboembolic pulmonary hypertension: State-of-the-art 2020. Pulm. Circ. 2021, 11, 20458940211007372. [Google Scholar] [CrossRef] [PubMed]
- Hecker, F.; Keller, H.; Vasa-Nicotera, M.; Iken, S.; Holubec, T. Percutaneous dual-outflow extracorporeal membrane oxygenation support in secondary right ventricular failure. JTCVS Tech. 2022, 13, 125–127. [Google Scholar] [CrossRef]
- Leopold, J.A.; Maron, B.A. Precision Medicine in Pulmonary Arterial Hypertension. Circ. Res. 2019, 124, 832–833. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Zhang, X.; Ahmad, A. Epigenetic regulation of pulmonary inflamation. Semin. Cell Dev. Biol. 2024, 154, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Khirfan, G.; Tonelli, A.R.; Ramsey, J.; Sahay, S. Palliative care in pulmonary arterial hypertension: An underutilised treatment. Eur. Respir. Rev. 2018, 27, 180069. [Google Scholar] [CrossRef]
- Christiansen, D.; Porter, S.; Hurlburt, L.; Weiss, A.; Granton, J.; Wentlandt, K. Pulmonary Arterial Hypertension: A Palliative Medicine Review of the Disease, Its Therapies, and Drug Interactions. J. Pain. Symptom Manag. 2020, 59, 932–943. [Google Scholar] [CrossRef]
- Loisel, F.; Provost, B.; Haddad, F.; Guihaire, J.; Amsallem, M.; Vrtovec, B.; Fadel, E.; Uzan, G.; Mercier, O. Stem cell therapy targeting the right ventricle in pulmonary arterial hypertension: Is it a potential avenue of therapy? Pulm. Circ. 2018, 8, 2045893218755979. [Google Scholar] [CrossRef]
- Brouwer, K.M.; Hoogenkamp, H.R.; Daamen, W.F.; van Kuppevelt, T.H. Regenerative medicine for the respiratory system: Distant future or tomorrow’s treatment? Am. J. Respir. Crit. Care Med. 2013, 187, 468–475. [Google Scholar] [CrossRef]
- Mason, C.; Dunnill, P. A brief definition of regenerative medicine. Regen. Med. 2008, 3, 1–5. [Google Scholar] [CrossRef]
- Weissman, I.L. Stem cells: Units of development, units of regeneration, and units in evolution. Cell 2000, 100, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Bisserier, M.; Pradhan, N.; Hadri, L. Current and emerging therapeutic approaches to pulmonary hypertension. Rev. Cardiovasc. Med. 2020, 21, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Young, R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 2008, 132, 567–582. [Google Scholar] [CrossRef]
- Dulak, J.; Szade, K.; Szade, A.; Nowak, W.; Józkowicz, A. Adult stem cells: Hopes and hypes of regenerative medicine. Acta Biochim. Pol. 2015, 62, 329–337. [Google Scholar] [CrossRef]
- Zheng, R.; Xu, T.; Wang, X.; Yang, L.; Wang, J.; Huang, X. Stem cell therapy in pulmonary hypertension: Current practice and future opportunities. Eur. Respir. Rev. 2023, 32, 230112. [Google Scholar] [CrossRef]
- Oh, S.; Jung, J.-H.; Ahn, K.-J.; Jang, A.Y.; Byun, K.; Yang, P.C.; Chung, W.-J. Stem Cell and Exosome Therapy in Pulmonary Hypertension. Korean Circ. J. 2022, 52, 110–122. [Google Scholar] [CrossRef]
- Pu, X.; Du, L.; Hu, Y.; Fan, Y.; Xu, Q. Stem/Progenitor Cells and Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.W.; Sun, Z. Stem cell therapy for pulmonary arterial hypertension: An update. J. Heart Lung Transplant. 2022, 41, 692–703. [Google Scholar] [CrossRef]
- Dierick, F.; Solinc, J.; Bignard, J.; Soubrier, F.; Nadaud, S. Progenitor/Stem Cells in Vascular Remodeling during Pulmonary Arterial Hypertension. Cells 2021, 10, 1338. [Google Scholar] [CrossRef]
- Yang, J.X.; Pan, Y.Y.; Zhao, Y.Y.; Wang, X.X. Endothelial progenitor cell-based therapy for pulmonary arterial hypertension. Cell Transplant. 2013, 22, 1325–1336. [Google Scholar] [CrossRef]
- Nagaya, N.; Kangawa, K.; Kanda, M.; Uematsu, M.; Horio, T.; Fukuyama, N.; Hino, J.; Harada-Shiba, M.; Okumura, H.; Tabata, Y.; et al. Hybrid cell-gene therapy for pulmonary hypertension based on phagocytosing action of endothelial progenitor cells. Circulation 2003, 108, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.D.; Courtman, D.W.; Deng, Y.; Kugathasan, L.; Zhang, Q.; Stewart, D.J. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: Efficacy of combined cell and eNOS gene therapy in established disease. Circ. Res. 2005, 96, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Granton, J.; Langleben, D.; Kutryk, M.B.; Camack, N.; Galipeau, J.; Courtman, D.W.; Stewart, D.J. Endothelial NO-Synthase Gene-Enhanced Progenitor Cell Therapy for Pulmonary Arterial Hypertension: The PHACeT Trial. Circ. Res. 2015, 117, 645–654. [Google Scholar] [CrossRef]
- Wang, X.-X.; Zhang, F.-R.; Shang, Y.-P.; Zhu, J.-H.; Xie, X.-D.; Tao, Q.-M.; Zhu, J.-H.; Chen, J.-Z. Transplantation of Autologous Endothelial Progenitor Cells May Be Beneficial in Patients With Idiopathic Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 2007, 49, 1566–1571. [Google Scholar] [CrossRef] [PubMed]
- Farkas, L.; Kolb, M. Vascular repair and regeneration as a therapeutic target for pulmonary arterial hypertension. Respiration 2013, 85, 355–364. [Google Scholar] [CrossRef]
- Fukumitsu, M.; Suzuki, K. Mesenchymal stem/stromal cell therapy for pulmonary arterial hypertension: Comprehensive review of preclinical studies. J. Cardiol. 2019, 74, 304–312. [Google Scholar] [CrossRef]
- Huang, W.-C.; Ke, M.-W.; Cheng, C.-C.; Chiou, S.-H.; Wann, S.-R.; Shu, C.-W.; Chiou, K.-R.; Tseng, C.-J.; Pan, H.-W.; Mar, G.-Y.; et al. Therapeutic Benefits of Induced Pluripotent Stem Cells in Monocrotaline-Induced Pulmonary Arterial Hypertension. PLoS ONE 2016, 11, e0142476. [Google Scholar] [CrossRef]
- Nicholson, M.W.; Ting, C.-Y.; Chan, D.Z.H.; Cheng, Y.-C.; Lee, Y.-C.; Hsu, C.-C.; Huang, C.-Y.; Hsieh, P.C.H. Utility of iPSC-Derived Cells for Disease Modeling, Drug Development, and Cell Therapy. Cells 2022, 11, 1853. [Google Scholar] [CrossRef]
- Gonçalves, G.A.R.; Paiva, R.d.M.A. Gene therapy: Advances, challenges and perspectives. Einstein 2017, 15, 369–375. [Google Scholar] [CrossRef]
- Wang, M.T.; Charng, M.J.; Chi, P.L.; Cheng, C.C.; Hung, C.C.; Huang, W.C. Gene Mutation Annotation and Pedigree for Pulmonary Arterial Hypertension Patients in Han Chinese Patients. Glob. Heart 2021, 16, 70. [Google Scholar] [CrossRef]
- Fazal, S.; Bisserier, M.; Hadri, L. Molecular and Genetic Profiling for Precision Medicines in Pulmonary Arterial Hypertension. Cells 2021, 10, 638. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.M.; Holmes, M.D.; Danilov, S.M.; Reynolds, P.N. Targeted gene delivery of BMPR2 attenuates pulmonary hypertension. Eur. Respir. J. 2012, 39, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Harper, R.L.; Reynolds, A.M.; Bonder, C.S.; Reynolds, P.N. BMPR2 gene therapy for PAH acts via Smad and non-Smad signalling. Respirology 2016, 21, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Theilmann, A.L.; Hawke, L.G.; Hilton, L.R.; Whitford, M.K.M.; Cole, D.V.; Mackeil, J.L.; Dunham-Snary, K.J.; Mewburn, J.; James, P.D.; Maurice, D.H.; et al. Endothelial BMPR2 Loss Drives a Proliferative Response to BMP (Bone Morphogenetic Protein) 9 via Prolonged Canonical Signaling. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2605–2618. [Google Scholar] [CrossRef]
- Xu, B.; Xu, G.; Yu, Y.; Lin, J. The role of TGF-β or BMPR2 signaling pathway-related miRNA in pulmonary arterial hypertension and systemic sclerosis. Arthritis Res. Ther. 2021, 23, 288. [Google Scholar] [CrossRef]
- Benincasa, G.; DeMeo, D.L.; Glass, K.; Silverman, E.K.; Napoli, C. Epigenetics and pulmonary diseases in the horizon of precision medicine: A review. Eur. Respir. J. 2021, 57, 2003406. [Google Scholar] [CrossRef]
- Dave, J.; Jagana, V.; Janostiak, R.; Bisserier, M. Unraveling the epigenetic landscape of pulmonary arterial hypertension: Implications for personalized medicine development. J. Transl. Med. 2023, 21, 477. [Google Scholar] [CrossRef]
- Napoli, C.; Benincasa, G.; Loscalzo, J. Epigenetic Inheritance Underlying Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 653–664. [Google Scholar] [CrossRef]
- Johnson, K.B.; Wei, W.-Q.; Weeraratne, D.; Frisse, M.E.; Misulis, K.; Rhee, K.; Zhao, J.; Snowdon, J.L. Precision Medicine, AI, and the Future of Personalized Health Care. Clin. Transl. Sci. 2021, 14, 86–93. [Google Scholar] [CrossRef]
- Hemnes, A.; Rothman, A.M.K.; Swift, A.J.; Zisman, L.S. Role of biomarkers in evaluation, treatment and clinical studies of pulmonary arterial hypertension. Pulm. Circ. 2020, 10, 2045894020957234. [Google Scholar] [CrossRef]
- Rhodes, C.J.; Wharton, J.; Swietlik, E.M.; Harbaum, L.; Girerd, B.; Coghlan, J.G.; Lordan, J.; Church, C.; Pepke-Zaba, J.; Toshner, M.; et al. Using the Plasma Proteome for Risk Stratifying Patients with Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2022, 205, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Zolty, R. Novel Experimental Therapies for Treatment of Pulmonary Arterial Hypertension. J. Exp. Pharmacol. 2021, 13, 817–857. [Google Scholar] [CrossRef] [PubMed]
- Bassareo, P.P.; D’Alto, M. Metabolomics in Pulmonary Hypertension-A Useful Tool to Provide Insights into the Dark Side of a Tricky Pathology. Int. J. Mol. Sci. 2023, 24, 13227. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean Pulmonary Artery Pressure (mPAP) | ≥20 mmHg |
| Pulmonary Arterial Wedge Pressure (PAWP) | ≤15 mmHg |
| Pulmonary Vascular Resistance (PVR) | ≥2 WU (Wood units) |
|
| Drugs | Abnormal Liver Function | Peripheral Edema | Anemia |
|---|---|---|---|
| Bosentan | RR 2.93. (95% CI, 1.78–4.84) | RR 1.32. (95% CI, 0.87–2.00) | RR 1.39. (95% CI, 0.67–2.86) |
| Ambrisentan | RR 0.13. (95% CI, 0.01–1.13) | RR 1.62. (95% CI, 1.23–2.13) | RR 1.58. (95% CI, 0.88–2.82) |
| Macitentan | RR 0.78. (95% CI, 0.37–1.64) | RR 0.95. (95% CI, 0.68–1.31) | RR 3.42. (95% CI, 1.65–7.07) |
| Feature | PDE-5 Inhibitors (Sildenafil, Tadalafil) | sGC Stimulators (Riociguat) |
|---|---|---|
| Mechanism | Inhibits cGMP breakdown (NO-dependent) | Directly stimulates sGC (NO-independent) |
| Key indications | PAH (Group 1) | PAH (Group 1), CTEPH (group 4) |
| Dosing | Sildenafil (20 mg Three times daily), Tadalafil (40 mg daily) | Riociguat (0.5–2.5 mg Three times daily) |
| Combination allowed? | Not with sGC stimulators | Not with PDE-5 inhibitors |
| Nitrate contraindication? | Yes | Yes |
| M/T FDC | Macitentan | Treatment Effect | |
| Reduction in PVR | 45% | 23% | 29% reduction |
| Geometric mean ratio of change in PVR | 0.55 (95% CI = 0.50–0.60) | 0.77 (95% CI = 0.69–0.87) | 0.71 (95% CI = 0.61–0.82) |
| M/T FDC | Tadalafil | Treatment Effect | |
| Reduction in PVR | 44% | 22% | 28% reduction |
| Geometric mean ratio of change in PVR | 0.56 (95% CI = 0.52–0.60) | 0.78 (95% CI = 0.72–0.84) | 0.72 (95% CI = 0.64–0.80) |
| Medication | Formulation | Estimated Monthly Cost (USD) |
|---|---|---|
| Bosentan | Oral | 10,000–12,000 [54] |
| Sildenafil | Oral | 1000–2000 (brand); 200–400 (generic) [55] |
| Tadalafil | Oral | 1300–2000 [55] |
| Epoprostenol | Intravenous | 40,000–50,000 [56] |
| Treprostinil | Intravenous, subcutaneous, inhaled, oral | 20,000–40,000 [57] |
| Selexipag | Oral | 8000–10,000 [58] |
| Macitentan | Oral | 11,000–13,000 [59] |
| Riociguat | Oral | 8000–9000 [60] |
| Drug Class | Medications | Mechanism of Action | Some Related Clinical Trial(s) and Outcomes |
|---|---|---|---|
| Endothelin Receptor Antagonists (ERA) | Bosentan, Ambrisentan, Macitentan | Block endothelin-1 receptors (ET-A and/or ET-B), reducing vasoconstriction and proliferation | - SERAPHIN (Macitentan): Reduced morbidity and mortality [17]. - AMBITION (Ambrisentan and Tadalafil combination): Improved outcomes vs. monotherapy [52]. |
| Phosphodiesterase-5 Inhibitors (PDE-5i) | Sildenafil, Tadalafil | Inhibit PDE-5, increasing cGMP and promoting vasodilation | - SUPER-1 (Sildenafil): Improved 6MWD [22]. - PHIRST (Tadalafil): Improved 6 MWD and reduced clinical worsening [24]. |
| Soluble Guanylate Cyclase (sGC) Stimulators | Riociguat | Enhance sGC activity, increasing cGMP for vasodilation | - PATENT-1 (Riociguat): Improved 6 MWD and hemodynamics [29]. |
| Prostacyclin Analogues | Epoprostenol, Treprostinil (IV/SQ/Inhaled/Oral), Iloprost | Prostacyclin receptor activation, promoting vasodilation and antiproliferation | - REVEAL (Epoprostenol): Improved survival [69]. - FREEDOM-EV (Oral Treprostinil): Reduced clinical worsening [70]. |
| Prostacyclin Receptor Agonists | Selexipag | Selective IP receptor agonist, mimicking prostacyclin effects | - GRIPHON (Selexipag): Reduced morbidity/mortality [51]. |
| Activin Signaling Inhibitor | Sotatercept | Restores BMPR2 signaling, promoting vascular remodeling | STELLAR: Improved 6MWD, PVR, and risk profile [67]. |
| ERA + PDE-5i Combination pill | (Macitentan + Tadalafil) | Combines Macitentan (ERA) and Tadalafil (PDE-5i) | A DUE: Greater PVR and functional improvements vs. monotherapy [53]. |
| Subgroup | General Management Principles | Treatment Consideration for PAH | Additional Considerations |
|---|---|---|---|
| Pulmonary Arterial Hypertension Associated with Connective Tissue Disease (PAH-CTD) | - Treat underlying autoimmune disease like Systemic Sclerosis according to the latest guideline [71] - Close coordination with multispecialty; example, rheumatology | - PDE5i, ERA, prostacyclin analogs - Same treatment algorithm as for patients with Idiopathic PAH | - Monitor closely for exacerbations and progression of underlying CTD |
| Pulmonary Arterial Hypertension Associated with Drugs and Toxins (DPAH) | - Immediately discontinue the agent causing PAH | - Same basic principles as the treatment for IPAH; PAH-directed therapy considered in intermediate- to high-risk group [1] | - Low-risk patients recommended to be re-evaluated at 3–4 months after discontinuation of offending agent; if hemodynamics does not normalize treat with PAH-specific medications [1] |
| HIV-Associated Pulmonary Arterial Hypertension (HIV-PH) | - Early diagnosis and initiation of antiretroviral therapy (ART) - Ensure optimal viral load control and immune function | - Current recommendations for PAH-specific medications are based on IPAH data - Initial monotherapy for PAH considered and, if needed, sequential combination therapy | - Monitor carefully for drug interactions |
| PAH Associated with Portal Hypertension | - Echocardiogram in patients with clinical features of PH and underlying liver disease and/or portal hypertension - Echocardiogram in patients being considered for Transjugular Portosystemic shunt or liver transplantation - Management of underlying liver disease and portal hypertension at a specialty center | - Initial monotherapy with PAH medication followed by sequential combination as necessary [72] | - Liver transplantation consideration if PVR is normal or near normal |
| PAH Associated with Adult CHD | In ASD, VSD, PDA with PVR M 3 WU, shunt closure recommended [1] | Limited data on the use of PAH-specific drugs; PDE5i and ERA might show improvements in functional class and hemodynamics in Eisenmenger [73] | Heart–lung transplantation is an option in patients who are not responsive to medical treatment; availability of organ and mortality in first year post-transplantation a concern. |
| PAH Associated with Schistosomiasis | Leading cause of PAH in Asia, Africa, South America | Uses of PAH-specific medications have improved survival per registry data [74] | |
| Pulmonary Veno-Occlusive Disease (PVOD) | Combination of clinical, radiological, blood gas, pulmonary function test and genetic testing (EIF2AK4 mutation) recommended in PAH with suspicion of venous/capillary involvement | Risk of pulmonary edema with the use of pulmonary vasodilators | Referral to a transplant center for evaluation is recommended |
| Invasive therapy |
|
| Non-invasive therapy |
|
| Others |
|
| Title | Primary Outcome Measures | Time Frame |
|---|---|---|
| Positioning Imatinib for Pulmonary Arterial Hypertension (PIPAH) |
|
|
| Clinical Trial of 2-hydroxbenzylamine (2-HOBA) in Pulmonary Arterial Hypertension | Changes in acetylated Superoxide Dismutase 2 (SOD2) and Long-chain acyl-CoA dehydrogenase (LCAD) in plasma. | Baseline and 12-weeks |
| Apabetalone for Pulmonary Arterial Hypertension (APPROACH-2) | Placebo-corrected change from baseline in PVR at week 24 | Baseline and 24 weeks |
| Metabolic Remodeling in Pulmonary Arterial Hypertension (PAH) | Change in ratio of oxidative metabolism to glycolysis | Baseline and 6 months |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, M.; Paudyal, V.; Syed, S.K.; Thapa, R.; Kassam, N.; Surani, S. Management of Pulmonary Arterial Hypertension: Current Strategies and Future Prospects. Life 2025, 15, 430. https://doi.org/10.3390/life15030430
Sharma M, Paudyal V, Syed SK, Thapa R, Kassam N, Surani S. Management of Pulmonary Arterial Hypertension: Current Strategies and Future Prospects. Life. 2025; 15(3):430. https://doi.org/10.3390/life15030430
Chicago/Turabian StyleSharma, Munish, Vivek Paudyal, Saifullah Khalid Syed, Rubi Thapa, Nadeem Kassam, and Salim Surani. 2025. "Management of Pulmonary Arterial Hypertension: Current Strategies and Future Prospects" Life 15, no. 3: 430. https://doi.org/10.3390/life15030430
APA StyleSharma, M., Paudyal, V., Syed, S. K., Thapa, R., Kassam, N., & Surani, S. (2025). Management of Pulmonary Arterial Hypertension: Current Strategies and Future Prospects. Life, 15(3), 430. https://doi.org/10.3390/life15030430