
Pathophysiology, Clinical Heterogeneity, and Therapeutic Advances in Amyotrophic Lateral Sclerosis: A Comprehensive Review of Molecular Mechanisms, Diagnostic Challenges, and Multidisciplinary Management Strategies



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Clinical Manifestations
3. Genetics
4. Epidemiology
5. Pathophysiology
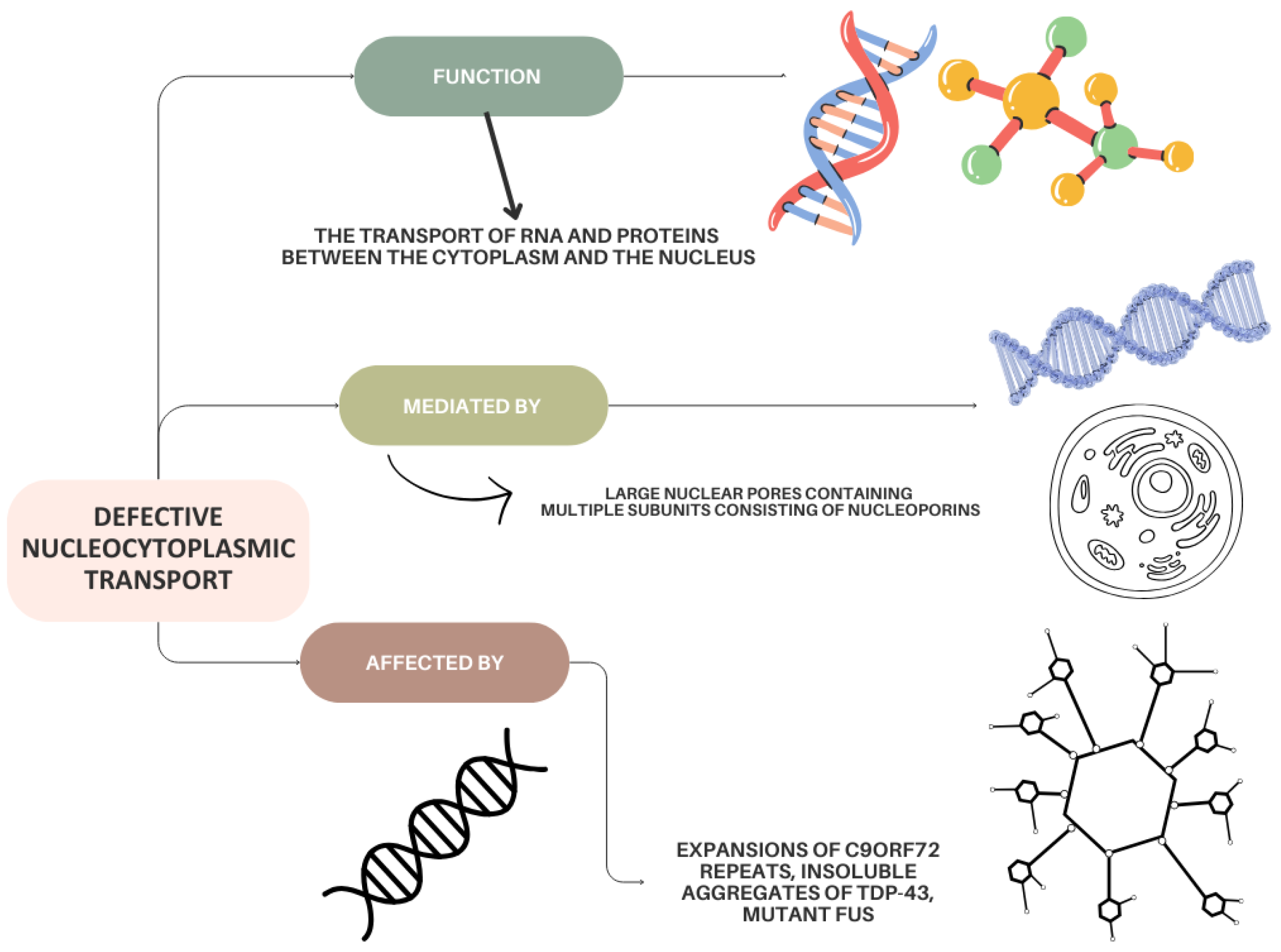
5.1. Defects in Nucleocytoplasmic Transport in ALS
5.2. C9orf72 Dipeptide Repeat Proteins and Neurotoxicity
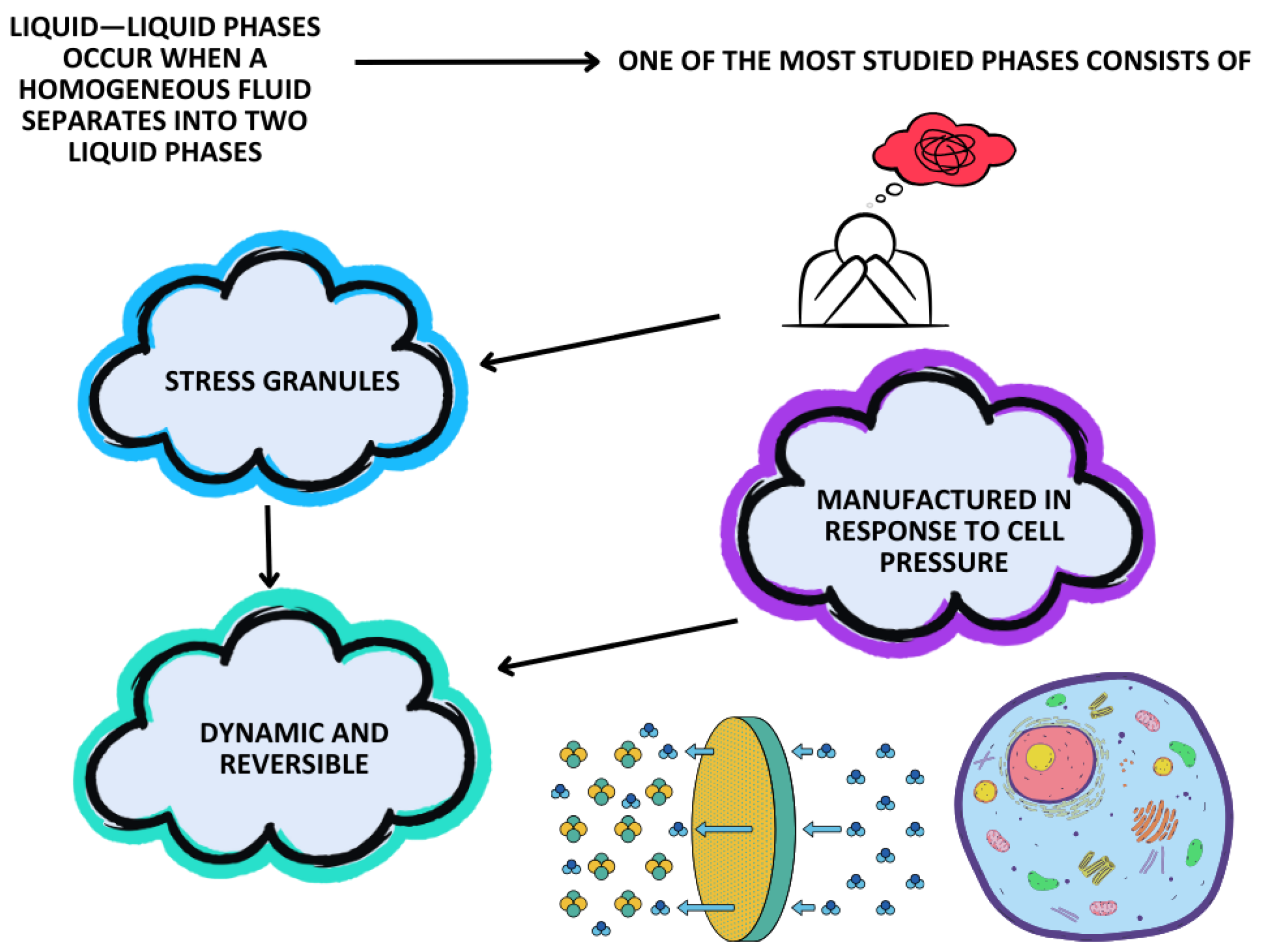
5.3. Liquid–Liquid Phase Separation
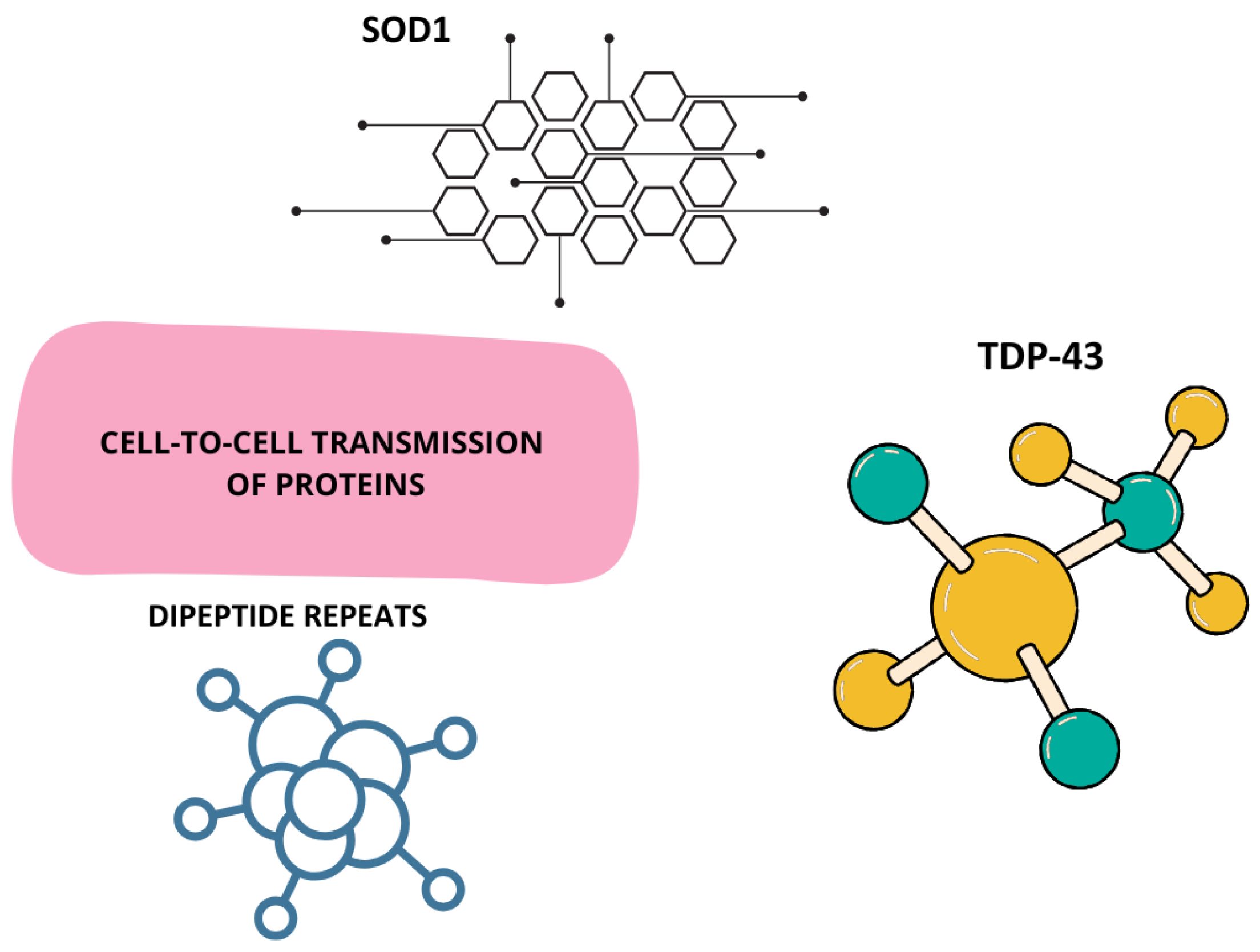
5.4. Cell-to-Cell Prion Transmission
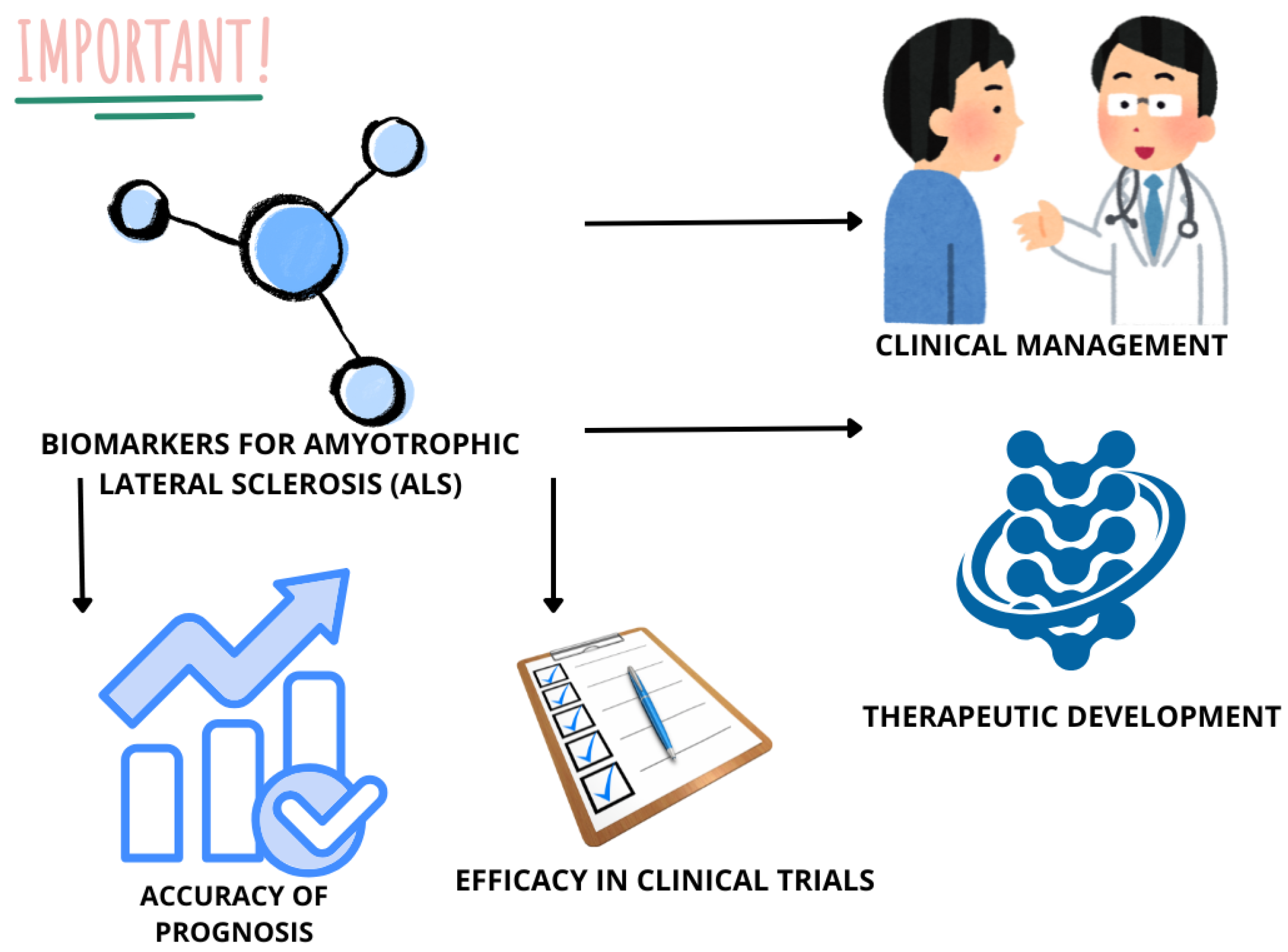
5.5. ALS Biomarkers
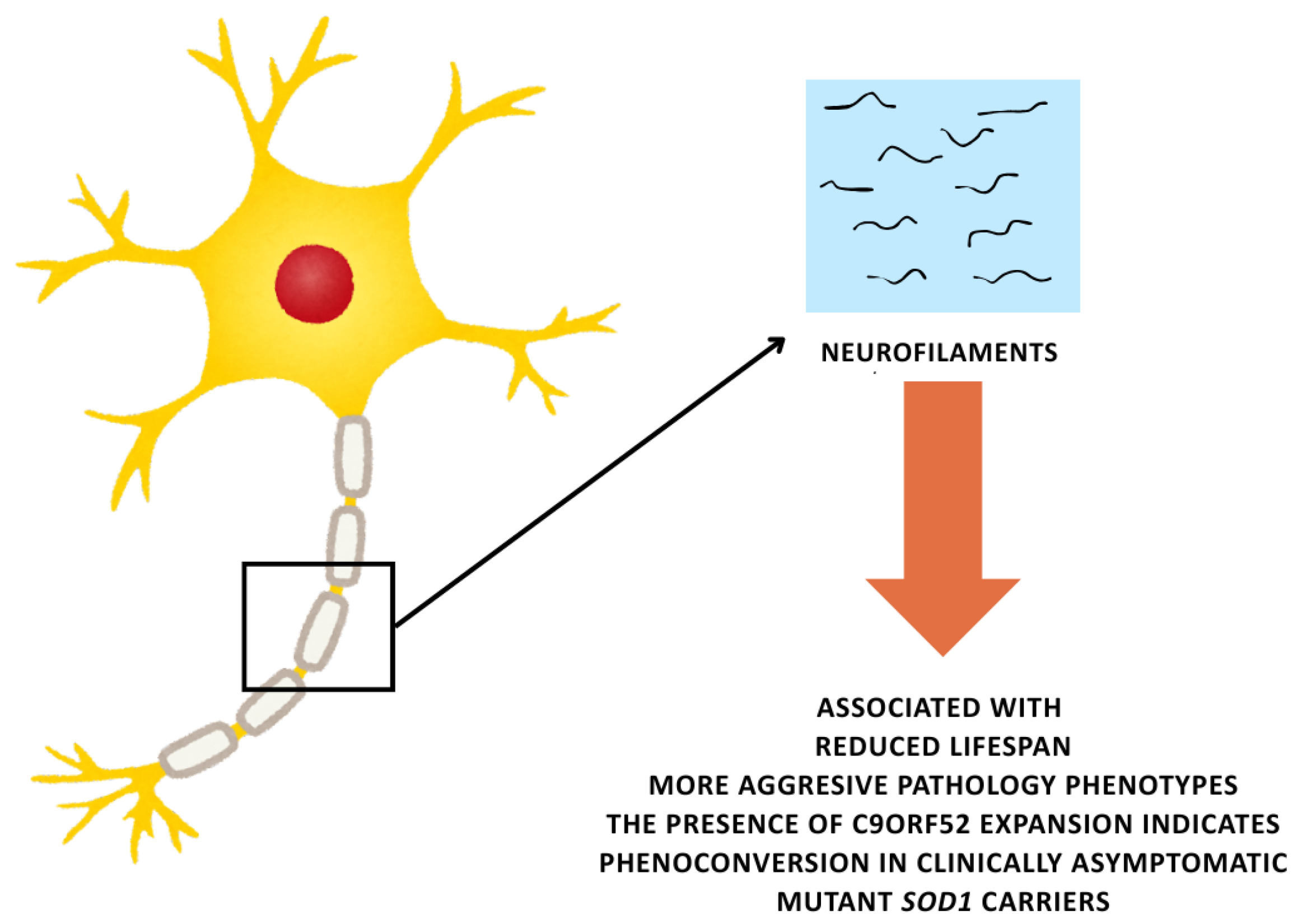
5.5.1. Neurofilaments
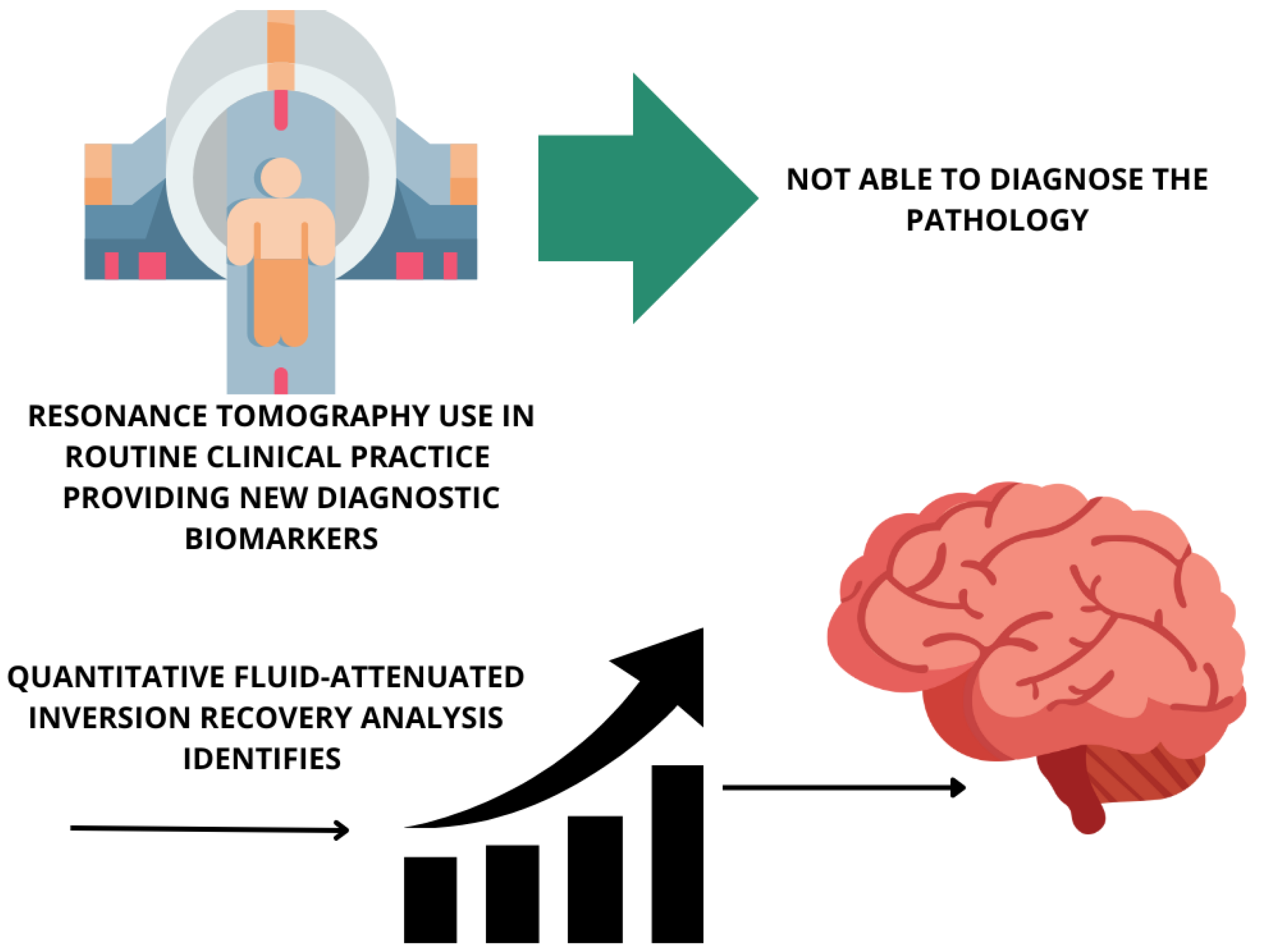
5.5.2. Brain Imaging
5.5.3. Emerging Biomarkers
6. Etiopathogenesis
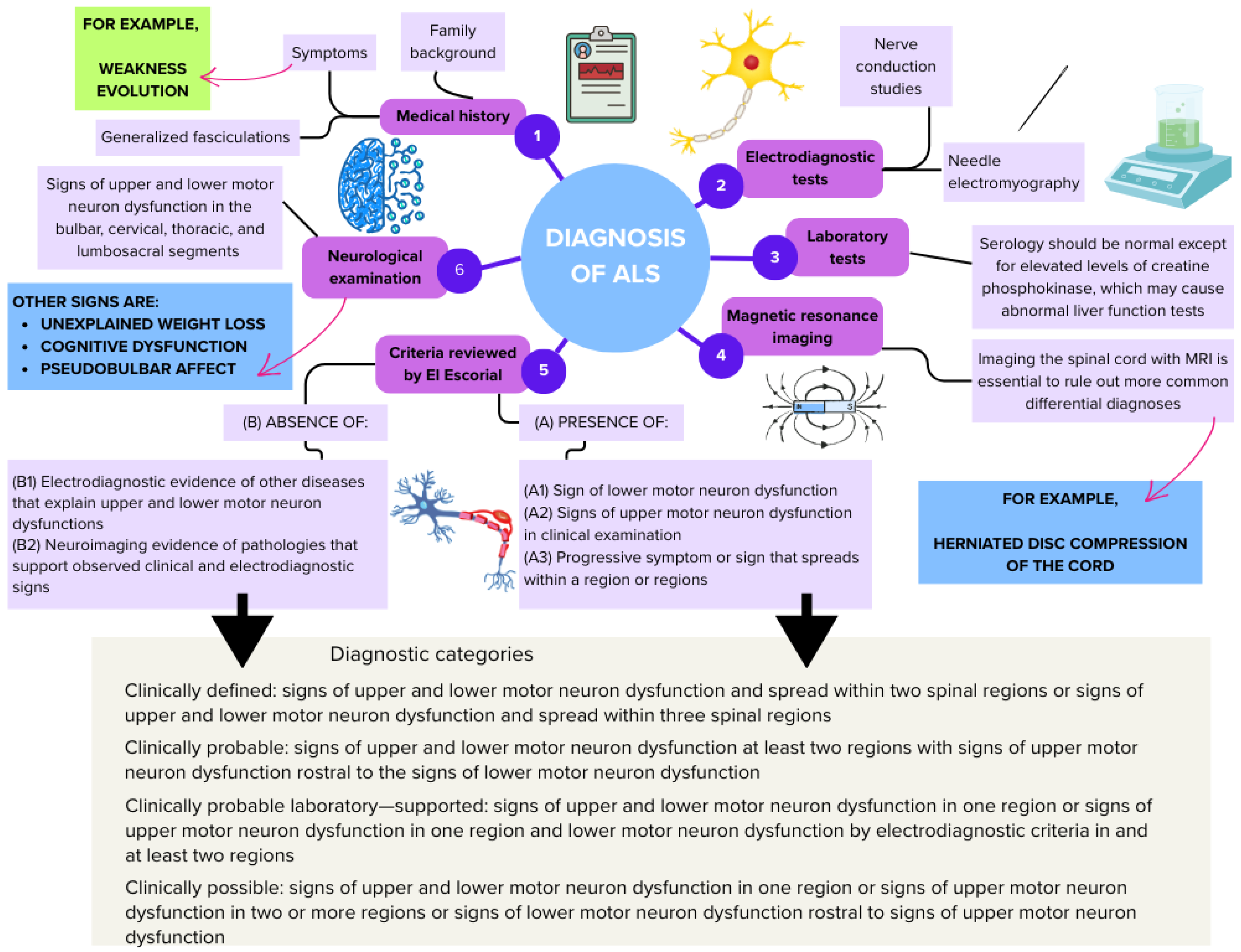
7. Diagnosis
8. From Symptom Onset to Disease Progression
8.1. ALS in the Early Stage
8.2. ALS in the Middle Phase
8.2.1. Musculoskeletal Symptoms
8.2.2. Spasticity and Cramps
8.2.3. Dysphagia and Sialorrhea/Secretion Management
8.2.4. Fatigue and Sleepiness
8.2.5. Dysarthria
8.2.6. Weakness of the Respiratory Muscles
8.3. Advanced ALS
9. Airway Clearance Strategies in ALS
9.1. Pathophysiology of Cough
9.2. Cough Insufficiency/Cough Increase
9.3. Manual Assistance
Sputum Mobilization and Salivary Secretion Management
10. ALS Forecast
11. Treatment
11.1. Pharmacological Treatment
11.2. Non-Pharmacological Treatment
11.2.1. Physiotherapy as Multidisciplinary Care
11.2.2. Exercise
12. Palliative Care
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic Lateral Sclerosis |
| FTD | Frontotemporal Dementia |
| C9orf72 | Chromosome 9 Open Reading Frame 72 |
| TDP-43 | TAR DNA-Binding Protein 43 |
| SOD1 | Superoxide Dismutase 1 |
| FUS | Fused in Sarcoma |
| NfL | Neurofilament Light Chain |
| pNfH | Phosphorylated Neurofilament Heavy Chain |
| GFAP | Glial Fibrillary Acidic Protein |
| CSF | Cerebrospinal Fluid |
| MRI | Magnetic Resonance Imaging |
| NIV | Non-Invasive Ventilation |
| FDA | Food and Drug Administration |
| RNA | Ribonucleic Acid |
| DNA | Deoxyribonucleic Acid |
| miRNA | MicroRNA |
| BDNF | Brain-Derived Neurotrophic Factor |
| GDNF | Glial Cell Line-Derived Neurotrophic Factor |
| AMX0035 | Sodium Phenylbutyrate/Taurursodiol |
| ALSFRS-R | ALS Functional Rating Scale-Revised |
| SIMOA | Single-Molecule Array |
References
- Fenoy, A. Scientific plurality and amyotrophic lateral sclerosis (ALS): A philosophical and historical perspective on Charcot’s texts. J. Hist. Neurosci. 2024, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nijs, M.; Van Damme, P. The genetics of amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2024, 37, 560–569. [Google Scholar] [CrossRef]
- Van Damme, P.; Al-Chalabi, A.; Andersen, P.M.; Chio, A.; Couratier, P.; De Carvalho, M.; Hardiman, O.; Kuzma-Kozakiewicz, M.; Ludolph, A.; McDermott, C.J.; et al. European Academy of Neurology (EAN) guideline on the management of amyotrophic lateral sclerosis in collaboration with European Reference Network for Neuromuscular Diseases (ERN EURO-NMD). Eur. J. Neurol. 2024, 31, e16264. [Google Scholar] [CrossRef]
- Eisen, A.; Vucic, S.; Kiernan, M.C. Amyotrophic lateral sclerosis represents corticomotoneuronal system failure. Muscle Nerve 2025, 71, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Nolan, M.; Scott, C.; Hof, P.R.; Ansorge, O. Betz cells of the primary motor cortex. J. Comp. Neurol. 2024, 532, e25567. [Google Scholar] [CrossRef] [PubMed]
- Lemon, R. The Corticospinal System and Amyotrophic Lateral Sclerosis: IFCN handbook chapter. Clin. Neurophysiol. 2024, 160, 56–67. [Google Scholar] [CrossRef]
- Ludolph, A.C.; Dietrich, J.; Dreyhaupt, J.; Kassubek, J.; Del Tredici, K.; Rosenbohm, A. Clinical spreading of muscle weakness in amyotrophic lateral sclerosis (ALS): A study in 910 patients. J. Neurol. 2024, 271, 5357–5367. [Google Scholar] [CrossRef]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chio, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Recent advances in the diagnosis and prognosis of amyotrophic lateral sclerosis. Lancet Neurol. 2022, 21, 480–493. [Google Scholar] [CrossRef]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chio, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 2022, 21, 465–479. [Google Scholar] [CrossRef]
- Brotman, R.G.; Moreno-Escobar, M.C.; Joseph, J.; Munakomi, S.; Pawar, G. Amyotrophic Lateral Sclerosis. In StatPearls; StatPearls: Treasure Island, FL, USA, 2025. [Google Scholar]
- Kaneko, K.; Hoskin, J.; Hodis, B. Primary Lateral Sclerosis. In StatPearls; StatPearls: Treasure Island, FL, USA, 2025. [Google Scholar]
- Kollstrom, A.M.; Christiansen, N.; Sandvig, A.; Sandvig, I. Dysregulation of synaptic transcripts underlies network abnormalities in ALS patient-derived motor neurons. Am. J. Physiol. Cell Physiol. 2025, 328, C1029–C1044. [Google Scholar] [CrossRef]
- Ramroop, H.; Cruz, R. Electrodiagnostic Evaluation of Motor Neuron Disease. In StatPearls; StatPearls: Treasure Island, FL, USA, 2025. [Google Scholar]
- Ravits, J.; Laurie, P.; Fan, Y.; Moore, D.H. Implications of ALS focality: Rostral-caudal distribution of lower motor neuron loss postmortem. Neurology 2007, 68, 1576–1582. [Google Scholar] [CrossRef]
- Ravits, J.; Paul, P.; Jorg, C. Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 2007, 68, 1571–1575. [Google Scholar] [CrossRef] [PubMed]
- Olesen, M.A.; Villavicencio-Tejo, F.; Cuevas-Espinoza, V.; Quintanilla, R.A. Unknown roles of tau pathology in neurological disorders. Challenges and new perspectives. Ageing Res. Rev. 2025, 103, 102594. [Google Scholar] [CrossRef]
- Pradat, P.F.; El Mendili, M.M. Neuroimaging to investigate multisystem involvement and provide biomarkers in amyotrophic lateral sclerosis. Biomed. Res. Int. 2014, 2014, 467560. [Google Scholar] [CrossRef] [PubMed]
- Goyal, N.A.; Berry, J.D.; Windebank, A.; Staff, N.P.; Maragakis, N.J.; van den Berg, L.H.; Genge, A.; Miller, R.; Baloh, R.H.; Kern, R.; et al. Addressing heterogeneity in amyotrophic lateral sclerosis CLINICAL TRIALS. Muscle Nerve 2020, 62, 156–166. [Google Scholar] [CrossRef]
- Chio, A.; Pagani, M.; Agosta, F.; Calvo, A.; Cistaro, A.; Filippi, M. Neuroimaging in amyotrophic lateral sclerosis: Insights into structural and functional changes. Lancet Neurol. 2014, 13, 1228–1240. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Ghirelli, A.; Spinelli, E.G.; Agosta, F. A comprehensive update on neuroimaging endpoints in amyotrophic lateral sclerosis. Expert Rev. Neurother. 2025, 25, 397–413. [Google Scholar] [CrossRef]
- Tan, H.H.G.; Nitert, A.D.; van Veenhuijzen, K.; Dukic, S.; van Zandvoort, M.J.E.; Hendrikse, J.; van Es, M.A.; Veldink, J.H.; Westeneng, H.J.; van den Berg, L.H. Neuroimaging correlates of domain-specific cognitive deficits in amyotrophic lateral sclerosis. Neuroimage Clin. 2025, 45, 103749. [Google Scholar] [CrossRef]
- Menke, R.A.; Agosta, F.; Grosskreutz, J.; Filippi, M.; Turner, M.R. Neuroimaging Endpoints in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2017, 14, 11–23. [Google Scholar] [CrossRef]
- Beeldman, E.; Govaarts, R.; de Visser, M.; Klein Twennaar, M.; van der Kooi, A.J.; van den Berg, L.H.; Veldink, J.H.; Pijnenburg, Y.A.L.; de Haan, R.J.; Schmand, B.A.; et al. Progression of cognitive and behavioural impairment in early amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2020, 91, 779–780. [Google Scholar] [CrossRef]
- Pender, N.; Pinto-Grau, M.; Hardiman, O. Cognitive and behavioural impairment in amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2020, 33, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Bersano, E.; Sarnelli, M.F.; Solara, V.; Iazzolino, B.; Peotta, L.; De Marchi, F.; Facchin, A.; Moglia, C.; Canosa, A.; Calvo, A.; et al. Decline of cognitive and behavioral functions in amyotrophic lateral sclerosis: A longitudinal study. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 373–379. [Google Scholar] [CrossRef]
- Crockford, C.; Newton, J.; Lonergan, K.; Chiwera, T.; Booth, T.; Chandran, S.; Colville, S.; Heverin, M.; Mays, I.; Pal, S.; et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology 2018, 91, e1370–e1380. [Google Scholar] [CrossRef]
- Cady, J.; Allred, P.; Bali, T.; Pestronk, A.; Goate, A.; Miller, T.M.; Mitra, R.D.; Ravits, J.; Harms, M.B.; Baloh, R.H. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann. Neurol. 2015, 77, 100–113. [Google Scholar] [CrossRef]
- Maurel, C.; Dangoumau, A.; Marouillat, S.; Brulard, C.; Chami, A.; Hergesheimer, R.; Corcia, P.; Blasco, H.; Andres, C.R.; Vourc’h, P. Causative Genes in Amyotrophic Lateral Sclerosis and Protein Degradation Pathways: A Link to Neurodegeneration. Mol. Neurobiol. 2018, 55, 6480–6499. [Google Scholar] [CrossRef]
- Mizielinska, S.; Hautbergue, G.M.; Gendron, T.F.; van Blitterswijk, M.; Hardiman, O.; Ravits, J.; Isaacs, A.M.; Rademakers, R. Amyotrophic lateral sclerosis caused by hexanucleotide repeat expansions in C9orf72: From genetics to therapeutics. Lancet. Neurol. 2025, 24, 261–274. [Google Scholar] [CrossRef]
- Moens, T.G.; Da Cruz, S.; Neumann, M.; Shelkovnikova, T.A.; Shneider, N.A.; Van Den Bosch, L. Amyotrophic lateral sclerosis caused by FUS mutations: Advances with broad implications. Lancet Neurol. 2025, 24, 166–178. [Google Scholar] [CrossRef] [PubMed]
- van Zundert, B.; Montecino, M. Epigenetics in Neurodegenerative Diseases. Subcell Biochem. 2025, 108, 73–109. [Google Scholar] [CrossRef] [PubMed]
- AlAnazi, A.; Alghadir, A.H.; Gabr, S.A. Handgrip Strength Exercises Modulate Shoulder Pain, Function, and Strength of Rotator Cuff Muscles of Patients with Primary Subacromial Impingement Syndrome. Biomed. Res. Int. 2022, 2022, 9151831. [Google Scholar] [CrossRef]
- Kiernan, J.A.; Hudson, A.J. Frontal lobe atrophy in motor neuron diseases. Brain 1994, 117 Pt 4, 747–757. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Lomen-Hoerth, C.; Anderson, T.; Miller, B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 2002, 59, 1077–1079. [Google Scholar] [CrossRef]
- Strong, M.J.; Grace, G.M.; Freedman, M.; Lomen-Hoerth, C.; Woolley, S.; Goldstein, L.H.; Murphy, J.; Shoesmith, C.; Rosenfeld, J.; Leigh, P.N.; et al. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 10, 131–146. [Google Scholar] [CrossRef]
- Strong, M.J.; Grace, G.M.; Orange, J.B.; Leeper, H.A.; Menon, R.S.; Aere, C. A prospective study of cognitive impairment in ALS. Neurology 1999, 53, 1665–1670. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.; Wu, L.; Zhang, X. Structural insights and milestones in TDP-43 research: A comprehensive review of its pathological and therapeutic advances. Int. J. Biol. Macromol. 2025, 306, 141677. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Mild cognitive impairment in amyotrophic lateral sclerosis: Current view. J. Neural. Transm. 2025, 132, 357–368. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Nicholson, K.; Murphy, A.; McDonnell, E.; Shapiro, J.; Simpson, E.; Glass, J.; Mitsumoto, H.; Forshew, D.; Miller, R.; Atassi, N. Improving symptom management for people with amyotrophic lateral sclerosis. Muscle Nerve 2018, 57, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Cairns, N.J.; Bigio, E.H.; Mackenzie, I.R.; Neumann, M.; Lee, V.M.; Hatanpaa, K.J.; White, C.L., 3rd; Schneider, J.A.; Grinberg, L.T.; Halliday, G.; et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: Consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007, 114, 5–22. [Google Scholar] [CrossRef]
- Metz, A.; Zeighami, Y.; Ducharme, S.; Villeneuve, S.; Dadar, M. Frontotemporal dementia subtyping using machine learning, multivariate statistics and neuroimaging. Brain Commun. 2025, 7, fcaf065. [Google Scholar] [CrossRef]
- Yoshida, M. Amyotrophic lateral sclerosis with dementia: The clinicopathological spectrum. Neuropathology 2004, 24, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Chow, T.W. What are the different initial presentations of frontotemporal dementia? J. Mol. Neurosci. 2011, 45, 710–712. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.; Heverin, M.; McLaughlin, R.L.; Hardiman, O. Lifetime Risk and Heritability of Amyotrophic Lateral Sclerosis. JAMA Neurol. 2019, 76, 1367–1374. [Google Scholar] [CrossRef]
- Olsen, C.G.; Malmberg, V.N.; Fahlstrom, M.; Alstadhaug, K.B.; Bjorna, I.K.; Braathen, G.J.; Brathen, G.; Demic, N.; Hallerstig, E.; Hogenesch, I.; et al. Amyotrophic lateral sclerosis caused by the C9orf72 expansion in Norway—Prevalence, ancestry, clinical characteristics and sociodemographic status. Amyotroph. Lateral Scler. Front. Degener. 2025, 26, 132–140. [Google Scholar] [CrossRef]
- Nakken, O.; Lindstrom, J.C.; Tysnes, O.B.; Holmoy, T. Assessing amyotrophic lateral sclerosis prevalence in Norway from 2009 to 2015 from compulsory nationwide health registers. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 303–310. [Google Scholar] [CrossRef]
- Ragonese, P.; Cellura, E.; Aridon, P.; D’Amelio, M.; Spataro, R.; Taiello, A.C.; Maimone, D.; La Bella, V.; Savettieri, G. Incidence of amyotrophic lateral sclerosis in Sicily: A population based study. Amyotroph. Lateral Scler. 2012, 13, 284–287. [Google Scholar] [CrossRef]
- Alonso, A.; Logroscino, G.; Jick, S.S.; Hernan, M.A. Incidence and lifetime risk of motor neuron disease in the United Kingdom: A population-based study. Eur. J. Neurol. 2009, 16, 745–751. [Google Scholar] [CrossRef]
- Chio, A.; Mora, G.; Calvo, A.; Mazzini, L.; Bottacchi, E.; Mutani, R.; PARALS. Epidemiology of ALS in Italy: A 10-year prospective population-based study. Neurology 2009, 72, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Traxinger, K.; Kelly, C.; Johnson, B.A.; Lyles, R.H.; Glass, J.D. Prognosis and epidemiology of amyotrophic lateral sclerosis: Analysis of a clinic population, 1997–2011. Neurol. Clin. Pract. 2013, 3, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Diagnosis, E.T.F.o.; Management of Amyotrophic Lateral, S.; Andersen, P.M.; Abrahams, S.; Borasio, G.D.; de Carvalho, M.; Chio, A.; Van Damme, P.; Hardiman, O.; Kollewe, K.; et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)--revised report of an EFNS task force. Eur. J. Neurol. 2012, 19, 360–375. [Google Scholar] [CrossRef]
- Calvo, A.; Moglia, C.; Lunetta, C.; Marinou, K.; Ticozzi, N.; Ferrante, G.D.; Scialo, C.; Soraru, G.; Trojsi, F.; Conte, A.; et al. Factors predicting survival in ALS: A multicenter Italian study. J. Neurol. 2017, 264, 54–63. [Google Scholar] [CrossRef]
- Marin, B.; Fontana, A.; Arcuti, S.; Copetti, M.; Boumediene, F.; Couratier, P.; Beghi, E.; Preux, P.M.; Logroscino, G. Age-specific ALS incidence: A dose-response meta-analysis. Eur. J. Epidemiol. 2018, 33, 621–634. [Google Scholar] [CrossRef]
- Mehta, P.; Kaye, W.; Raymond, J.; Punjani, R.; Larson, T.; Cohen, J.; Muravov, O.; Horton, K. Prevalence of Amyotrophic Lateral Sclerosis—United States, 2015. MMWR Morb. Mortal Wkly Rep. 2018, 67, 1285–1289. [Google Scholar] [CrossRef]
- Xu, L.; Liu, T.; Liu, L.; Yao, X.; Chen, L.; Fan, D.; Zhan, S.; Wang, S. Global variation in prevalence and incidence of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. 2020, 267, 944–953. [Google Scholar] [CrossRef]
- Collaborators, G.U.N.D.; Feigin, V.L.; Vos, T.; Alahdab, F.; Amit, A.M.L.; Barnighausen, T.W.; Beghi, E.; Beheshti, M.; Chavan, P.P.; Criqui, M.H.; et al. Burden of Neurological Disorders Across the US From 1990-2017: A Global Burden of Disease Study. JAMA Neurol. 2021, 78, 165–176. [Google Scholar] [CrossRef]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chio, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef]
- Chio, A.; Logroscino, G.; Traynor, B.J.; Collins, J.; Simeone, J.C.; Goldstein, L.A.; White, L.A. Global epidemiology of amyotrophic lateral sclerosis: A systematic review of the published literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef]
- Stevic, Z.; Kostic-Dedic, S.; Peric, S.; Dedic, V.; Basta, I.; Rakocevic-Stojanovic, V.; Lavrnic, D. Prognostic factors and survival of ALS patients from Belgrade, Serbia. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Brylev, L.; Ataulina, A.; Fominykh, V.; Parshikov, V.; Vorobyeva, A.; Istomina, E.; Shikhirimov, R.; Salikov, A.; Zakharova, M.; Guekht, A.; et al. The epidemiology of amyotrophic lateral sclerosis in Moscow (Russia). Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 410–415. [Google Scholar] [CrossRef]
- Demetriou, C.A.; Hadjivasiliou, P.M.; Kleopa, K.A.; Christou, Y.P.; Leonidou, E.; Kyriakides, T.; Zamba-Papanicolaou, E. Epidemiology of Amyotrophic Lateral Sclerosis in the Republic of Cyprus: A 25-Year Retrospective Study. Neuroepidemiology 2017, 48, 79–85. [Google Scholar] [CrossRef]
- Demetriou, C.A.; Hadjivasiliou, P.M.; Kleopa, K.A.; Christou, Y.P.; Leonidou, E.; Kyriakides, T.; Zamba-Papanicolaou, E. Retrospective longitudinal study of ALS in Cyprus: Clinical characteristics, management and survival. PLoS ONE 2019, 14, e0220246. [Google Scholar] [CrossRef]
- Logroscino, G.; Piccininni, M. Amyotrophic Lateral Sclerosis Descriptive Epidemiology: The Origin of Geographic Difference. Neuroepidemiology 2019, 52, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Erazo, D.; Luna, J.; Preux, P.M.; Boumediene, F.; Couratier, P. Epidemiological and genetic features of amyotrophic lateral sclerosis in Latin America and the Caribbean: A systematic review. Amyotroph. Lateral Scler. Front. Degener. 2022, 23, 4–15. [Google Scholar] [CrossRef]
- Mehta, P.; Antao, V.; Kaye, W.; Sanchez, M.; Williamson, D.; Bryan, L.; Muravov, O.; Horton, K.; Division of Toxicology and Human Health Sciences. Prevalence of amyotrophic lateral sclerosis-United States, 2010–2011. MMWR Suppl. 2014, 63, 1–14. [Google Scholar]
- Logroscino, G.; Traynor, B.J.; Hardiman, O.; Chio, A.; Mitchell, D.; Swingler, R.J.; Millul, A.; Benn, E.; Beghi, E.; Eurals Consortium. Incidence of amyotrophic lateral sclerosis in Europe. J. Neurol. Neurosurg. Psychiatry 2010, 81, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.A.; Lally, C.; Kupelian, V.; Flanders, W.D. Estimated Prevalence and Incidence of Amyotrophic Lateral Sclerosis and SOD1 and C9orf72 Genetic Variants. Neuroepidemiology 2021, 55, 342–353. [Google Scholar] [CrossRef]
- Wolfson, C.; Gauvin, D.E.; Ishola, F.; Oskoui, M. Global Prevalence and Incidence of Amyotrophic Lateral Sclerosis: A Systematic Review. Neurology 2023, 101, e613–e623. [Google Scholar] [CrossRef]
- Pang, W.; Hu, F. Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J. Neurochem. 2021, 157, 334–350. [Google Scholar] [CrossRef] [PubMed]
- Peltner, J.; Becker, C.; Wicherski, J.; Wortberg, S.; Aborageh, M.; Costa, I.; Ehrenstein, V.; Fernandes, J.; Hess, S.; Horvath-Puho, E.; et al. The EU project Real4Reg: Unlocking real-world data with AI. Health Res. Policy Syst. 2025, 23, 27. [Google Scholar] [CrossRef]
- Liu, Z.; Song, S.Y. Genomic and Transcriptomic Approaches Advance the Diagnosis and Prognosis of Neurodegenerative Diseases. Genes 2025, 16, 135. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.; Mannan, A.; Nauriyal, A.; Singh, T.G. Emerging targets in amyotrophic lateral sclerosis (ALS): The promise of ATP-binding cassette (ABC) transporter modulation. Behav. Brain Res. 2025, 476, 115242. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.P.; Van Broeckhoven, C.; van der Zee, J. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet 2018, 34, 404–423. [Google Scholar] [CrossRef]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C.; et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA 2019, 116, 4696–4705. [Google Scholar] [CrossRef]
- Morgan, S.; Shatunov, A.; Sproviero, W.; Jones, A.R.; Shoai, M.; Hughes, D.; Al Khleifat, A.; Malaspina, A.; Morrison, K.E.; Shaw, P.J.; et al. A comprehensive analysis of rare genetic variation in amyotrophic lateral sclerosis in the UK. Brain 2017, 140, 1611–1618. [Google Scholar] [CrossRef]
- Kim, H.J.; Taylor, J.P. Lost in Transportation: Nucleocytoplasmic Transport Defects in ALS and Other Neurodegenerative Diseases. Neuron 2017, 96, 285–297. [Google Scholar] [CrossRef]
- Deng, F.Y.; Zhu, G.L.; Ou, K.L.; Zhu, L.H.; Jia, Q.Q.; Wang, X.; Guo, M.W.; Li, B.; Li, S.H.; Li, X.J.; et al. Ribosome-associated pathological TDP-43 alters the expression of multiple mRNAs in the monkey brain. Zool Res. 2025, 46, 263–276. [Google Scholar] [CrossRef]
- Mengistu, D.Y.; Terribili, M.; Pellacani, C.; Ciapponi, L.; Marzullo, M. Epigenetic regulation of TDP-43: Potential implications for amyotrophic lateral sclerosis. Front. Mol. Med. 2025, 5, 1530719. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Kumar, M.S.; Ramesh, N.; Anderson, E.N.; Nguyen, A.T.; Kim, B.; Cheung, S.; McDonough, J.A.; Skarnes, W.C.; Lopez-Gonzalez, R.; et al. Interactions between ALS-linked FUS and nucleoporins are associated with defects in the nucleocytoplasmic transport pathway. Nat. Neurosci. 2021, 24, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Yamashita, T.; Nakano, Y.; Morihara, R.; Li, X.; Feng, T.; Liu, X.; Huang, Y.; Fukui, Y.; Hishikawa, N.; et al. Aberrant distributions of nuclear pore complex proteins in ALS mice and ALS patients. Neuroscience 2017, 350, 158–168. [Google Scholar] [CrossRef]
- Chou, C.C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Guo, L.; Gonzales, P.K.; Gendron, T.F.; Wu, Y.; Jansen-West, K.; O’Raw, A.D.; Pickles, S.R.; Prudencio, M.; Carlomagno, Y.; et al. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 2019, 363, eaav2606. [Google Scholar] [CrossRef]
- Ito, T.; Ohuchi, K.; Kurita, H.; Murakami, T.; Takizawa, S.; Fujimaki, A.; Murata, J.; Oida, Y.; Hozumi, I.; Kitaichi, K.; et al. Activated Fibroblast Growth Factor Receptor 1 Mitigated Poly-PR-Induced Oxidative Stress and Protein Translational Impairment. Biol. Pharm. Bull 2025, 48, 93–100. [Google Scholar] [CrossRef]
- Kim, K.M.; Girdhar, A.; Cicardi, M.E.; Kankate, V.; Hayashi, M.; Yang, R.; Carey, J.L.; Fare, C.M.; Shorter, J.; Cingolani, G.; et al. NLS-binding deficient Kapbeta2 reduces neurotoxicity via selective interaction with C9orf72-ALS/FTD dipeptide repeats. Commun. Biol. 2025, 8, 2. [Google Scholar] [CrossRef]
- Pakravan, D.; Orlando, G.; Bercier, V.; Van Den Bosch, L. Role and therapeutic potential of liquid-liquid phase separation in amyotrophic lateral sclerosis. J. Mol. Cell Biol. 2021, 13, 15–28. [Google Scholar] [CrossRef]
- McCaig, C.D. Neurological Diseases can be Regulated by Phase Separation. Rev. Physiol. Biochem. Pharmacol. 2025, 187, 273–338. [Google Scholar] [CrossRef]
- Pokrishevsky, E.; Grad, L.I.; Cashman, N.R. TDP-43 or FUS-induced misfolded human wild-type SOD1 can propagate intercellularly in a prion-like fashion. Sci. Rep. 2016, 6, 22155. [Google Scholar] [CrossRef] [PubMed]
- Westergard, T.; Jensen, B.K.; Wen, X.; Cai, J.; Kropf, E.; Iacovitti, L.; Pasinelli, P.; Trotti, D. Cell-to-Cell Transmission of Dipeptide Repeat Proteins Linked to C9orf72-ALS/FTD. Cell Rep. 2016, 17, 645–652. [Google Scholar] [CrossRef]
- Khosravi, B.; LaClair, K.D.; Riemenschneider, H.; Zhou, Q.; Frottin, F.; Mareljic, N.; Czuppa, M.; Farny, D.; Hartmann, H.; Michaelsen, M.; et al. Cell-to-cell transmission of C9orf72 poly-(Gly-Ala) triggers key features of ALS/FTD. EMBO J. 2020, 39, e102811. [Google Scholar] [CrossRef] [PubMed]
- Sackmann, C.; Sackmann, V.; Hallbeck, M. TDP-43 Is Efficiently Transferred Between Neuron-Like Cells in a Manner Enhanced by Preservation of Its N-Terminus but Independent of Extracellular Vesicles. Front. Neurosci. 2020, 14, 540. [Google Scholar] [CrossRef]
- Baek, Y.; Kim, H.; Lee, D.; Kim, D.; Jo, E.; Roh, S.H.; Ha, N.C. Structural insights into the role of reduced cysteine residues in SOD1 amyloid filament formation. Proc. Natl. Acad. Sci. USA 2025, 122, e2408582122. [Google Scholar] [CrossRef]
- Dong, S.; Liu, X.; Zhou, Y.; Li, J.; Qi, Z.; Wang, Z.; Yang, W.; Chen, X. Prognostic Value of Cerebrospinal Fluid and Serum Neurofilament Light Chain in Amyotrophic Lateral Sclerosis: A Correlation Study. Brain Behav. 2025, 15, e70256. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, F.; Garofalo, M.; Di Gerlando, R.; Rizzo, B.; Bordoni, M.; Scarian, E.; Viola, C.; Bettoni, V.; Fiamingo, G.; Tornabene, D.; et al. Whole transcriptome analysis of unmutated sporadic ALS patients’ peripheral blood reveals phenotype-specific gene expression signature. Neurobiol. Dis. 2025, 206, 106823. [Google Scholar] [CrossRef]
- Benatar, M.; Ostrow, L.W.; Lewcock, J.W.; Bennett, F.; Shefner, J.; Bowser, R.; Larkin, P.; Bruijn, L.; Wuu, J. Biomarker Qualification for Neurofilament Light Chain in Amyotrophic Lateral Sclerosis: Theory and Practice. Ann. Neurol. 2024, 95, 211–216. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Turner, M.R. Neurofilament light chain in drug development for amyotrophic lateral sclerosis: A critical appraisal. Brain 2023, 146, 2711–2716. [Google Scholar] [CrossRef]
- Benatar, M.; Zhang, L.; Wang, L.; Granit, V.; Statland, J.; Barohn, R.; Swenson, A.; Ravits, J.; Jackson, C.; Burns, T.M.; et al. Validation of serum neurofilaments as prognostic and potential pharmacodynamic biomarkers for ALS. Neurology 2020, 95, e59–e69. [Google Scholar] [CrossRef]
- Huang, F.; Zhu, Y.; Hsiao-Nakamoto, J.; Tang, X.; Dugas, J.C.; Moscovitch-Lopatin, M.; Glass, J.D.; Brown, R.H., Jr.; Ladha, S.S.; Lacomis, D.; et al. Longitudinal biomarkers in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Lombardi, V.; Malaspina, A. Neurofilament light: A candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann. Neurol. 2018, 84, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; O’Reilly, E.J.; Molsberry, S.; Kolonel, L.N.; Le Marchand, L.; Paganoni, S.; Schwarzschild, M.A.; Benkert, P.; Kuhle, J.; Ascherio, A. Prediagnostic Neurofilament Light Chain Levels in Amyotrophic Lateral Sclerosis. Neurology 2021, 97, e1466–e1474. [Google Scholar] [CrossRef] [PubMed]
- Gille, B.; De Schaepdryver, M.; Goossens, J.; Dedeene, L.; De Vocht, J.; Oldoni, E.; Goris, A.; Van Den Bosch, L.; Depreitere, B.; Claeys, K.G.; et al. Serum neurofilament light chain levels as a marker of upper motor neuron degeneration in patients with Amyotrophic Lateral Sclerosis. Neuropathol. Appl. Neurobiol. 2019, 45, 291–304. [Google Scholar] [CrossRef]
- Mondesert, E.; Delaby, C.; De La Cruz, E.; Kuhle, J.; Benkert, P.; Pradeilles, N.; Duchiron, M.; Morchikh, M.; Camu, W.; Cristol, J.P.; et al. Comparative Performances of 4 Serum NfL Assays, pTau181, and GFAP in Patients With Amyotrophic Lateral Sclerosis. Neurology 2025, 104, e213400. [Google Scholar] [CrossRef]
- Chatterjee, M.; Ozdemir, S.; Fritz, C.; Mobius, W.; Kleineidam, L.; Mandelkow, E.; Biernat, J.; Dogdu, C.; Peters, O.; Cosma, N.C.; et al. Plasma extracellular vesicle tau and TDP-43 as diagnostic biomarkers in FTD and ALS. Nat. Med. 2024, 30, 1771–1783. [Google Scholar] [CrossRef]
- Sugimoto, K.; Han, Y.; Song, Y.; Gao, Y. Correlational Analysis of ALS Progression and Serum NfL Measured by Simoa Assay in Chinese Patients. Front. Neurol. 2020, 11, 579094. [Google Scholar] [CrossRef]
- Fabes, J.; Matthews, L.; Filippini, N.; Talbot, K.; Jenkinson, M.; Turner, M.R. Quantitative FLAIR MRI in Amyotrophic Lateral Sclerosis. Acad. Radiol. 2017, 24, 1187–1194. [Google Scholar] [CrossRef]
- Thomas, E.V.; Han, C.; Kim, W.J.; Asress, S.; Li, Y.; Taylor, J.A.; Gearing, M.; Fournier, C.N.; McEachin, Z.T.; Seyfried, N.T.; et al. ALS plasma biomarkers reveal neurofilament and pTau correlate with disease onset and progression. Ann. Clin. Transl. Neurol. 2025. [Google Scholar] [CrossRef]
- Frohlich, A.; Pfaff, A.L.; Bubb, V.J.; Quinn, J.P.; Koks, S. Transcriptomic profiling of cerebrospinal fluid identifies ALS pathway enrichment and RNA biomarkers in MND individuals. Exp. Biol. Med. 2023, 248, 2325–2331. [Google Scholar] [CrossRef]
- Krishnan, G.; Raitcheva, D.; Bartlett, D.; Prudencio, M.; McKenna-Yasek, D.M.; Douthwright, C.; Oskarsson, B.E.; Ladha, S.; King, O.D.; Barmada, S.J.; et al. Poly(GR) and poly(GA) in cerebrospinal fluid as potential biomarkers for C9ORF72-ALS/FTD. Nat. Commun. 2022, 13, 2799. [Google Scholar] [CrossRef]
- Ansari, A.; Thibault, P.A.; Salapa, H.E.; Clarke, J.W.E.; Levin, M.C. Mutations in hnRNP A1 drive neurodegeneration and alternative RNA splicing of neuronal gene targets. Neurobiol. Dis. 2025, 206, 106814. [Google Scholar] [CrossRef]
- Deshaies, J.E.; Shkreta, L.; Moszczynski, A.J.; Sidibe, H.; Semmler, S.; Fouillen, A.; Bennett, E.R.; Bekenstein, U.; Destroismaisons, L.; Toutant, J.; et al. TDP-43 regulates the alternative splicing of hnRNP A1 to yield an aggregation-prone variant in amyotrophic lateral sclerosis. Brain 2018, 141, 1320–1333. [Google Scholar] [CrossRef] [PubMed]
- Noh, M.Y.; Kwon, M.S.; Oh, K.W.; Nahm, M.; Park, J.; Jin, H.K.; Bae, J.S.; Son, B.; Kim, S.H. miRNA-214 to predict progression and survival in ALS. J. Neurol. Neurosurg. Psychiatry 2025. [Google Scholar] [CrossRef] [PubMed]
- Waller, R.; Goodall, E.F.; Milo, M.; Cooper-Knock, J.; Da Costa, M.; Hobson, E.; Kazoka, M.; Wollff, H.; Heath, P.R.; Shaw, P.J.; et al. Serum miRNAs miR-206, 143–3p and 374b-5p as potential biomarkers for amyotrophic lateral sclerosis (ALS). Neurobiol. Aging 2017, 55, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Gschwendtberger, T.; Thau-Habermann, N.; von der Ohe, J.; Luo, T.; Hass, R.; Petri, S. Protective effects of EVs/exosomes derived from permanently growing human MSC on primary murine ALS motor neurons. Neurosci. Lett. 2023, 816, 137493. [Google Scholar] [CrossRef]
- Moreno-Garcia, L.; Miana-Mena, F.J.; Moreno-Martinez, L.; de la Torre, M.; Lunetta, C.; Tarlarini, C.; Zaragoza, P.; Calvo, A.C.; Osta, R. Inflammasome in ALS Skeletal Muscle: NLRP3 as a Potential Biomarker. Int. J. Mol. Sci. 2021, 22, 2523. [Google Scholar] [CrossRef]
- Si, Y.; Kazamel, M.; Benatar, M.; Wuu, J.; Kwon, Y.; Kwan, T.; Jiang, N.; Kentrup, D.; Faul, C.; Alesce, L.; et al. FGF23, a novel muscle biomarker detected in the early stages of ALS. Sci. Rep. 2021, 11, 12062. [Google Scholar] [CrossRef]
- Golini, E.; Rigamonti, M.; Iannello, F.; De Rosa, C.; Scavizzi, F.; Raspa, M.; Mandillo, S. A Non-invasive Digital Biomarker for the Detection of Rest Disturbances in the SOD1G93A Mouse Model of ALS. Front. Neurosci. 2020, 14, 896. [Google Scholar] [CrossRef]
- Straczkiewicz, M.; Burke, K.M.; Calcagno, N.; Premasiri, A.; Vieira, F.G.; Onnela, J.P.; Berry, J.D. Free-living monitoring of ALS progression in upper limbs using wearable accelerometers. J. Neuroeng. Rehabil. 2024, 21, 223. [Google Scholar] [CrossRef]
- Troger, J.; Baltes, J.; Baykara, E.; Kasper, E.; Kring, M.; Linz, N.; Robin, J.; Schafer, S.; Schneider, A.; Hermann, A. PROSA-a multicenter prospective observational study to develop low-burden digital speech biomarkers in ALS and FTD. Amyotroph. Lateral Scler. Front. Degener. 2023, 24, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Troger, J.; Dorr, F.; Schwed, L.; Linz, N.; Konig, A.; Thies, T.; Barbe, M.T.; Orozco-Arroyave, J.R.; Rusz, J. An automatic measure for speech intelligibility in dysarthrias-validation across multiple languages and neurological disorders. Front. Digit. Health 2024, 6, 1440986. [Google Scholar] [CrossRef]
- de Jong, S.; Huisman, M.; Sutedja, N.; van der Kooi, A.; de Visser, M.; Schelhaas, J.; van der Schouw, Y.; Veldink, J.; van den Berg, L. Endogenous female reproductive hormones and the risk of amyotrophic lateral sclerosis. J. Neurol. 2013, 260, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Manning, J.T.; Bundred, P.E. The ratio of 2nd to 4th digit length: A new predictor of disease predisposition? Med. Hypotheses 2000, 54, 855–857. [Google Scholar] [CrossRef]
- Vivekananda, U.; Manjalay, Z.R.; Ganesalingam, J.; Simms, J.; Shaw, C.E.; Leigh, P.N.; Turner, M.R.; Al-Chalabi, A. Low index-to-ring finger length ratio in sporadic ALS supports prenatally defined motor neuronal vulnerability. J. Neurol. Neurosurg. Psychiatry 2011, 82, 635–637. [Google Scholar] [CrossRef]
- de Jong, S.W.; Huisman, M.H.; Sutedja, N.A.; van der Kooi, A.J.; de Visser, M.; Schelhaas, H.J.; Fischer, K.; Veldink, J.H.; van den Berg, L.H. Smoking, alcohol consumption, and the risk of amyotrophic lateral sclerosis: A population-based study. Am. J. Epidemiol. 2012, 176, 233–239. [Google Scholar] [CrossRef]
- Bishop, J.Y.; Santiago-Torres, J.E.; Rimmke, N.; Flanigan, D.C. Smoking Predisposes to Rotator Cuff Pathology and Shoulder Dysfunction: A Systematic Review. Arthroscopy 2015, 31, 1598–1605. [Google Scholar] [CrossRef]
- Benatar, M.; Heiman-Patterson, T.D.; Cooper-Knock, J.; Brickman, D.; Casaletto, K.B.; Goutman, S.A.; Vinceti, M.; Dratch, L.; Arias, J.J.; Swidler, J.; et al. Guidance for clinical management of pathogenic variant carriers at elevated genetic risk for ALS/FTD. J. Neurol. Neurosurg. Psychiatry 2025, 96, 209–218. [Google Scholar] [CrossRef]
- Beard, J.D.; Kamel, F. Military service, deployments, and exposures in relation to amyotrophic lateral sclerosis etiology and survival. Epidemiol. Rev. 2015, 37, 55–70. [Google Scholar] [CrossRef]
- Qureshi, M.M.; Hayden, D.; Urbinelli, L.; Ferrante, K.; Newhall, K.; Myers, D.; Hilgenberg, S.; Smart, R.; Brown, R.H.; Cudkowicz, M.E. Analysis of factors that modify susceptibility and rate of progression in amyotrophic lateral sclerosis (ALS). Amyotroph. Lateral Scler. 2006, 7, 173–182. [Google Scholar] [CrossRef]
- Horner, R.D.; Kamins, K.G.; Feussner, J.R.; Grambow, S.C.; Hoff-Lindquist, J.; Harati, Y.; Mitsumoto, H.; Pascuzzi, R.; Spencer, P.S.; Tim, R.; et al. Occurrence of amyotrophic lateral sclerosis among Gulf War veterans. Neurology 2003, 61, 742–749. [Google Scholar] [CrossRef]
- Niazi, F.A.; Riggs, J.E. Association of ALS and Military Service: Reflection of Survival Bias due to the “Healthy Soldier Effect”? Mil. Med. 2020, 185, e5–e7. [Google Scholar] [CrossRef] [PubMed]
- Hamidou, B.; Couratier, P.; Besancon, C.; Nicol, M.; Preux, P.M.; Marin, B. Epidemiological evidence that physical activity is not a risk factor for ALS. Eur. J. Epidemiol. 2014, 29, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Arras, C.; Kylies, J.; Viezens, L.; Leonhardt, L.G. Conservative treatment of injuries to the cervical spine: Mobilization or immobilization. Unfallchirurgie 2025, 128, 96–102. [Google Scholar] [CrossRef]
- Huisman, M.H.; Seelen, M.; de Jong, S.W.; Dorresteijn, K.R.; van Doormaal, P.T.; van der Kooi, A.J.; de Visser, M.; Schelhaas, H.J.; van den Berg, L.H.; Veldink, J.H. Lifetime physical activity and the risk of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 976–981. [Google Scholar] [CrossRef]
- Armon, C.; Albert, S.M. A blow to the head trauma—ALS hypothesis. Neurology 2015, 84, 1728–1729. [Google Scholar] [CrossRef]
- Cao, G.; Wang, S.; Yu, J.; Wang, X.; Shi, X.; Yang, L.; Zhang, X.; Tong, P.; Tan, H. Outcomes of combined single-bundle anterior cruciate ligament reconstruction and anterolateral structure reconstruction through a modified single femoral tunnel. Int. Orthop. 2025, 49, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Falcao de Campos, C.; Gromicho, M.; Uysal, H.; Grosskreutz, J.; Kuzma-Kozakiewicz, M.; Oliveira Santos, M.; Pinto, S.; Petri, S.; Swash, M.; de Carvalho, M. Delayed Diagnosis and Diagnostic Pathway of ALS Patients in Portugal: Where Can We Improve? Front. Neurol. 2021, 12, 761355. [Google Scholar] [CrossRef]
- Williams, J.R.; Fitzhenry, D.; Grant, L.; Martyn, D.; Kerr, D.A. Diagnosis pathway for patients with amyotrophic lateral sclerosis: Retrospective analysis of the US Medicare longitudinal claims database. BMC Neurol. 2013, 13, 160. [Google Scholar] [CrossRef]
- Dave, K.D.; Oskarsson, B.; Yersak, J.; Krauss, R.; Heiman-Patterson, T.; Lomen-Hoerth, C.; Selig, W.K.D.; Halpern Paul, I.; Schaeffer, M.; Garcia-Trujillo, B.; et al. Contributions of neurologists to diagnostic timelines of ALS and thinkALS as an early referral instrument for clinicians. Amyotroph. Lateral Scler. Front. Degener. 2024, 1–10. [Google Scholar] [CrossRef]
- Babu, S.; Pioro, E.P.; Li, J.; Li, Y. Optimizing muscle selection for electromyography in amyotrophic lateral sclerosis. Muscle Nerve 2017, 56, 36–44. [Google Scholar] [CrossRef]
- Shayya, L.; Babu, S.; Pioro, E.P.; Li, J.; Li, Y. Distal Predominance of Electrodiagnostic Abnormalities in Early-Stage Amyotrophic Lateral Sclerosis. Muscle Nerve 2018, 58, 389–395. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L.; World Federation of Neurology Research Group on Motor Neuron, D. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor. Neuron. Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Karam, C.; Joyce, N.; Bedlack, R.; Carter, G.T. Comprehensive rehabilitative care across the spectrum of amyotrophic lateral sclerosis. NeuroRehabilitation 2015, 37, 53–68. [Google Scholar] [CrossRef]
- Maessen, M.; Veldink, J.H.; van den Berg, L.H.; Schouten, H.J.; van der Wal, G.; Onwuteaka-Philipsen, B.D. Requests for euthanasia: Origin of suffering in ALS, heart failure, and cancer patients. J. Neurol. 2010, 257, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Gosselt, I.K.; Nijboer, T.C.W.; Van Es, M.A. An overview of screening instruments for cognition and behavior in patients with ALS: Selecting the appropriate tool for clinical practice. Amyotroph. Lateral Scler. Front.l Degener. 2020, 21, 324–336. [Google Scholar] [CrossRef]
- Didcote, L.; Vitoratou, S.; Al-Chalabi, A.; Goldstein, L.H. Predicting ALS informant distress from cognitive and behavioural change in people with ALS. J. Neurol. 2025, 272, 144. [Google Scholar] [CrossRef] [PubMed]
- Dal Bello-Haas, V.; Kloos, A.D.; Mitsumoto, H. Physical therapy for a patient through six stages of amyotrophic lateral sclerosis. Phys. Ther. 1998, 78, 1312–1324. [Google Scholar] [CrossRef]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef]
- Dubbioso, R.; Spisto, M.; Hausdorff, J.M.; Aceto, G.; Iuzzolino, V.V.; Senerchia, G.; De Marco, S.; Marcuccio, L.; Femiano, C.; Iodice, R.; et al. Cognitive impairment is associated with gait variability and fall risk in amyotrophic lateral sclerosis. Eur. J. Neurol. 2023, 30, 3056–3067. [Google Scholar] [CrossRef]
- Lukac, M.; Luben, H.; Martin, A.E.; Simmons, Z.; Geronimo, A. Spatial-Temporal Analysis of Gait in Amyotrophic Lateral Sclerosis Using Foot-Worn Inertial Sensors: An Observational Study. Digit. Biomark. 2024, 8, 22–29. [Google Scholar] [CrossRef]
- Garcia-Casanova, P.H.; Vazquez-Costa, J.F. Advances in the early diagnosis of amyotrophic lateral sclerosis. Expert Rev. Neurother. 2025, 25, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Altayb Ismail, M.A.; Daffalla, I.; Singh, T.; Siddique, Q.R.; Almadhoun, M.; Irfan, R.; Saqib, M.; Haris, M.; Khan, Z.; Fernandes, J.G.F.; et al. Efficacy of Aerobic and Stretching Exercises in Managing Willis-Ekbom Disease (Restless Leg Syndrome) Among Hemodialysis Patients. Cureus 2024, 16, e71470. [Google Scholar] [CrossRef]
- Young, C.A.; Chaouch, A.; McDermott, C.J.; Al-Chalabi, A.; Chhetri, S.K.; Bidder, C.; Edmonds, E.; Ellis, C.; Annadale, J.; Wilde, L.; et al. Determinants and progression of stigma in amyotrophic lateral sclerosis/motor neuron disease. Amyotroph. Lateral Scler. Front. Degener. 2025, 1–11. [Google Scholar] [CrossRef]
- Pizzimenti, A.; Aragona, M.; Onesti, E.; Inghilleri, M. Depression, pain and quality of life in patients with amyotrophic lateral sclerosis: A cross-sectional study. Funct. Neurol. 2013, 28, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Rivera, I.; Ajroud-Driss, S.; Casey, P.; Heller, S.; Allen, J.; Siddique, T.; Sufit, R. Prevalence and characteristics of pain in early and late stages of ALS. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 369–372. [Google Scholar] [CrossRef]
- Ho, D.T.; Ruthazer, R.; Russell, J.A. Shoulder pain in amyotrophic lateral sclerosis. J. Clin. Neuromuscul. Dis. 2011, 13, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, X.; Ding, X.; Song, M.; Sui, K. Analysis of clinical and electrophysiological characteristics of 150 patients with amyotrophic lateral sclerosis in China. Neurol. Sci. 2019, 40, 363–369. [Google Scholar] [CrossRef]
- Dietrich, M.; Hartung, H.P.; Albrecht, P. Neuroprotective Properties of 4-Aminopyridine. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e976. [Google Scholar] [CrossRef]
- Firstenfeld, A.J. A positive effect of Cerebrolysin on motor functions and spasticity in ALS with limb or bulbar onset is questionable. J. Med. Life 2024, 17, 243. [Google Scholar] [CrossRef]
- Ribeiro, S. Iyengar yoga therapy as an intervention for cramp management in individuals with amyotrophic lateral sclerosis: Three case reports. J. Altern. Complement. Med. 2014, 20, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Desport, J.C.; Preux, P.M.; Truong, T.C.; Vallat, J.M.; Sautereau, D.; Couratier, P. Nutritional status is a prognostic factor for survival in ALS patients. Neurology 1999, 53, 1059–1063. [Google Scholar] [CrossRef]
- James, E.; Ellis, C.; Brassington, R.; Sathasivam, S.; Young, C.A. Treatment for sialorrhea (excessive saliva) in people with motor neuron disease/amyotrophic lateral sclerosis. Cochrane. Database. Syst. Rev. 2022, 5, CD006981. [Google Scholar] [CrossRef]
- Gibbons, C.J.; Thornton, E.W.; Young, C.A. The patient experience of fatigue in motor neurone disease. Front. Psychol. 2013, 4, 788. [Google Scholar] [CrossRef]
- Lo Coco, D.; Piccoli, F.; La Bella, V. Restless legs syndrome in patients with amyotrophic lateral sclerosis. Mov. Disord. 2010, 25, 2658–2661. [Google Scholar] [CrossRef]
- Lo Coco, D.; La Bella, V. Fatigue, sleep, and nocturnal complaints in patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2012, 19, 760–763. [Google Scholar] [CrossRef]
- Dubbioso, R.; Spisto, M.; Verde, L.; Iuzzolino, V.V.; Senerchia, G.; Salvatore, E.; De Pietro, G.; De Falco, I.; Sannino, G. Voice signals database of ALS patients with different dysarthria severity and healthy controls. Sci. Data 2024, 11, 800. [Google Scholar] [CrossRef]
- Stegmann, G.; Charles, S.; Liss, J.; Shefner, J.; Rutkove, S.; Berisha, V. A speech-based prognostic model for dysarthria progression in ALS. Amyotroph. Lateral Scler. Front. Degener. 2023, 24, 599–604. [Google Scholar] [CrossRef]
- Tomik, B.; Guiloff, R.J. Dysarthria in amyotrophic lateral sclerosis: A review. Amyotroph. Lateral Scler. 2010, 11, 4–15. [Google Scholar] [CrossRef]
- Kleinerova, J.; Garcia-Gallardo, A.; Tacheva, A.; Bede, P. Subcortical grey matter involvement in ALS and PLS—Vulnerable hubs of cortico-cortical and cortico-basal circuits: Extrapyramidal, cognitive, bulbar and respiratory correlates. Amyotroph. Lateral Scler. Front. Degener. 2025, 26, 1–4. [Google Scholar] [CrossRef]
- Hermann, W.; Langner, S.; Freigang, M.; Fischer, S.; Storch, A.; Gunther, R.; Hermann, A. Affection of Respiratory Muscles in ALS and SMA. J. Clin. Med. 2022, 11, 1163. [Google Scholar] [CrossRef] [PubMed]
- Faull, C.; Rowe Haynes, C.; Oliver, D. Issues for palliative medicine doctors surrounding the withdrawal of non-invasive ventilation at the request of a patient with motor neurone disease: A scoping study. BMJ Support Palliat Care 2014, 4, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Gouveia, C.; Araujo, L.; Freitas, S.; Correia, J.; Passos, V.; Camacho, G.; Gomes, L.; Fragoeiro, H.; Camacho, C.; Chambino, B. A Palliative Care Approach to Amyotrophic Lateral Sclerosis. Cureus 2023, 15, e51048. [Google Scholar] [CrossRef]
- Washington, K.T.; Mechling, C.A.; Pitzer, K.A.; Maiser, S.; Mehta, A.K. Identifying the Unmet Needs of People Living With Amyotrophic Lateral Sclerosis: A National Survey to Inform Interdisciplinary Palliative Care. Am. J. Hosp. Palliat Care 2025, 42, 326–333. [Google Scholar] [CrossRef]
- Lee, K.K.; Davenport, P.W.; Smith, J.A.; Irwin, R.S.; McGarvey, L.; Mazzone, S.B.; Birring, S.S.; Panel, C.E.C. Global Physiology and Pathophysiology of Cough: Part 1: Cough Phenomenology—CHEST Guideline and Expert Panel Report. Chest 2021, 159, 282–293. [Google Scholar] [CrossRef] [PubMed]
- McGarvey, L.; Rubin, B.K.; Ebihara, S.; Hegland, K.; Rivet, A.; Irwin, R.S.; Bolser, D.C.; Chang, A.B.; Gibson, P.G.; Mazzone, S.B.; et al. Global Physiology and Pathophysiology of Cough: Part 2. Demographic and Clinical Considerations: CHEST Expert Panel Report. Chest 2021, 160, 1413–1423. [Google Scholar] [CrossRef]
- Britton, D.; Benditt, J.O.; Merati, A.L.; Miller, R.M.; Stepp, C.E.; Boitano, L.; Hu, A.; Ciol, M.A.; Yorkston, K.M. Associations between laryngeal and cough dysfunction in motor neuron disease with bulbar involvement. Dysphagia 2014, 29, 637–646. [Google Scholar] [CrossRef]
- Ferraro, P.M.; Mollar, E.; Melissari, L.; Buscema, M.; Bagnoli, E.; Cabona, C.; Gemelli, C.; Vignolo, M.; Maranzana, C.; Marogna, M.; et al. Longitudinal respiratory trajectories in motor neuron disease phenotypes: Multiparametric characterization and clinical management. Respir. Med. 2025, 239, 108003. [Google Scholar] [CrossRef]
- Hopkins, L.C.; Tatarian, G.T.; Pianta, T.F. Management of ALS: Respiratory care. Neurology 1996, 47, S123–S125. [Google Scholar] [CrossRef]
- Tankere, P.; Cascarano, E.; Saint Raymond, C.; Mallaret, M.; Toribio Ruiz, C.; Herquelot, E.; Denis, H.; Cals Maurette, M.; Tamisier, R.; Pepin, J.L. Care trajectories and adherence to respiratory management recommendations in persons living with amyotrophic lateral sclerosis: A ten-year cohort study in a French tertiary university centre. Amyotroph. Lateral Scler. Front. Degener. 2025, 1–9. [Google Scholar] [CrossRef]
- Chatwin, M.; Toussaint, M.; Goncalves, M.R.; Sheers, N.; Mellies, U.; Gonzales-Bermejo, J.; Sancho, J.; Fauroux, B.; Andersen, T.; Hov, B.; et al. Airway clearance techniques in neuromuscular disorders: A state of the art review. Respir. Med. 2018, 136, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Cazzolli, P.A.; Brooks, B.R.; Nakayama, Y.; Lewarski, J.S.; McKim, D.A.; Holt, S.L.; Chatburn, R.L. The Oral Secretion Scale and Prognostic Factors for Survival in Subjects With Amyotrophic Lateral Sclerosis. Respir. Care 2020, 65, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Benditt, J.O. Saliva, Swallowing, and Breathing: The Ultimate Challenge of Amyotrophic Lateral Sclerosis. Respir. Care 2020, 65, 1221–1222. [Google Scholar] [CrossRef]
- Plowman, E.K.; Watts, S.A.; Robison, R.; Tabor, L.; Dion, C.; Gaziano, J.; Vu, T.; Gooch, C. Voluntary Cough Airflow Differentiates Safe Versus Unsafe Swallowing in Amyotrophic Lateral Sclerosis. Dysphagia 2016, 31, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Sales de Campos, P.; Olsen, W.L.; Wymer, J.P.; Smith, B.K. Respiratory therapies for Amyotrophic Lateral Sclerosis: A state of the art review. Chron. Respir. Dis. 2023, 20, 14799731231175915. [Google Scholar] [CrossRef]
- van Eijk, R.P.A.; de Jongh, A.D.; Nikolakopoulos, S.; McDermott, C.J.; Eijkemans, M.J.C.; Roes, K.C.B.; van den Berg, L.H. An old friend who has overstayed their welcome: The ALSFRS-R total score as primary endpoint for ALS clinical trials. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 300–307. [Google Scholar] [CrossRef]
- van Eijk, R.P.A.; Nikolakopoulos, S.; Roes, K.C.B.; Kendall, L.; Han, S.S.; Lavrov, A.; Epstein, N.; Kliest, T.; de Jongh, A.D.; Westeneng, H.J.; et al. Innovating Clinical Trials for Amyotrophic Lateral Sclerosis: Challenging the Established Order. Neurology 2021, 97, 528–536. [Google Scholar] [CrossRef]
- Pirola, A.; De Mattia, E.; Lizio, A.; Sannicolo, G.; Carraro, E.; Rao, F.; Sansone, V.; Lunetta, C. The prognostic value of spirometric tests in Amyotrophic Lateral Sclerosis patients. Clin. Neurol. Neurosurg. 2019, 184, 105456. [Google Scholar] [CrossRef]
- Olofsson, J.; Bergstrom, S.; Mravinacova, S.; Klappe, U.; Oijerstedt, L.; Zetterberg, H.; Blennow, K.; Ingre, C.; Nilsson, P.; Manberg, A. Cerebrospinal fluid levels of NfM in relation to NfL and pNfH as prognostic markers in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2025, 26, 113–123. [Google Scholar] [CrossRef]
- Eshima, J.; O’Connor, S.A.; Marschall, E.; Consortium, N.A.; Bowser, R.; Plaisier, C.L.; Smith, B.S. Molecular subtypes of ALS are associated with differences in patient prognosis. Nat. Commun. 2023, 14, 95. [Google Scholar] [CrossRef]
- Foldvari, K.M.; Stolee, P.; Neiterman, E.; Boscart, V.; Tong, C. “...but I know something’s not right here”: Exploring the diagnosis and disclosure experiences of persons living with ALS. PLoS ONE 2024, 19, e0301249. [Google Scholar] [CrossRef]
- Lajoie, I.; Canadian, A.L.S.N.C.; Kalra, S.; Dadar, M. Regional Cerebral Atrophy Contributes to Personalized Survival Prediction in Amyotrophic Lateral Sclerosis: A Multicentre, Machine Learning, Deformation-Based Morphometry Study. Ann. Neurol. 2025. [Google Scholar] [CrossRef] [PubMed]
- Foster, L.A.; Salajegheh, M.K. Motor Neuron Disease: Pathophysiology, Diagnosis, and Management. Am. J. Med. 2019, 132, 32–37. [Google Scholar] [CrossRef]
- Bourke, S.C.; McColl, E.; Shaw, P.J.; Gibson, G.J. Validation of quality of life instruments in ALS. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2004, 5, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Burke, T.; Galvin, M.; Pinto-Grau, M.; Lonergan, K.; Madden, C.; Mays, I.; Carney, S.; Hardiman, O.; Pender, N. Caregivers of patients with amyotrophic lateral sclerosis: Investigating quality of life, caregiver burden, service engagement, and patient survival. J. Neurol. 2017, 264, 898–904. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Lacomblez, L.; Bensimon, G.; Leigh, P.N.; Guillet, P.; Powe, L.; Durrleman, S.; Delumeau, J.C.; Meininger, V. A confirmatory dose-ranging study of riluzole in ALS. ALS/Riluzole Study Group-II. Neurology 1996, 47, S242–S250. [Google Scholar] [CrossRef] [PubMed]
- Takei, K.; Takahashi, F.; Liu, S.; Tsuda, K.; Palumbo, J. Post-hoc analysis of randomised, placebo-controlled, double-blind study (MCI186-19) of edaravone (MCI-186) in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 49–54. [Google Scholar] [CrossRef]
- Hamad, A.A.; Alkhawaldeh, I.M.; Nashwan, A.J.; Meshref, M.; Imam, Y. Tofersen for SOD1 amyotrophic lateral sclerosis: A systematic review and meta-analysis. Neurol. Sci. 2025. [Google Scholar] [CrossRef]
- Ludolph, A.; Wiesenfarth, M. Tofersen and other antisense oligonucleotides in ALS. Ther. Adv. Neurol. Disord. 2025, 18, 17562864251313915. [Google Scholar] [CrossRef]
- Naveed, A.; Usmani, W.A.; Vandara, M.P.; Karmani, V.K. Tofersen for Amyotrophic Lateral Sclerosis: A Step Forward or Another False Hope? J. Coll. Physicians Surg. Pak. 2025, 35, 259–260. [Google Scholar] [CrossRef]
- Smith, S.E.; McCoy-Gross, K.; Malcolm, A.; Oranski, J.; Markway, J.W.; Miller, T.M.; Bucelli, R.C. Tofersen treatment leads to sustained stabilization of disease in SOD1 ALS in a “real-world” setting. Ann. Clin. Transl. Neurol. 2025, 12, 311–319. [Google Scholar] [CrossRef]
- Gil, J.; Funalot, B.; Verschueren, A.; Danel-Brunaud, V.; Camu, W.; Vandenberghe, N.; Desnuelle, C.; Guy, N.; Camdessanche, J.P.; Cintas, P.; et al. Causes of death amongst French patients with amyotrophic lateral sclerosis: A prospective study. Eur. J. Neurol. 2008, 15, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Sojdeh, S.; Safarkhani, M.; Daneshgar, H.; Aldhaher, A.; Heidari, G.; Nazarzadeh Zare, E.; Iravani, S.; Zarrabi, A.; Rabiee, N. Promising breakthroughs in amyotrophic lateral sclerosis treatment through nanotechnology’s unexplored frontier. Eur. J. Med. Chem. 2025, 282, 117080. [Google Scholar] [CrossRef]
- Fang, T.; Al Khleifat, A.; Meurgey, J.H.; Jones, A.; Leigh, P.N.; Bensimon, G.; Al-Chalabi, A. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: A retrospective analysis of data from a dose-ranging study. Lancet Neurol. 2018, 17, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.A.; Jackson, C.E.; Heiman-Patterson, T.D.; Bettica, P.; Brooks, B.R.; Pioro, E.P. Real-world evidence of riluzole effectiveness in treating amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Jiang, S.; Xu, R. Clinical features and progress in diagnosis and treatment of amyotrophic lateral sclerosis. Ann. Med. 2024, 56, 2399962. [Google Scholar] [CrossRef]
- Vucic, S.; Lin, C.S.; Cheah, B.C.; Murray, J.; Menon, P.; Krishnan, A.V.; Kiernan, M.C. Riluzole exerts central and peripheral modulating effects in amyotrophic lateral sclerosis. Brain 2013, 136, 1361–1370. [Google Scholar] [CrossRef]
- Edaravone Als 16 Study, G. A post-hoc subgroup analysis of outcomes in the first phase III clinical study of edaravone (MCI-186) in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 11–19. [Google Scholar] [CrossRef]
- Shefner, J.; Heiman-Patterson, T.; Pioro, E.P.; Wiedau-Pazos, M.; Liu, S.; Zhang, J.; Agnese, W.; Apple, S. Long-term edaravone efficacy in amyotrophic lateral sclerosis: Post-hoc analyses of Study 19 (MCI186-19). Muscle Nerve 2020, 61, 218–221. [Google Scholar] [CrossRef]
- Lunetta, C.; Moglia, C.; Lizio, A.; Caponnetto, C.; Dubbioso, R.; Giannini, F.; Mata, S.; Mazzini, L.; Sabatelli, M.; Siciliano, G.; et al. The Italian multicenter experience with edaravone in amyotrophic lateral sclerosis. J. Neurol. 2020, 267, 3258–3267. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, D.; Makam, A.N.; Suh, K.; McKenna, A.; Carlson, J.J.; Richardson, M.; Rind, D.M.; Pearson, S.D. The effectiveness and value of AMX0035 and oral edaravone for amyotrophic lateral sclerosis: A summary from the Institute for Clinical and Economic Review’s Midwest Comparative Effectiveness Public Advisory Council. J. Manag. Care Spec. Pharm. 2023, 29, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Thisted, R.A.; Appel, S.H.; Bradley, W.G.; Olney, R.K.; Berg, J.E.; Pope, L.E.; Smith, R.A.; Group, A.-A.S. Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: A randomized trial. Neurology 2004, 63, 1364–1370. [Google Scholar] [CrossRef]
- Xu, Q.; Cho, J.; Ben Chaouch, Z.; Lo, A.W. Incorporating patient preferences and burden-of-disease in evaluating ALS drug candidate AMX0035: A Bayesian decision analysis perspective. Amyotroph. Lateral Scler. Front. Degener. 2023, 24, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Bowser, R.; An, J.; Mehta, L.; Chen, J.; Timmons, J.; Cudkowicz, M.; Paganoni, S. Effect of sodium phenylbutyrate and taurursodiol on plasma concentrations of neuroinflammatory biomarkers in amyotrophic lateral sclerosis: Results from the CENTAUR trial. J. Neurol. Neurosurg. Psychiatry 2024, 95, 605–608. [Google Scholar] [CrossRef]
- Paganoni, S.; Hendrix, S.; Dickson, S.P.; Knowlton, N.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; et al. Effect of sodium phenylbutyrate/taurursodiol on tracheostomy/ventilation-free survival and hospitalisation in amyotrophic lateral sclerosis: Long-term results from the CENTAUR trial. J. Neurol. Neurosurg. Psychiatry 2022, 93, 871–875. [Google Scholar] [CrossRef]
- Paganoni, S.; Hendrix, S.; Dickson, S.P.; Knowlton, N.; Macklin, E.A.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; et al. Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve 2021, 63, 31–39. [Google Scholar] [CrossRef]
- Paganoni, S.; Watkins, C.; Cawson, M.; Hendrix, S.; Dickson, S.P.; Knowlton, N.; Timmons, J.; Manuel, M.; Cudkowicz, M. Survival analyses from the CENTAUR trial in amyotrophic lateral sclerosis: Evaluating the impact of treatment crossover on outcomes. Muscle Nerve 2022, 66, 136–141. [Google Scholar] [CrossRef]
- Ketabforoush, A.; Faghihi, F.; Azedi, F.; Ariaei, A.; Habibi, M.A.; Khalili, M.; Ashtiani, B.H.; Joghataei, M.T.; Arnold, W.D. Sodium Phenylbutyrate and Tauroursodeoxycholic Acid: A Story of Hope Turned to Disappointment in Amyotrophic Lateral Sclerosis Treatment. Clin. Drug. Investig. 2024, 44, 495–512. [Google Scholar] [CrossRef]
- Sulek, A. Secretome—The role of extracellular vesicles in the pathogenesis and therapy of neurodegenerative diseases. Postep. Psychiatr. Neurol. 2024, 33, 147–162. [Google Scholar] [CrossRef]
- Faller, K.M.E.; Chaytow, H.; Gillingwater, T.H. Targeting common disease pathomechanisms to treat amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2025, 21, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Ruffo, P.; Traynor, B.J.; Conforti, F.L. Advancements in genetic research and RNA therapy strategies for amyotrophic lateral sclerosis (ALS): Current progress and future prospects. J. Neurol. 2025, 272, 233. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.F.; Jiang, X.C.; Gao, J.Q. Present insights into the progress in gene therapy delivery systems for central nervous system diseases. Int. J. Pharm. 2025, 669, 125069. [Google Scholar] [CrossRef]
- Arnold, C. Tailored treatment for ALS poised to move ahead. Nat. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Andrews, J.; Farhan, S. Recent advances in the genetics of familial and sporadic ALS. Int. Rev. Neurobiol. 2024, 176, 49–74. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Brown, R.H., Jr. Finding a Treatment for ALS—Will Gene Editing Cut It? N. Engl. J. Med. 2018, 378, 1454–1456. [Google Scholar] [CrossRef]
- Gupta, D.; Vagha, S.; Dhingra, H.; Shirsath, H. Advances in Understanding and Treating Amyotrophic Lateral Sclerosis (ALS): A Comprehensive Review. Cureus 2023, 15, e48691. [Google Scholar] [CrossRef]
- Cappella, M.; Ciotti, C.; Cohen-Tannoudji, M.; Biferi, M.G. Gene Therapy for ALS-A Perspective. Int. J. Mol. Sci. 2019, 20, 4388. [Google Scholar] [CrossRef]
- Miller, T.M.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: A phase 1, randomised, first-in-man study. Lancet Neurol. 2013, 12, 435–442. [Google Scholar] [CrossRef]
- Gendron, T.F.; Chew, J.; Stankowski, J.N.; Hayes, L.R.; Zhang, Y.J.; Prudencio, M.; Carlomagno, Y.; Daughrity, L.M.; Jansen-West, K.; Perkerson, E.A.; et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaai7866. [Google Scholar] [CrossRef]
- Korobeynikov, V.A.; Lyashchenko, A.K.; Blanco-Redondo, B.; Jafar-Nejad, P.; Shneider, N.A. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat. Med. 2022, 28, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Akyuz, E.; Aslan, F.S.; Gokce, E.; Ilmaz, O.; Topcu, F.; Kakac, S. Extracellular vesicle and CRISPR gene therapy: Current applications in Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and Huntington’s disease. Eur. J. Neurosci. 2024, 60, 6057–6090. [Google Scholar] [CrossRef]
- Naveed, M.; Aqib Shabbir, M.; Aziz, T.; Hurraira, H.M.; Fatima Zaidi, S.; Athar, R.; Chattha, H.A.; Alharbi, M.; Alshammari, A.; Alasmari, A.F. CRISPR-Cas9 guided rna based model for the treatment of Amyotrophic Lateral Sclerosis: A progressive neurodegenerative disorder. Acta. Biochim. Pol. 2023, 70, 643–653. [Google Scholar] [CrossRef]
- Shi, Y.; Zhao, Y.; Lu, L.; Gao, Q.; Yu, D.; Sun, M. CRISPR/Cas9: Implication for modeling and therapy of amyotrophic lateral sclerosis. Front. Neurosci. 2023, 17, 1223777. [Google Scholar] [CrossRef]
- Gaj, T.; Ojala, D.S.; Ekman, F.K.; Byrne, L.C.; Limsirichai, P.; Schaffer, D.V. In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci. Adv. 2017, 3, eaar3952. [Google Scholar] [CrossRef]
- Mueller, C.; Berry, J.D.; McKenna-Yasek, D.M.; Gernoux, G.; Owegi, M.A.; Pothier, L.M.; Douthwright, C.L.; Gelevski, D.; Luppino, S.D.; Blackwood, M.; et al. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N. Engl. J. Med. 2020, 383, 151–158. [Google Scholar] [CrossRef]
- Deng, L.; Chu, Z.; Liu, P.; Li, B.; Lei, G.; Li, S.; Ma, Y.; Dang, Y. Effects of brain-derived neurotrophic factor and adeno-associated viral vector on morphine-induced condition through target concentration changes in the ventral tegmental area and nucleus accumbens. Behav. Brain Res. 2023, 445, 114385. [Google Scholar] [CrossRef] [PubMed]
- Petrou, P.; Argov, A.; Lennon, V.A.; Gotkine, M.; Kassis, I.; Vaknin-Dembinsky, A.; Ben-Hur, T.; Offen, D.; Abramsky, O.; Melamed, E.; et al. Rare combination of myasthenia and motor neuronopathy, responsive to Msc-Ntf stem cell therapy. Muscle Nerve 2014, 49, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Boido, M.; Piras, A.; Valsecchi, V.; Spigolon, G.; Mareschi, K.; Ferrero, I.; Vizzini, A.; Temi, S.; Mazzini, L.; Fagioli, F.; et al. Human mesenchymal stromal cell transplantation modulates neuroinflammatory milieu in a mouse model of amyotrophic lateral sclerosis. Cytotherapy 2014, 16, 1059–1072. [Google Scholar] [CrossRef]
- Forostyak, S.; Homola, A.; Turnovcova, K.; Svitil, P.; Jendelova, P.; Sykova, E. Intrathecal delivery of mesenchymal stromal cells protects the structure of altered perineuronal nets in SOD1 rats and amends the course of ALS. Stem. Cells 2014, 32, 3163–3172. [Google Scholar] [CrossRef]
- Marconi, S.; Bonaconsa, M.; Scambi, I.; Squintani, G.M.; Rui, W.; Turano, E.; Ungaro, D.; D’Agostino, S.; Barbieri, F.; Angiari, S.; et al. Systemic treatment with adipose-derived mesenchymal stem cells ameliorates clinical and pathological features in the amyotrophic lateral sclerosis murine model. Neuroscience 2013, 248, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Milanese, M.; Principato, M.C.; Morando, S.; Bonifacino, T.; Vergani, L.; Giunti, D.; Voci, A.; Carminati, E.; Giribaldi, F.; et al. Intravenous mesenchymal stem cells improve survival and motor function in experimental amyotrophic lateral sclerosis. Mol. Med. 2012, 18, 794–804. [Google Scholar] [CrossRef]
- Vercelli, A.; Mereuta, O.M.; Garbossa, D.; Muraca, G.; Mareschi, K.; Rustichelli, D.; Ferrero, I.; Mazzini, L.; Madon, E.; Fagioli, F. Human mesenchymal stem cell transplantation extends survival, improves motor performance and decreases neuroinflammation in mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 31, 395–405. [Google Scholar] [CrossRef]
- Zhao, C.P.; Zhang, C.; Zhou, S.N.; Xie, Y.M.; Wang, Y.H.; Huang, H.; Shang, Y.C.; Li, W.Y.; Zhou, C.; Yu, M.J.; et al. Human mesenchymal stromal cells ameliorate the phenotype of SOD1-G93A ALS mice. Cytotherapy 2007, 9, 414–426. [Google Scholar] [CrossRef]
- Glass, J.D.; Boulis, N.M.; Johe, K.; Rutkove, S.B.; Federici, T.; Polak, M.; Kelly, C.; Feldman, E.L. Lumbar intraspinal injection of neural stem cells in patients with amyotrophic lateral sclerosis: Results of a phase I trial in 12 patients. Stem. Cells 2012, 30, 1144–1151. [Google Scholar] [CrossRef]
- Lewis, C.M.; Suzuki, M. Therapeutic applications of mesenchymal stem cells for amyotrophic lateral sclerosis. Stem. Cell Res. Ther. 2014, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, L.; Ferrero, I.; Luparello, V.; Rustichelli, D.; Gunetti, M.; Mareschi, K.; Testa, L.; Stecco, A.; Tarletti, R.; Miglioretti, M.; et al. Mesenchymal stem cell transplantation in amyotrophic lateral sclerosis: A Phase I clinical trial. Exp. Neurol. 2010, 223, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, L.; Gelati, M.; Profico, D.C.; Soraru, G.; Ferrari, D.; Copetti, M.; Muzi, G.; Ricciolini, C.; Carletti, S.; Giorgi, C.; et al. Results from Phase I Clinical Trial with Intraspinal Injection of Neural Stem Cells in Amyotrophic Lateral Sclerosis: A Long-Term Outcome. Stem. Cells Transl. Med. 2019, 8, 887–897. [Google Scholar] [CrossRef]
- Barczewska, M.; Grudniak, M.; Maksymowicz, S.; Siwek, T.; Oldak, T.; Jezierska-Wozniak, K.; Gladysz, D.; Maksymowicz, W. Safety of intrathecal injection of Wharton’s jelly-derived mesenchymal stem cells in amyotrophic lateral sclerosis therapy. Neural. Regen. Res. 2019, 14, 313–318. [Google Scholar] [CrossRef]
- Nabavi, S.M.; Arab, L.; Jarooghi, N.; Bolurieh, T.; Abbasi, F.; Mardpour, S.; Azimyian, V.; Moeininia, F.; Maroufizadeh, S.; Sanjari, L.; et al. Safety, Feasibility of Intravenous and Intrathecal Injection of Autologous Bone Marrow Derived Mesenchymal Stromal Cells in Patients with Amyotrophic Lateral Sclerosis: An Open Label Phase I Clinical Trial. Cell J. 2019, 20, 592–598. [Google Scholar] [CrossRef]
- Oh, K.W.; Noh, M.Y.; Kwon, M.S.; Kim, H.Y.; Oh, S.I.; Park, J.; Kim, H.J.; Ki, C.S.; Kim, S.H. Repeated Intrathecal Mesenchymal Stem Cells for Amyotrophic Lateral Sclerosis. Ann. Neurol. 2018, 84, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Petrou, P.; Gothelf, Y.; Argov, Z.; Gotkine, M.; Levy, Y.S.; Kassis, I.; Vaknin-Dembinsky, A.; Ben-Hur, T.; Offen, D.; Abramsky, O.; et al. Safety and Clinical Effects of Mesenchymal Stem Cells Secreting Neurotrophic Factor Transplantation in Patients With Amyotrophic Lateral Sclerosis: Results of Phase 1/2 and 2a Clinical Trials. JAMA Neurol. 2016, 73, 337–344. [Google Scholar] [CrossRef]
- Staff, N.P.; Madigan, N.N.; Morris, J.; Jentoft, M.; Sorenson, E.J.; Butler, G.; Gastineau, D.; Dietz, A.; Windebank, A.J. Safety of intrathecal autologous adipose-derived mesenchymal stromal cells in patients with ALS. Neurology 2016, 87, 2230–2234. [Google Scholar] [CrossRef]
- Scott, S.; Kranz, J.E.; Cole, J.; Lincecum, J.M.; Thompson, K.; Kelly, N.; Bostrom, A.; Theodoss, J.; Al-Nakhala, B.M.; Vieira, F.G.; et al. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph. Lateral Scler. 2008, 9, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Trudler, D.; Ghatak, S.; Lipton, S.A. Emerging hiPSC Models for Drug Discovery in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 8196. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Kitaoka, S.; Tsukita, K.; Naitoh, M.; Takahashi, K.; Yamamoto, T.; Adachi, F.; Kondo, T.; Okita, K.; Asaka, I.; et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 2012, 4, 145ra104. [Google Scholar] [CrossRef]
- Miller, R.G.; Jackson, C.E.; Kasarskis, E.J.; England, J.D.; Forshew, D.; Johnston, W.; Kalra, S.; Katz, J.S.; Mitsumoto, H.; Rosenfeld, J.; et al. Practice parameter update: The care of the patient with amyotrophic lateral sclerosis: Multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2009, 73, 1227–1233. [Google Scholar] [CrossRef]
- Mayadev, A.S.; Weiss, M.D.; Distad, B.J.; Krivickas, L.S.; Carter, G.T. The amyotrophic lateral sclerosis center: A model of multidisciplinary management. Phys. Med. Rehabil. Clin. N. Am. 2008, 19, 619–631. [Google Scholar] [CrossRef]
- Lero, C.M.; Yang, A.; Everett, E.; Pitzer, K.A.; McCoy Gross, K.; Washington, K.T. Associations Between End-Stage ALS Care and Specialty Palliative Care: A Hypothesis-Generating Study. Muscle Nerve 2025, 71, 632–638. [Google Scholar] [CrossRef]
- Walsh, S.; Simmons, Z.; Miyamoto, S.; Geronimo, A. A nurse coaching intervention to improve support to individuals living with ALS. Amyotroph. Lateral Scler. Front. Degener. 2025, 26, 22–28. [Google Scholar] [CrossRef]
- Macpherson, C.E.; Bassile, C.C. Pulmonary Physical Therapy Techniques to Enhance Survival in Amyotrophic Lateral Sclerosis: A Systematic Review. J. Neurol. Phys. Ther. 2016, 40, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Aboussouan, L.S. Mechanisms of exercise limitation and pulmonary rehabilitation for patients with neuromuscular disease. Chron. Respir. Dis. 2009, 6, 231–249. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, J.P.; Silvestre, R.; Pinto, A.C.; de Carvalho, M. Exercise and amyotrophic lateral sclerosis. Neurol. Sci. 2012, 33, 9–15. [Google Scholar] [CrossRef]
- Thakore, N.J.; Drawert, B.J.; Lapin, B.R.; Pioro, E.P. Progressive arm muscle weakness in ALS follows the same sequence regardless of onset site: Use of TOMS, a novel analytic method to track limb strength. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2021, 22, 380–387. [Google Scholar] [CrossRef]
- Wimmer, N.; Muller, H.P.; Metze, P.; Rasche, V.; Ludolph, A.C.; Kassubek, J. The central pattern of weakness of ALS: Morphological correlates in whole-body muscle MRI. Ann. Clin. Transl. Neurol. 2024, 11, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Jawdat, O.; Rucker, J.; Nakano, T.; Takeno, K.; Statland, J.; Pasnoor, M.; Dimachkie, M.M.; Sabus, C.; Badawi, Y.; Hunt, S.L.; et al. Resistance exercise in early-stage ALS patients, ALSFRS-R, Sickness Impact Profile ALS-19, and muscle transcriptome: A pilot study. Sci. Rep. 2024, 14, 21729. [Google Scholar] [CrossRef]
- An, M.H.; You, S.C.; Park, R.W.; Lee, S. Using an Extended Technology Acceptance Model to Understand the Factors Influencing Telehealth Utilization After Flattening the COVID-19 Curve in South Korea: Cross-sectional Survey Study. JMIR Med. Inform. 2021, 9, e25435. [Google Scholar] [CrossRef]
- Kilmer, D.D. Response to aerobic exercise training in humans with neuromuscular disease. Am. J. Phys. Med. Rehabil. 2002, 81, S148–S150. [Google Scholar] [CrossRef]
- Dalbello-Haas, V.; Florence, J.M.; Krivickas, L.S. Therapeutic exercise for people with amyotrophic lateral sclerosis or motor neuron disease. Cochrane. Database. Syst. Rev. 2008, 5, CD005229. [Google Scholar] [CrossRef]
- Voet, N.B.; van der Kooi, E.L.; Riphagen, I.; Lindeman, E.; van Engelen, B.G.; Geurts, A. Strength training and aerobic exercise training for muscle disease. Cochrane Database Syst. Rev. 2010, 1, CD003907. [Google Scholar] [CrossRef]
- Voet, N.B.M. Exercise in neuromuscular disorders: A promising intervention. Acta. Myol. 2019, 38, 207–214. [Google Scholar] [PubMed]
- Rahmati, M.; Malakoutinia, F. Aerobic, resistance and combined exercise training for patients with amyotrophic lateral sclerosis: A systematic review and meta-analysis. Physiotherapy 2021, 113, 12–28. [Google Scholar] [CrossRef]
- Zhu, Y.; Xu, Y.; Xuan, R.; Huang, J.; Istvan, B.; Fekete, G.; Gu, Y. Mixed Comparison of Different Exercise Interventions for Function, Respiratory, Fatigue, and Quality of Life in Adults with Amyotrophic Lateral Sclerosis: Systematic Review and Network Meta-Analysis. Front. Aging Neurosci. 2022, 14, 919059. [Google Scholar] [CrossRef]
- Su, W.M.; Cheng, Y.F.; Jiang, Z.; Duan, Q.Q.; Yang, T.M.; Shang, H.F.; Chen, Y.P. Predictors of survival in patients with amyotrophic lateral sclerosis: A large meta-analysis. EBioMedicine 2021, 74, 103732. [Google Scholar] [CrossRef] [PubMed]
- Penders, Y.W.H.; Bopp, M.; Zellweger, U.; Bosshard, G.; Swiss Medical End-of-Life Decisions Study, G. Continuing, Withdrawing, and Withholding Medical Treatment at the End of Life and Associated Characteristics: A Mortality Follow-back Study. J. Gen. Intern. Med. 2020, 35, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Hogden, A.; Greenfield, D.; Nugus, P.; Kiernan, M.C. What influences patient decision-making in amyotrophic lateral sclerosis multidisciplinary care? A study of patient perspectives. Patient Prefer Adherence 2012, 6, 829–838. [Google Scholar] [CrossRef]
- Ozanne, A.; Graneheim, U.H. Understanding the incomprehensible—Patients’ and spouses’ experiences of comprehensibility before, at and after diagnosis of amyotrophic lateral sclerosis. Scand. J. Caring Sci. 2018, 32, 663–671. [Google Scholar] [CrossRef]
- Seeber, A.A.; Hijdra, A.; Vermeulen, M.; Willems, D.L. Discussions about treatment restrictions in chronic neurologic diseases: A structured review. Neurology 2012, 78, 590–597. [Google Scholar] [CrossRef]
- Johnston, W.S.; Hoskins, K.; McCluskey, L. Amyotrophic lateral sclerosis: Ethical challenges. Neurology 2011, 76, S1–S5. [Google Scholar] [CrossRef]
- Rigotti, L. Freedom to choose treatments in incurable disease.Summary. The lack of knowledge and application of legal and ethical norms have often justified behaviors of many health workers not adequate to the care needs of people with ALS or other incurable diseases, highlighting their cultural unpreparedness. The narration of the experience and reflections of a palliativist doctor with an ALS patient can be reason for mainly ethical considerations. Recenti. Prog. Med. 2020, 111, 670–672. [Google Scholar] [CrossRef]
- Stanzel, S.B.; Spiesshoefer, J.; Trudzinski, F.; Cornelissen, C.; Kabitz, H.J.; Fuchs, H.; Boentert, M.; Mathes, T.; Michalsen, A.; Hirschfeld, S.; et al. S3 Guideline: Treating Chronic Respiratory Failure with Non-invasive Ventilation. Pneumologie 2025, 79, 25–79. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Sánchez, M.; Ramírez-Expósito, M.J.; Martínez-Martos, J.M. Pathophysiology, Clinical Heterogeneity, and Therapeutic Advances in Amyotrophic Lateral Sclerosis: A Comprehensive Review of Molecular Mechanisms, Diagnostic Challenges, and Multidisciplinary Management Strategies. Life 2025, 15, 647. https://doi.org/10.3390/life15040647
González-Sánchez M, Ramírez-Expósito MJ, Martínez-Martos JM. Pathophysiology, Clinical Heterogeneity, and Therapeutic Advances in Amyotrophic Lateral Sclerosis: A Comprehensive Review of Molecular Mechanisms, Diagnostic Challenges, and Multidisciplinary Management Strategies. Life. 2025; 15(4):647. https://doi.org/10.3390/life15040647
Chicago/Turabian StyleGonzález-Sánchez, María, María Jesús Ramírez-Expósito, and José Manuel Martínez-Martos. 2025. "Pathophysiology, Clinical Heterogeneity, and Therapeutic Advances in Amyotrophic Lateral Sclerosis: A Comprehensive Review of Molecular Mechanisms, Diagnostic Challenges, and Multidisciplinary Management Strategies" Life 15, no. 4: 647. https://doi.org/10.3390/life15040647
APA StyleGonzález-Sánchez, M., Ramírez-Expósito, M. J., & Martínez-Martos, J. M. (2025). Pathophysiology, Clinical Heterogeneity, and Therapeutic Advances in Amyotrophic Lateral Sclerosis: A Comprehensive Review of Molecular Mechanisms, Diagnostic Challenges, and Multidisciplinary Management Strategies. Life, 15(4), 647. https://doi.org/10.3390/life15040647