
Pathogenesis of Focal Segmental Glomerulosclerosis and Minimal Change Disease: Insights from Glomerular Proteomics


 , ,
, ,  , , ,
, , , 
 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Storage
2.2. Samples Preparation for Bottom-Up Proteomics Profiling
2.3. LC-MS Analysis
2.4. Differential Protein Abundance Analysis
2.5. Network Visualization and Enrichment Analysis
2.6. Immunohistochemistry
3. Results
3.1. Comparative Proteomic Analysis of Glomeruli in FSGS and MCD Patients
3.2. Differently Expressed Proteins
3.3. Immunohistochemistry Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| FSGS | Focal segmental glomerulosclerosis |
| MCD | Minimal change disease |
| GBM | Glomerular basement membrane |
| UBE2K | Ubiquitin-conjugating enzyme 2K |
| CFHR1 | Complement factor H-related protein-1 |
| SD | Slid diaphragm |
| FP | Foot process |
| FA | Focal adhesion |
| FPE | Foot process effacement |
| MPGN | Membranoproliferative glomerulonephritis |
| FFPE | Formalin-fixed paraffin-embedded |
| TEM | Transmission electron microscopy |
| eGFR | Estimated glomerular filtration rate |
| RNase | Ribonuclease |
| DNase | Deoxyribonuclease |
| LCM | Laser capture microdissection |
| CNI | Calcineurin inhibitors |
| CFS | Cyclophosphamide |
| HPA | Human Protein Atlas |
| MAC | membrane attack complex |
| ECM | Extracellular matrix |
| FSGS-NOS | Focal segmental glomerulosclerosis not other specified |
| MAC | Membrane attack complex |
| UPS | Ubiquitin–proteasome system |
| ER | Endoplasmic reticulum |
| MN | Membranous nephropathy |
| CypA | Cyclophilin A |
| MYL3 | Myosin light chain-3 |
| MME | Neprilysin |
| DEPs | Differentially expressed proteins |
| PPI | Protein–protein interaction |
| FDR | False discovery rate |
References
- Perico, L.; Conti, S.; Benigni, A.; Remuzzi, G. Podocyte–Actin Dynamics in Health and Disease. Nat. Rev. Nephrol. 2016, 12, 692–710. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Qiu, Y.; Zhang, C. Cytoskeleton Rearrangement in Podocytopathies: An Update. Int. J. Mol. Sci. 2024, 25, 647. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, K.; Kurihara, H.; Sakai, T. Actin Filament Organization of Foot Processes in Rat Podocytes. J. Histochem. Cytochem. 2003, 51, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Husain, S. Role of Podocyte in Kidney Disease. Front. Biosci.-Landmark 2024, 29, 250. [Google Scholar] [CrossRef]
- Ye, Q.; Lan, B.; Liu, H.; Persson, P.B.; Lai, E.Y.; Mao, J. A Critical Role of the Podocyte Cytoskeleton in the Pathogenesis of Glomerular Proteinuria and Autoimmune Podocytopathies. Acta Physiol. 2022, 235, e13850. [Google Scholar] [CrossRef]
- Ravaglia, F.; Melica, M.E.; Angelotti, M.L.; De Chiara, L.; Romagnani, P.; Lasagni, L. The Pathology Lesion Patterns of Podocytopathies: How and Why? Front. Cell Dev. Biol. 2022, 10, 838272. [Google Scholar] [CrossRef]
- Rovin, B.H.; Adler, S.G.; Barratt, J.; Bridoux, F.; Burdge, K.A.; Chan, T.M.; Cook, H.T.; Fervenza, F.C.; Gibson, K.L.; Glassock, R.J.; et al. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int. 2021, 100, S1–S276. [Google Scholar] [CrossRef]
- Thomas, D.B.; Franceschini, N.; Hogan, S.L.; ten Holder, S.; Jennette, C.E.; Falk, R.J.; Jennette, J.C. Clinical and Pathologic Characteristics of Focal Segmental Glomerulosclerosis Pathologic Variants. Kidney Int. 2006, 69, 920–926. [Google Scholar] [CrossRef]
- D’Agati, V. Pathologic Classification of Focal Segmental Glomerulosclerosis. Semin. Nephrol. 2003, 23, 117–134. [Google Scholar] [CrossRef]
- Sethi, S.; Fervenza, F.C. Membranoproliferative Glomerulonephritis: Pathogenetic Heterogeneity and Proposal for a New Classification. Semin. Nephrol. 2011, 31, 341–348. [Google Scholar] [CrossRef]
- Sethi, S.; Madden, B. Mapping Antigens of Membranous Nephropathy: Almost There. Kidney Int. 2023, 103, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Beck, L.H.; Zeng, C.; Chen, Z.; Li, S.; Zuo, K.; Salant, D.J.; Liu, Z. Anti-Phospholipase A2 Receptor Antibody in Membranous Nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Fervenza, F. Pathology of Renal Diseases Associated with Dysfunction of the Alternative Pathway of Complement: C3 Glomerulopathy and Atypical Hemolytic Uremic Syndrome (AHUS). Semin. Thromb. Hemost. 2014, 40, 416–421. [Google Scholar] [CrossRef]
- Smith, A.; Iablokov, V.; Mazza, M.; Guarnerio, S.; Denti, V.; Ivanova, M.; Stella, M.; Piga, I.; Chinello, C.; Heijs, B.; et al. Detecting Proteomic Indicators to Distinguish Diabetic Nephropathy from Hypertensive Nephrosclerosis by Integrating Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry Imaging with High-Mass Accuracy Mass Spectrometry. Kidney Blood Press. Res. 2020, 45, 233–248. [Google Scholar] [CrossRef]
- Ivanova, M.; Dyadyk, O.; Ivanov, D.; Clerici, F.; Smith, A.; Magni, F. Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry Imaging to Uncover Protein Alterations Associated with the Progression of IgA Nephropathy. Virchows Arch. 2020, 476, 903–914. [Google Scholar] [CrossRef]
- Bărar, A.A.; Pralea, I.-E.; Maslyennikov, Y.; Munteanu, R.; Berindan-Neagoe, I.; Pîrlog, R.; Rusu, I.; Nuțu, A.; Rusu, C.C.; Moldovan, D.T.; et al. Minimal Change Disease: Pathogenetic Insights from Glomerular Proteomics. Int. J. Mol. Sci. 2024, 25, 5613. [Google Scholar] [CrossRef]
- Bărar, A.A.; Pralea, I.E.; Berindan-Neagoe, I.; Pirlog, R.; Nutu, A.; Maslyennikov, Y.; Potra, A.R.; Iuga, C.A.; Kacso, I.M. Proteomic Patterns in Glomerular Research, a Laser Capture Microdissection and Liquid Chromatography-Tandem Mass Spectrometry Approach. Rev. Rom. Med. Lab. 2023, 31, 263–274. [Google Scholar] [CrossRef]
- Merchant, M.L.; Barati, M.T.; Caster, D.J.; Hata, J.L.; Hobeika, L.; Coventry, S.; Brier, M.E.; Wilkey, D.W.; Li, M.; Rood, I.M.; et al. Proteomic Analysis Identifies Distinct Glomerular Extracellular Matrix in Collapsing Focal Segmental Glomerulosclerosis. J. Am. Soc. Nephrol. 2020, 31, 1883–1904. [Google Scholar] [CrossRef]
- Ni, J.; Tian, S.; Bai, L.; Lv, Q.; Liu, J.; Liu, J.; Fang, Y.; Zhai, Y.; Shen, Q.; Rao, J.; et al. Comparative Proteomic Analysis of Children FSGS FFPE Tissues. BMC Pediatr. 2022, 22, 707. [Google Scholar] [CrossRef]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-Pot, Solid-Phase-Enhanced Sample Preparation for Proteomics Experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef]
- Distler, U.; Kuharev, J.; Navarro, P.; Tenzer, S. Label-Free Quantification in Ion Mobility–Enhanced Data-Independent Acquisition Proteomics. Nat. Protoc. 2016, 11, 795–812. [Google Scholar] [CrossRef]
- Pang, Z.; Zhou, G.; Ewald, J.; Chang, L.; Hacariz, O.; Basu, N.; Xia, J. Using MetaboAnalyst 5.0 for LC–HRMS Spectra Processing, Multi-Omics Integration and Covariate Adjustment of Global Metabolomics Data. Nat. Protoc. 2022, 17, 1735–1761. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Stevens, L.A.; Schmid, C.H.; Zhang, Y.; Castro , A.F., III; Feldman, H.I.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Greene, T.; et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009, 150, 604. [Google Scholar] [CrossRef] [PubMed]
- Nell, D.; Wolf, R.; Podgorny, P.M.; Kuschnereit, T.; Kuschnereit, R.; Dabers, T.; Stracke, S.; Schmidt, T. Complement Activation in Nephrotic Glomerular Diseases. Biomedicines 2024, 12, 455. [Google Scholar] [CrossRef]
- Kimoto, Y.; Horiuchi, T. The Complement System and ANCA Associated Vasculitis in the Era of Anti-Complement Drugs. Front. Immunol. 2022, 13, 926044. [Google Scholar] [CrossRef]
- Li, X.; Ding, F.; Zhang, X.; Li, B.; Ding, J. The Expression Profile of Complement Components in Podocytes. Int. J. Mol. Sci. 2016, 17, 471. [Google Scholar] [CrossRef]
- Zoshima, T.; Hara, S.; Yamagishi, M.; Pastan, I.; Matsusaka, T.; Kawano, M.; Nagata, M. Possible Role of Complement Factor H in Podocytes in Clearing Glomerular Subendothelial Immune Complex Deposits. Sci. Rep. 2019, 9, 7857. [Google Scholar] [CrossRef]
- Mühlig, A.K.; Keir, L.S.; Abt, J.C.; Heidelbach, H.S.; Horton, R.; Welsh, G.I.; Meyer-Schwesinger, C.; Licht, C.; Coward, R.J.; Fester, L.; et al. Podocytes Produce and Secrete Functional Complement C3 and Complement Factor H. Front. Immunol. 2020, 11, 01833. [Google Scholar] [CrossRef]
- Angeletti, A.; Cantarelli, C.; Petrosyan, A.; Andrighetto, S.; Budge, K.; D’Agati, V.D.; Hartzell, S.; Malvi, D.; Donadei, C.; Thurman, J.M.; et al. Loss of Decay-Accelerating Factor Triggers Podocyte Injury and Glomerulosclerosis. J. Exp. Med. 2020, 217, e20191699. [Google Scholar] [CrossRef]
- Thurman, J.M.; Wong, M.; Renner, B.; Frazer-Abel, A.; Giclas, P.C.; Joy, M.S.; Jalal, D.; Radeva, M.K.; Gassman, J.; Gipson, D.S.; et al. Complement Activation in Patients with Focal Segmental Glomerulosclerosis. PLoS ONE 2015, 10, e0136558. [Google Scholar] [CrossRef]
- Cambier, A.; Patey, N.; Royal, V.; Gougeon, F.; Genest, D.S.; Brachemi, S.; Bollée, G.; Merlen, C.; Bonnefoy, A.; Lapeyraque, A.-L.; et al. A Prospective Study on Complement Activation Distinguishes Focal Segmental Glomerulosclerosis from Minimal Change Disease. Kidney Int. Rep. 2024, 9, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xie, J.; Zhang, X.; Tong, J.; Hao, X.; Ren, H.; Wang, W.; Chen, N. Serum C3 and Renal Outcome in Patients with Primary Focal Segmental Glomerulosclerosis. Sci. Rep. 2017, 7, 4095. [Google Scholar] [CrossRef]
- Huang, J.; Cui, Z.; Gu, Q.; Zhang, Y.; Qu, Z.; Wang, X.; Wang, F.; Cheng, X.; Meng, L.; Liu, G.; et al. Complement Activation Profile of Patients with Primary Focal Segmental Glomerulosclerosis. PLoS ONE 2020, 15, e0234934. [Google Scholar] [CrossRef]
- Zagorec, N.; Horvatić, I.; Šenjug, P.; Sović, S.; Galešić, K.; Galešić Ljubanović, D. The Lectin Pathway–A Dominant Pattern of the Complement System Activation in Primary Focal Segmental Glomerulosclerosis? Kidney Int. Rep. 2024, 9, 1925–1926. [Google Scholar] [CrossRef]
- van de Lest, N.A.; Zandbergen, M.; Wolterbeek, R.; Kreutz, R.; Trouw, L.A.; Dorresteijn, E.M.; Bruijn, J.A.; Bajema, I.M.; Scharpfenecker, M.; Chua, J.S. Glomerular C4d Deposition Can Precede the Development of Focal Segmental Glomerulosclerosis. Kidney Int. 2019, 96, 738–749. [Google Scholar] [CrossRef]
- Borza, D.-B. Glomerular Basement Membrane Heparan Sulfate in Health and Disease: A Regulator of Local Complement Activation. Matrix Biol. 2017, 57–58, 299–310. [Google Scholar] [CrossRef]
- Kopp, A.; Hebecker, M.; Svobodová, E.; Józsi, M. Factor H: A Complement Regulator in Health and Disease, and a Mediator of Cellular Interactions. Biomolecules 2012, 2, 46–75. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Lauer, N.; Hartmann, A.; Stippa, S.; Keilhauer, C.N.; Oppermann, M.; Pandey, M.K.; Köhl, J.; Zipfel, P.F.; Weber, B.H.F.; et al. An Imbalance of Human Complement Regulatory Proteins CFHR1, CFHR3 and Factor H Influences Risk for Age-Related Macular Degeneration (AMD). Hum. Mol. Genet. 2010, 19, 4694–4704. [Google Scholar] [CrossRef]
- Goicoechea de Jorge, E.; Caesar, J.J.E.; Malik, T.H.; Patel, M.; Colledge, M.; Johnson, S.; Hakobyan, S.; Morgan, B.P.; Harris, C.L.; Pickering, M.C.; et al. Dimerization of Complement Factor H-Related Proteins Modulates Complement Activation in Vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 4685–4690. [Google Scholar] [CrossRef]
- Skerka, C.; Pradel, G.; Halder, L.D.; Zipfel, P.F.; Zipfel, S.L.H.; Strauß, O. Factor H-related Protein 1: A Complement Regulatory Protein and Guardian of Necrotic-type Surfaces. Br. J. Pharmacol. 2021, 178, 2823–2831. [Google Scholar] [CrossRef] [PubMed]
- Skerka, C.; Chen, Q.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement Factor H Related Proteins (CFHRs). Mol. Immunol. 2013, 56, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Chugh, S.S.; Clement, L.C. “Idiopathic” Minimal Change Nephrotic Syndrome: A Podocyte Mystery Nears the End. Am. J. Physiol.-Ren. Physiol. 2023, 325, F685–F694. [Google Scholar] [CrossRef]
- Wendt, R.; Siwy, J.; He, T.; Latosinska, A.; Wiech, T.; Zipfel, P.F.; Tserga, A.; Vlahou, A.; Rupprecht, H.; Catanese, L.; et al. Molecular Mapping of Urinary Complement Peptides in Kidney Diseases. Proteomes 2021, 9, 49. [Google Scholar] [CrossRef]
- Mastellos, D.C.; Hajishengallis, G.; Lambris, J.D. A Guide to Complement Biology, Pathology and Therapeutic Opportunity. Nat. Rev. Immunol. 2024, 24, 118–141. [Google Scholar] [CrossRef]
- Nagata, M. Podocyte Injury and Its Consequences. Kidney Int. 2016, 89, 1221–1230. [Google Scholar] [CrossRef]
- Beeken, M.; Lindenmeyer, M.T.; Blattner, S.M.; Radón, V.; Oh, J.; Meyer, T.N.; Hildebrand, D.; Schlüter, H.; Reinicke, A.T.; Knop, J.-H.; et al. Alterations in the Ubiquitin Proteasome System in Persistent but Not Reversible Proteinuric Diseases. J. Am. Soc. Nephrol. 2014, 25, 2511–2525. [Google Scholar] [CrossRef]
- Cybulsky, A.V. Endoplasmic Reticulum Stress in Proteinuric Kidney Disease. Kidney Int. 2010, 77, 187–193. [Google Scholar] [CrossRef]
- Kitzler, T.M.; Papillon, J.; Guillemette, J.; Wing, S.S.; Cybulsky, A.V. Complement Modulates the Function of the Ubiquitin–Proteasome System and Endoplasmic Reticulum-Associated Degradation in Glomerular Epithelial Cells. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2012, 1823, 1007–1016. [Google Scholar] [CrossRef]
- Liu, W.; Tang, X.; Qi, X.; Fu, X.; Ghimire, S.; Ma, R.; Li, S.; Zhang, N.; Si, H. The Ubiquitin Conjugating Enzyme: An Important Ubiquitin Transfer Platform in Ubiquitin-Proteasome System. Int. J. Mol. Sci. 2020, 21, 2894. [Google Scholar] [CrossRef]
- Cybulsky, A.V. The Intersecting Roles of Endoplasmic Reticulum Stress, Ubiquitin–Proteasome System, and Autophagy in the Pathogenesis of Proteinuric Kidney Disease. Kidney Int. 2013, 84, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R. Endoplasmic Reticulum: The Master Regulator of Stress Responses in Glomerular Diseases. In An Update on Glomerulopathies—Etiology and Pathogenesis; InTech: London, UK, 2011. [Google Scholar] [CrossRef]
- Meyer-Schwesinger, C.; Meyer, T.; Münster, S.; Klug, P.; Saleem, M.; Helmchen, U.; Stahl, R. A New Role for the Neuronal Ubiquitin C-terminal Hydrolase-L1 (UCH-L1) in Podocyte Process Formation and Podocyte Injury in Human Glomerulopathies. J. Pathol. 2009, 217, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Schwesinger, C.; Meyer, T.N.; Sievert, H.; Hoxha, E.; Sachs, M.; Klupp, E.-M.; Münster, S.; Balabanov, S.; Carrier, L.; Helmchen, U.; et al. Ubiquitin C-Terminal Hydrolase-L1 Activity Induces Polyubiquitin Accumulation in Podocytes and Increases Proteinuria in Rat Membranous Nephropathy. Am. J. Pathol. 2011, 178, 2044–2057. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Fukusumi, Y.; Ivanov, V.; Zhang, Y.; Kawachi, H. Tacrolimus Ameliorates Podocyte Injury by Restoring FK506 Binding Protein 12 (FKBP12) at Actin Cytoskeleton. FASEB J. 2021, 35, e21983. [Google Scholar] [CrossRef]
- Hadpech, S.; Thongboonkerd, V. Current Update on Theranostic Roles of Cyclophilin A in Kidney Diseases. Theranostics 2022, 12, 4067–4080. [Google Scholar] [CrossRef]
- Haydak, J.; Azeloglu, E.U. Role of Biophysics and Mechanobiology in Podocyte Physiology. Nat. Rev. Nephrol. 2024, 20, 371–385. [Google Scholar] [CrossRef]
- Lennon, R.; Byron, A.; Humphries, J.D.; Randles, M.J.; Carisey, A.; Murphy, S.; Knight, D.; Brenchley, P.E.; Zent, R.; Humphries, M.J. Global Analysis Reveals the Complexity of the Human Glomerular Extracellular Matrix. J. Am. Soc. Nephrol. 2014, 25, 939–951. [Google Scholar] [CrossRef]
- De Luca, F.; Kha, M.; Swärd, K.; Johansson, M.E. Identification of ARMH4 and WIPF3 as Human Podocyte Proteins with Potential Roles in Immunomodulation and Cytoskeletal Dynamics. PLoS ONE 2023, 18, e0280270. [Google Scholar] [CrossRef]
- Zhao, M.; Bai, M.; Ding, G.; Zhang, Y.; Huang, S.; Jia, Z.; Zhang, A. Angiotensin II Stimulates the NLRP3 Inflammasome to Induce Podocyte Injury and Mitochondrial Dysfunction. Kidney Dis. 2018, 4, 83–94. [Google Scholar] [CrossRef]
- Kaltenecker, C.C.; Domenig, O.; Kopecky, C.; Antlanger, M.; Poglitsch, M.; Berlakovich, G.; Kain, R.; Stegbauer, J.; Rahman, M.; Hellinger, R.; et al. Critical Role of Neprilysin in Kidney Angiotensin Metabolism. Circ. Res. 2020, 127, 593–606. [Google Scholar] [CrossRef]
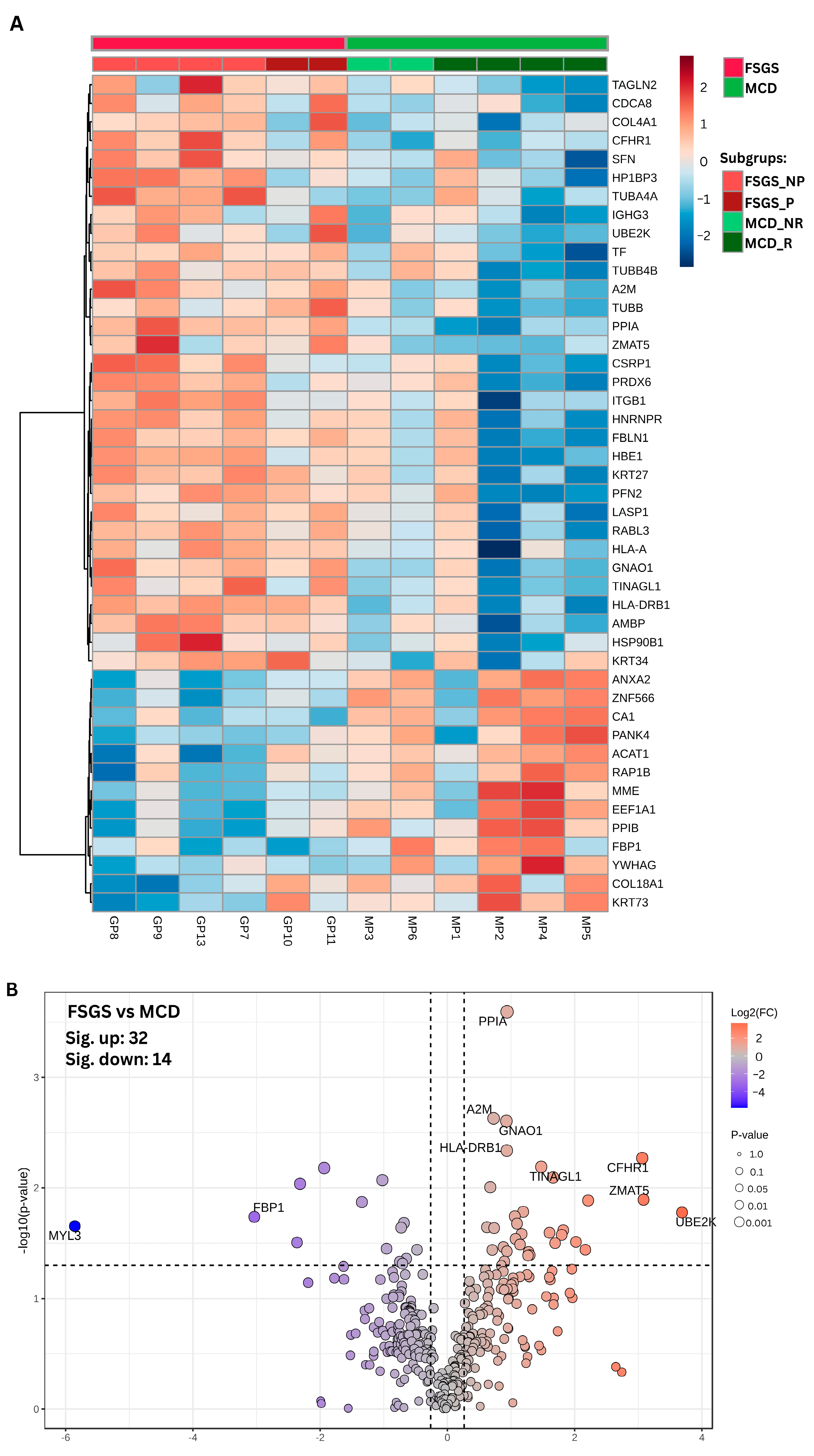
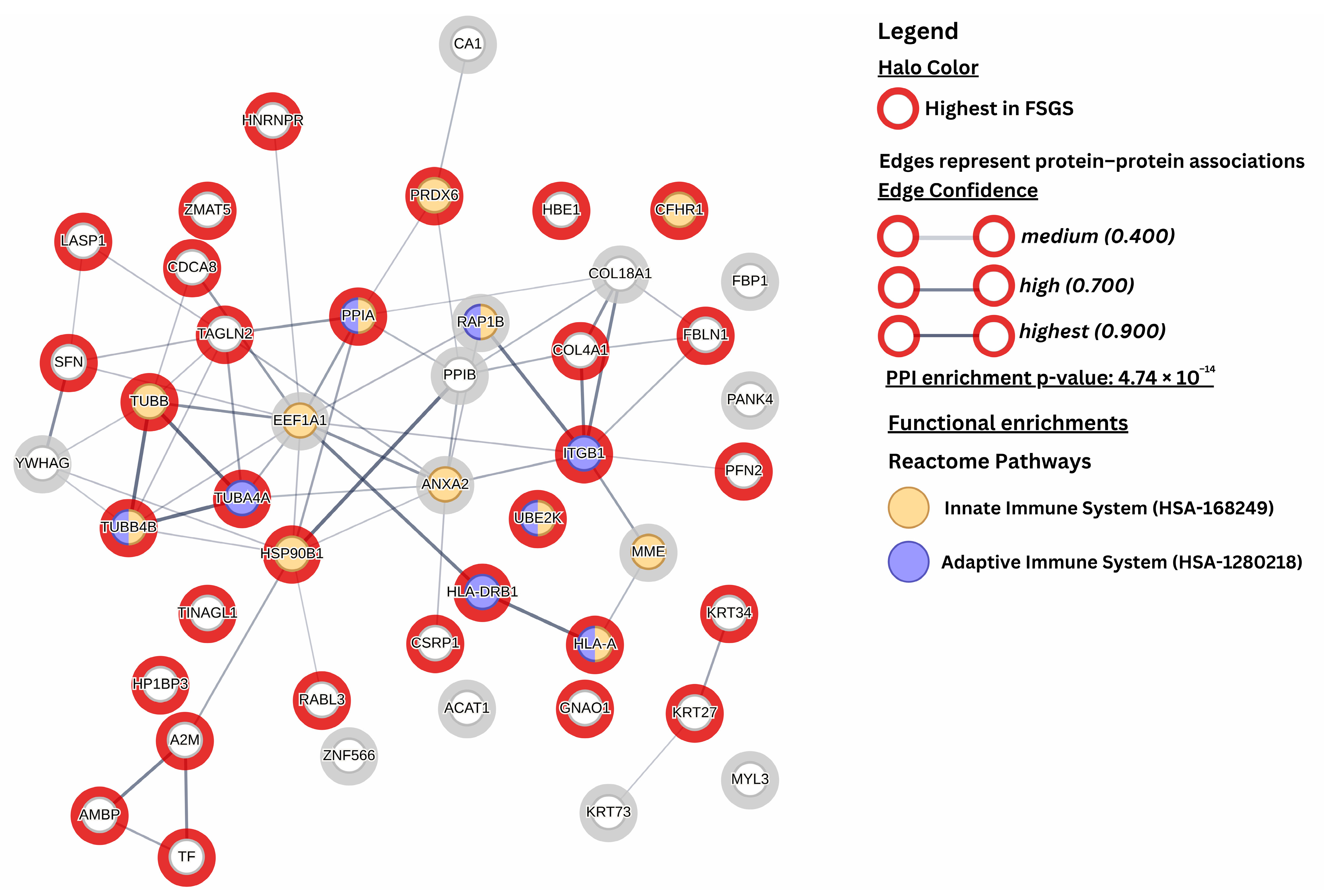



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | MP1 | MP2 | MP3 | MP4 | MP5 | MP6 | GP7 | GP8 | GP9 | GP10 | GP11 | GP13 | MCD (n = 6) | FSGS (n = 6) | p-Value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Previous childhood NS-RC | Yes | No | No | Yes | No | No | No | No | No | No | No | No | |||
| Previous immunosupression | Co/CNI | No | No | Co/CNI | No | No | No | No | No | No | No | No | |||
| Sex (males), n (%) | F | M | M | M | F | M | F | M | M | F | F | M | 4 (66.7) | 3 (50) | 1 |
| Age (years) ** | 20 | 51 | 18 | 20 | 66 | 59 | 36 | 27 | 41 | 57 | 25 | 60 | 39 ± 22.07 | 41 ± 14.79 | 0.857 |
| Serum creatinine (mg/dL) ** | 0.90 | 1.34 | 1.07 | 0.82 | 0.71 | 0.62 | 1.10 | 0.98 | 1.18 | 1.07 | 1.71 | 1.81 | 0.91 ± 0.26 | 1.31 ± 0.36 | 0.052 |
| eGFR (mL/min/1.73) ** | 94 | 64 | 103 | 128 | 92 | 109 | 67 | 108 | 80 | 61 | 42 | 32 | 98.33 ± 21.23 | 65 ± 27.25 | 0.040 |
| Serum albumin (g/dL) * | 3.15 | 1.46 | 1.61 | 1.46 | 1.6 | 3.21 | 1.50 | 2.74 | 2.80 | 3.00 | 3.77 | 1.91 | 1.61 (1.46; 3.15) | 2.77 (1.91; 3) | 0.394 |
| Total proteins (g/dL) ** | 5.82 | 3.44 | 3.89 | 3.38 | 4.2 | 5.82 | 5.00 | 5.03 | 5.45 | 5.82 | 6.41 | 5.08 | 4.43 ± 1.12 | 5.47 ± 0.56 | 0.070 |
| Cholesterol (mg/dL) ** | 315 | 400 | 564 | 300 | 290 | 283 | 300 | 229 | 270 | 325 | 188 | 412 | 358.67 ± 109.23 | 287.33 ± 78.35 | 0.223 |
| Hemoglobin (g/dL) ** | 15.1 | 15.6 | 18.1 | 14.9 | 12.0 | 11.6 | 15.1 | 13.9 | 14.5 | 16.2 | 12.9 | 11.2 | 14.55 ± 2.42 | 13.97 ± 1.75 | 0.643 |
| Hematuria, n (%) | Yes | Yes | No | Yes | No | Yes | No | Yes | No | Yes | Yes | Yes | 4 (80) | 4 (66.7) | 1 |
| Proteinuria (g/24 h) | 3.90 | 22.52 | 12.78 | 21.41 | 14.50 | 2.54 | 10.00 | 14.78 | 8.50 | 4.80 | 3.20 | 15.50 | 12.94 ± 8.44 | 9.46 ± 5.04 | 0.406 |
| C3 (g/L) ** | 1.49 | 2.18 | MD | 1.78 | 1.82 | 1.33 | 1.12 | 1.21 | 1.52 | 1.52 | 1.60 | 1.64 | 1.72 ± 0.33 | 1.44 ± 0.22 | 0.117 |
| C4 (g/L) ** | 0.42 | 0.51 | MD | 0.59 | 0.44 | 0.42 | 0.51 | 0.45 | 0.43 | 0.58 | 0.45 | 0.52 | 0.48 ± 0.07 | 0.49 ± 0.05 | 0.730 |
| Immunosupresive Line 1 | Co | Co | Co | Co | Co | Co | Co | Co | Co | Co | Co | Co | |||
| Response to treatment | No | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes | No | Yes | Yes | |||
| Relapse | - | Yes | No | No | Yes | No | Yes | No | - | No | No | ||||
| Immunospresive Line 2 | CNI | CFS | - | - | CNI | - | CNI | CNI | - | CNI | CNI | - | |||
| Response to treatment | Yes | Yes | - | - | Yes | - | Yes | Yes | - | No | No | - | |||
| Relapse | - | - | - | - | Yes | - | - | - | - | - | - | - | |||
| Immunospresive Line 3 | - | - | - | - | CFS | - | - | - | - | - | - | - | |||
| Response to treatment | - | - | - | - | Yes | - | - | - | - | - | - | - | |||
| Last follow up (months) | 84 | 73 | 12 | 10 | 156 | 12 | 40 | 70 | 65 | 38 | 31 | 6 | |||
| Last known serum creatinine (mg/dL) | 0.94 | 0.88 | 0.78 | 0.8 | 1.26 | 0.51 | 0.95 | 0.88 | 0 | ESRD | ESRD | 1.58 | |||
| Last known proteinuria (g/24 h) | 1.6 | 0.19 | 0.12 | 0.24 | 0.31 | 0.4 | 0.58 | 0.54 | 0.3 | ESRD | ESRD | 0.42 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maslyennikov, Y.; Pralea, I.-E.; Bărar, A.A.; Rusu, C.C.; Moldovan, D.T.; Potra, A.R.; Tirinescu, D.; Țicală, M.; Urs, A.; Zamfir, P.; et al. Pathogenesis of Focal Segmental Glomerulosclerosis and Minimal Change Disease: Insights from Glomerular Proteomics. Life 2025, 15, 527. https://doi.org/10.3390/life15040527
Maslyennikov Y, Pralea I-E, Bărar AA, Rusu CC, Moldovan DT, Potra AR, Tirinescu D, Țicală M, Urs A, Zamfir P, et al. Pathogenesis of Focal Segmental Glomerulosclerosis and Minimal Change Disease: Insights from Glomerular Proteomics. Life. 2025; 15(4):527. https://doi.org/10.3390/life15040527
Chicago/Turabian StyleMaslyennikov, Yuriy, Ioana-Ecaterina Pralea, Andrada Alina Bărar, Crina Claudia Rusu, Diana Tania Moldovan, Alina Ramona Potra, Dacian Tirinescu, Maria Țicală, Alexandra Urs, Paula Zamfir, and et al. 2025. "Pathogenesis of Focal Segmental Glomerulosclerosis and Minimal Change Disease: Insights from Glomerular Proteomics" Life 15, no. 4: 527. https://doi.org/10.3390/life15040527
APA StyleMaslyennikov, Y., Pralea, I.-E., Bărar, A. A., Rusu, C. C., Moldovan, D. T., Potra, A. R., Tirinescu, D., Țicală, M., Urs, A., Zamfir, P., Boțan, E., Mureșan, X.-M., Pîrv, S., Nuțu, A., Berindan-Neagoe, I., Iuga, C.-A., & Kacso, I. M. (2025). Pathogenesis of Focal Segmental Glomerulosclerosis and Minimal Change Disease: Insights from Glomerular Proteomics. Life, 15(4), 527. https://doi.org/10.3390/life15040527