

Epigenetic Mechanisms of Obesity: Insights from Transgenic Animal Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Ideological Underpinnings of Obesity
3. Central Mechanisms Governing Body Energy Homeostasis
3.1. The Arcuate Nucleus: Integration Site for Body Energy Homeostasis
3.2. The Lateral Hypothalamus: The “Hunger Center”
3.3. The Paraventricular Nucleus of the Hypothalamus: The Central Site for Homeostatic and Autonomic Function
3.4. The Ventromedial Hypothalamus: The “Satiety Center”
4. The Central Melanocortin System
5. The Complex Issue of Obesity and the Impact of Ultra-Processed Foods
6. How Environmental Factors Fundamentally Alter Our Epigenetic Landscape
6.1. Why Have Obesity Rates Risen Despite a Stable Genome?
6.2. Epigenetics: The Interface Between Genes and Environment
6.3. Early Life Exposure to Obesogenic Diets and Epigenetic Programming
6.4. Diet-Induced Epigenetic Changes in Adulthood
6.5. The Persistence of Epigenetic Memory in Obesity
6.6. Bridging Animal Research and Human Clinical Studies
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AP | Area postrema |
| ARC | Arcuate nucleus of the hypothalamus |
| ACTH | Adrenocorticotropic hormone |
| AgRP | Agouti-related peptide |
| BMI | Body Mass Index |
| CART | Cocaine- and amphetamine-regulated transcript |
| CDC | Centers for Disease Control |
| CNS | Central nervous system |
| DIO | Diet-induced obesity |
| DNMT | DNA methyltransferase |
| GLP1 | Glucagon-like peptide 1 |
| HDAC | Histone deacetylase |
| HPA | Hypothalamic–pituitary–adrenal |
| LH | Lateral hypothalamus |
| LPBN | Lateral parabrachial nucleus |
| MCH | Melanin-concentrating hormone |
| MeCP2 | Methyl-CpG binding protein 2 |
| MSH | Melanocyte-stimulating hormone |
| MCR | Melanocortin receptor |
| nPE1/2 | Neural POMC enhancer 1/2 |
| NPY | Neuropeptide Y |
| NTS | Nucleus of the solitary tract |
| PACAP | Pituitary adenylate cyclase-activating peptide |
| PGC1α | Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-alpha |
| POMC | Pro-opiomelanocortin |
| PTHrP | Parathyroid Hormone |
| PVH | Paraventricular nucleus of the hypothalamus |
| SF1 | Steroidogenic factor-1 |
| SIRT1 | Sirtuin 1 |
| TET | Ten-eleven translocation |
| TBRS | Tatton–Brown–Rahman Syndrome |
| TRH | Thyroid-releasing hormone |
| UCP1 | Uncoupling Protein 1 |
| UPF | Ultra-processed foods |
| VMH | Ventromedial hypothalamus |
| WAT | White adipose tissue |
| WHO | World Health Organization |
References
- Centers for Disease Control and Prevention. National Health and Nutrition Examination Survey, 2017–2018; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2017.
- Ward, Z.J.; Long, M.W.; Resch, S.C.; Giles, C.M.; Cradock, A.L.; Gortmaker, S.L. Simulation of Growth Trajectories of Childhood Obesity into Adulthood. N. Engl. J. Med. 2017, 377, 2145–2153. [Google Scholar] [CrossRef]
- AMA Adopts New Policy Clarifying Role of BMI as a Measure in Medicine. Available online: https://www.ama-assn.org/press-center/press-releases/ama-adopts-new-policy-clarifying-role-bmi-measure-medicine (accessed on 12 February 2025).
- Yuen, M.M.A. Health Complications of Obesity: 224 Obesity-Associated Comorbidities from a Mechanistic Perspective. Gastroenterol. Clin. N. Am. 2023, 52, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Fulton, S.; Decarie-Spain, L.; Fioramonti, X.; Guiard, B.; Nakajima, S. The menace of obesity to depression and anxiety prevalence. Trends Endocrinol. Metab. 2022, 33, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D. Perspective: Obesity-an unexplained epidemic. Am. J. Clin. Nutr. 2022, 115, 1445–1450. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Ronn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef]
- Obri, A.; Serra, D.; Herrero, L.; Mera, P. The role of epigenetics in the development of obesity. Biochem. Pharmacol. 2020, 177, 113973. [Google Scholar] [CrossRef]
- Tomlinson, D.J.; Erskine, R.M.; Morse, C.I.; Winwood, K.; Onambele-Pearson, G. The impact of obesity on skeletal muscle strength and structure through adolescence to old age. Biogerontology 2016, 17, 467–483. [Google Scholar] [CrossRef]
- Noria, S.F.; Shelby, R.D.; Atkins, K.D.; Nguyen, N.T.; Gadde, K.M. Weight Regain After Bariatric Surgery: Scope of the Problem, Causes, Prevention, and Treatment. Curr. Diabetes Rep. 2023, 23, 31–42. [Google Scholar] [CrossRef]
- Sjostrom, L.; Lindroos, A.K.; Peltonen, M.; Torgerson, J.; Bouchard, C.; Carlsson, B.; Dahlgren, S.; Larsson, B.; Narbro, K.; Sjostrom, C.D.; et al. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N. Engl. J. Med. 2004, 351, 2683–2693. [Google Scholar] [CrossRef]
- Meguid, M.M.; Glade, M.J.; Middleton, F.A. Weight regain after Roux-en-Y: A significant 20% complication related to PYY. Nutrition 2008, 24, 832–842. [Google Scholar] [CrossRef]
- Bernard, C. Bernard Lectures on the Phenomena Common to Animals and Plants; J. Librairie: Paris, France, 1878. [Google Scholar]
- Cannon, W.B. Organization for physiological homeostasis. Physiol. Rev. IX 1929, 9, 399–431. [Google Scholar] [CrossRef]
- Sterling, P. Allostasis: A model of predictive regulation. Physiol. Behav. 2012, 106, 5–15. [Google Scholar] [CrossRef]
- McEwen, B.S. Protection and damage from acute and chronic stress: Allostasis and allostatic overload and relevance to the pathophysiology of psychiatric disorders. Ann. N. Y. Acad. Sci. 2004, 1032, 1–7. [Google Scholar] [CrossRef]
- McEwen, B.S. Stress, adaptation, and disease. Allostasis and allostatic load. Ann. N. Y. Acad. Sci. 1998, 840, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Caradonna, S.G.; Paul, M.R.; Marrocco, J. An allostatic epigenetic memory on chromatin footprints after double-hit acute stress. Neurobiol. Stress 2022, 20, 100475. [Google Scholar] [CrossRef]
- Saper, C.B. (Ed.) Cytoarchitecture of the Human Hypothalamus; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Yin, W.; Gore, A.C. The hypothalamic median eminence and its role in reproductive aging. Ann. N. Y. Acad. Sci. 2010, 1204, 113–122. [Google Scholar] [CrossRef]
- Cowley, M.A.; Pronchuk, N.; Fan, W.; Dinulescu, D.M.; Colmers, W.F.; Cone, R.D. Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: Evidence of a cellular basis for the adipostat. Neuron 1999, 24, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.Y.; van den Pol, A.N. Agouti-related peptide and MC3/4 receptor agonists both inhibit excitatory hypothalamic ventromedial nucleus neurons. J. Neurosci. 2008, 28, 5433–5449. [Google Scholar] [CrossRef]
- Krude, H.; Biebermann, H.; Luck, W.; Horn, R.; Brabant, G.; Gruters, A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 1998, 19, 155–157. [Google Scholar] [CrossRef]
- Farooqi, I.S. Monogenic human obesity. Front. Horm. Res. 2008, 36, 1–11. [Google Scholar]
- Mendiratta, M.S.; Yang, Y.; Balazs, A.E.; Willis, A.S.; Eng, C.M.; Karaviti, L.P.; Potocki, L. Early onset obesity and adrenal insufficiency associated with a homozygous POMC mutation. Int. J. Pediatr. Endocrinol. 2011, 2011, 5. [Google Scholar] [CrossRef]
- Ranadive, S.A.; Vaisse, C. Lessons from extreme human obesity: Monogenic disorders. Endocrinol. Metab. Clin. N. Am. 2008, 37, 733–751. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chhabra, K.H.; Thompson, Z.; Jones, G.L.; Kiran, S.; Shangguan, G.; Low, M.J. Hypothalamic POMC deficiency increases circulating adiponectin despite obesity. Mol. Metab. 2020, 35, 100957. [Google Scholar] [CrossRef]
- Anand, B.K.; Brobeck, J.R. Localization of a “feeding center” in the hypothalamus of the rat. Proc. Soc. Exp. Biol. Med. 1951, 77, 323–324. [Google Scholar] [CrossRef]
- Waterson, M.J.; Horvath, T.L. Neuronal Regulation of Energy Homeostasis: Beyond the Hypothalamus and Feeding. Cell Metab. 2015, 22, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.M.; Anand, B.K. Increase of food intake induced by electrical stimulation of the lateral hypothalamus. Am. J. Physiol. 1953, 172, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Stanley, B.G.; Ha, L.H.; Spears, L.C.; Dee, M.G., 2nd. Lateral hypothalamic injections of glutamate, kainic acid, D,L-alpha-amino-3-hydroxy-5-methyl-isoxazole propionic acid or N-methyl-D-aspartic acid rapidly elicit intense transient eating in rats. Brain Res. 1993, 613, 88–95. [Google Scholar] [CrossRef]
- Kelly, J.; Rothstein, J.; Grossman, S.P. GABA and hypothalamic feeding systems. I. Topographic analysis of the effects of microinjections of muscimol. Physiol. Behav. 1979, 23, 1123–1134. [Google Scholar] [CrossRef]
- Stuber, G.D.; Wise, R.A. Lateral hypothalamic circuits for feeding and reward. Nat. Neurosci. 2016, 19, 198–205. [Google Scholar] [CrossRef]
- Bonnavion, P.; Mickelsen, L.E.; Fujita, A.; de Lecea, L.; Jackson, A.C. Hubs and spokes of the lateral hypothalamus: Cell types, circuits and behaviour. J. Physiol. 2016, 594, 6443–6462. [Google Scholar] [CrossRef]
- Sharpe, M.J. The cognitive (lateral) hypothalamus. Trends Cogn. Sci. 2024, 28, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Berridge, K.C.; Valenstein, E.S. What psychological process mediates feeding evoked by electrical stimulation of the lateral hypothalamus? Behav. Neurosci. 1991, 105, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Stratford, T.R.; Kelley, A.E. Evidence of a functional relationship between the nucleus accumbens shell and lateral hypothalamus subserving the control of feeding behavior. J. Neurosci. 1999, 19, 11040–11048. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T. Roles of orexins in the regulation of body weight homeostasis. Obes. Res. Clin. Pract. 2014, 8, e414–e420. [Google Scholar] [CrossRef]
- Jones, B.E.; Hassani, O.K. The role of Hcrt/Orx and MCH neurons in sleep-wake state regulation. Sleep 2013, 36, 1769–1772. [Google Scholar] [CrossRef]
- Muschamp, J.W.; Hollander, J.A.; Thompson, J.L.; Voren, G.; Hassinger, L.C.; Onvani, S.; Kamenecka, T.M.; Borgland, S.L.; Kenny, P.J.; Carlezon, W.A., Jr. Hypocretin (orexin) facilitates reward by attenuating the antireward effects of its cotransmitter dynorphin in ventral tegmental area. Proc. Natl. Acad. Sci. USA 2014, 111, E1648–E1655. [Google Scholar] [CrossRef]
- Furutani, N.; Hondo, M.; Kageyama, H.; Tsujino, N.; Mieda, M.; Yanagisawa, M.; Shioda, S.; Sakurai, T. Neurotensin co-expressed in orexin-producing neurons in the lateral hypothalamus plays an important role in regulation of sleep/wakefulness states. PLoS ONE 2013, 8, e62391. [Google Scholar] [CrossRef]
- Hahn, J.D.; Swanson, L.W. Distinct patterns of neuronal inputs and outputs of the juxtaparaventricular and suprafornical regions of the lateral hypothalamic area in the male rat. Brain Res. Rev. 2010, 64, 14–103. [Google Scholar] [CrossRef]
- Elias, C.F.; Lee, C.E.; Kelly, J.F.; Ahima, R.S.; Kuhar, M.; Saper, C.B.; Elmquist, J.K. Characterization of CART neurons in the rat and human hypothalamus. J. Comp. Neurol. 2001, 432, 1–19. [Google Scholar] [CrossRef]
- Hanriot, L.; Camargo, N.; Courau, A.C.; Leger, L.; Luppi, P.H.; Peyron, C. Characterization of the melanin-concentrating hormone neurons activated during paradoxical sleep hypersomnia in rats. J. Comp. Neurol. 2007, 505, 147–157. [Google Scholar] [CrossRef]
- Qin, C.; Li, J.; Tang, K. The Paraventricular Nucleus of the Hypothalamus: Development, Function, and Human Diseases. Endocrinology 2018, 159, 3458–3472. [Google Scholar] [CrossRef] [PubMed]
- Swanson, L.W.; Sawchenko, P.E. Hypothalamic integration: Organization of the paraventricular and supraoptic nuclei. Annu. Rev. Neurosci. 1983, 6, 269–324. [Google Scholar] [CrossRef] [PubMed]
- Sutton, A.K.; Pei, H.; Burnett, K.H.; Myers, M.G., Jr.; Rhodes, C.J.; Olson, D.P. Control of food intake and energy expenditure by Nos1 neurons of the paraventricular hypothalamus. J. Neurosci. 2014, 34, 15306–15318. [Google Scholar] [CrossRef]
- Shah, B.P.; Vong, L.; Olson, D.P.; Koda, S.; Krashes, M.J.; Ye, C.; Yang, Z.; Fuller, P.M.; Elmquist, J.K.; Lowell, B.B. MC4R-expressing glutamatergic neurons in the paraventricular hypothalamus regulate feeding and are synaptically connected to the parabrachial nucleus. Proc. Natl. Acad. Sci. USA 2014, 111, 13193–13198. [Google Scholar] [CrossRef]
- Sawchenko, P.E.; Brown, E.R.; Chan, R.K.; Ericsson, A.; Li, H.Y.; Roland, B.L.; Kovacs, K.J. The paraventricular nucleus of the hypothalamus and the functional neuroanatomy of visceromotor responses to stress. Prog. Brain Res. 1996, 107, 201–222. [Google Scholar]
- Grzeda, E.; Ziarniak, K.; Sliwowska, J.H. The paraventricular nucleus of the hypothalamus—The concertmaster of autonomic control. Focus on blood pressure regulation. Acta Neurobiol. Exp. 2023, 83, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Madara, J.C.; Steger, J.S.; Krashes, M.J.; Balthasar, N.; Campbell, J.N.; Resch, J.M.; Conley, N.J.; Garfield, A.S.; Lowell, B.B. The Paraventricular Hypothalamus Regulates Satiety and Prevents Obesity via Two Genetically Distinct Circuits. Neuron 2019, 102, 653–667.e6. [Google Scholar] [CrossRef]
- Khodai, T.; Luckman, S.M. Ventromedial Nucleus of the Hypothalamus Neurons Under the Magnifying Glass. Endocrinology 2021, 162, bqab141. [Google Scholar] [CrossRef]
- Hannibal, J. Pituitary adenylate cyclase-activating peptide in the rat central nervous system: An immunohistochemical and in situ hybridization study. J. Comp. Neurol. 2002, 453, 389–417. [Google Scholar] [CrossRef]
- Yamada, K.; Emson, P.; Hokfelt, T. Immunohistochemical mapping of nitric oxide synthase in the rat hypothalamus and colocalization with neuropeptides. J. Chem. Neuroanat. 1996, 10, 295–316. [Google Scholar] [CrossRef]
- Ikeda, Y.; Luo, X.; Abbud, R.; Nilson, J.H.; Parker, K.L. The nuclear receptor steroidogenic factor 1 is essential for the formation of the ventromedial hypothalamic nucleus. Mol. Endocrinol. 1995, 9, 478–486. [Google Scholar] [PubMed]
- Hetherington, A.W.; Ranson, S.W. Hypothalamic lesions and adiposity in the rat. Anat. Rec. 1940, 78, 149–172. [Google Scholar] [CrossRef]
- Dhillon, H.; Zigman, J.M.; Ye, C.; Lee, C.E.; McGovern, R.A.; Tang, V.; Kenny, C.D.; Christiansen, L.M.; White, R.D.; Edelstein, E.A.; et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 2006, 49, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Majdic, G.; Young, M.; Gomez-Sanchez, E.; Anderson, P.; Szczepaniak, L.S.; Dobbins, R.L.; McGarry, J.D.; Parker, K.L. Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology 2002, 143, 607–614. [Google Scholar] [CrossRef]
- Anand, B.K.; Chhina, G.S.; Sharma, K.N.; Dua, S.; Singh, B. Activity of Single Neurons in the Hypothalamic Feeding Centers: Effect of Glucose. Am. J. Physiol. 1964, 207, 1146–1154. [Google Scholar] [CrossRef]
- Shimazu, T.; Fukuda, A.; Ban, T. Reciprocal influences of the ventromedial and lateral hypothalamic nuclei on blood glucose level and liver glycogen content. Nature 1966, 210, 1178–1179. [Google Scholar] [CrossRef]
- Frohman, L.A.; Bernardis, L.L. Effect of hypothalamic stimulation on plasma glucose, insulin, and glucagon levels. Am. J. Physiol. 1971, 221, 1596–1603. [Google Scholar] [CrossRef]
- Sweeney, P.; Gimenez, L.E.; Hernandez, C.C.; Cone, R.D. Targeting the central melanocortin system for the treatment of metabolic disorders. Nat. Rev. Endocrinol. 2023, 19, 507–519. [Google Scholar] [CrossRef]
- Fenselau, H.; Campbell, J.N.; Verstegen, A.M.; Madara, J.C.; Xu, J.; Shah, B.P.; Resch, J.M.; Yang, Z.; Mandelblat-Cerf, Y.; Livneh, Y. A rapidly acting glutamatergic ARC-->PVH satiety circuit postsynaptically regulated by alpha-MSH. Nat. Neurosci. 2017, 20, 42–51. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Drop, S.; Clements, A.; Keogh, J.M.; Biernacka, J.; Lowenbein, S.; Challis, B.G.; O’Rahilly, S. Heterozygosity for a POMC-null mutation and increased obesity risk in humans. Diabetes 2006, 55, 2549–2553. [Google Scholar] [CrossRef]
- Farooqi, I.S. Monogenic human obesity syndromes. Handb. Clin. Neurol. 2021, 181, 301–310. [Google Scholar] [PubMed]
- Mountjoy, K.G. Pro-Opiomelanocortin (POMC) Neurones, POMC-Derived Peptides, Melanocortin Receptors and Obesity: How Understanding of this System has Changed Over the Last Decade. J. Neuroendocrinol. 2015, 27, 406–418. [Google Scholar] [CrossRef] [PubMed]
- Challis, B.G.; Coll, A.P.; Yeo, G.S.; Pinnock, S.B.; Dickson, S.L.; Thresher, R.R.; Dixon, J.; Zahn, D.; Rochford, J.J.; White, A.; et al. Mice lacking pro-opiomelanocortin are sensitive to high-fat feeding but respond normally to the acute anorectic effects of peptide-YY(3-36). Proc. Natl. Acad. Sci. USA 2004, 101, 4695–4700. [Google Scholar] [CrossRef]
- Lam, D.D.; de Souza, F.S.; Nasif, S.; Yamashita, M.; Lopez-Leal, R.; Otero-Corchon, V.; Meece, K.; Sampath, H.; Mercer, A.J.; Wardlaw, S.L. Partially redundant enhancers cooperatively maintain Mammalian pomc expression above a critical functional threshold. PLoS Genet. 2015, 11, e1004935. [Google Scholar] [CrossRef]
- Takahashi, K.A.; Cone, R.D. Fasting induces a large, leptin-dependent increase in the intrinsic action potential frequency of orexigenic arcuate nucleus neuropeptide Y/Agouti-related protein neurons. Endocrinology 2005, 146, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Luquet, S.; Perez, F.A.; Hnasko, T.S.; Palmiter, R.D. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science 2005, 310, 683–685. [Google Scholar] [CrossRef]
- Atasoy, D.; Betley, J.N.; Su, H.H.; Sternson, S.M. Deconstruction of a neural circuit for hunger. Nature 2012, 488, 172–177. [Google Scholar] [CrossRef]
- Rossi, M.; Kim, M.S.; Morgan, D.G.; Small, C.J.; Edwards, C.M.; Sunter, D.; Abusnana, S.; Goldstone, A.P.; Russell, S.H.; Stanley, S.A.; et al. A C-terminal fragment of Agouti-related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating hormone in vivo. Endocrinology 1998, 139, 4428–4431. [Google Scholar] [CrossRef]
- Krashes, M.J.; Koda, S.; Ye, C.; Rogan, S.C.; Adams, A.C.; Cusher, D.S.; Maratos-Flier, E.; Roth, B.L.; Lowell, B.B. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Investig. 2011, 121, 1424–1428. [Google Scholar] [CrossRef]
- Aponte, Y.; Atasoy, D.; Sternson, S.M. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat. Neurosci. 2011, 14, 351–355. [Google Scholar] [CrossRef]
- Ward, Z.J.; Bleich, S.N.; Cradock, A.L.; Barrett, J.L.; Giles, C.M.; Flax, C.; Long, M.W.; Gortmaker, S.L. Projected U.S. State-Level Prevalence of Adult Obesity and Severe Obesity. N. Engl. J. Med. 2019, 381, 2440–2450. [Google Scholar] [CrossRef] [PubMed]
- Estimates of Funding for Various Research, Condition, and Disease Categories (RCDC). 2024. Available online: https://report.nih.gov/funding/categorical-spending#/ (accessed on 18 February 2025).
- Lutz, T.A.; Woods, S.C. Overview of animal models of obesity. Curr. Protoc. Pharmacol. 2012, 58, 5–61. [Google Scholar] [CrossRef] [PubMed]
- Crimarco, A.; Landry, M.J.; Gardner, C.D. Ultra-processed Foods, Weight Gain, and Co-morbidity Risk. Curr. Obes. Rep. 2022, 11, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Valicente, V.M.; Peng, C.H.; Pacheco, K.N.; Lin, L.; Kielb, E.I.; Dawoodani, E.; Abdollahi, A.; Mattes, R.D. Ultraprocessed Foods and Obesity Risk: A Critical Review of Reported Mechanisms. Adv. Nutr. 2023, 14, 718–738. [Google Scholar] [CrossRef]
- Juul, F.; Parekh, N.; Martinez-Steele, E.; Monteiro, C.A.; Chang, V.W. Ultra-processed food consumption among US adults from 2001 to 2018. Am. J. Clin. Nutr. 2022, 115, 211–221. [Google Scholar] [CrossRef]
- Wang, L.; Martinez Steele, E.; Du, M.; Pomeranz, J.L.; O’Connor, L.E.; Herrick, K.A.; Luo, H.; Zhang, X.; Mozaffarian, D.; Zhang, F.F. Trends in Consumption of Ultraprocessed Foods Among US Youths Aged 2–19 Years, 1999–2018. JAMA 2021, 326, 519–530. [Google Scholar] [CrossRef]
- Monteiro, C.A.; Moubarac, J.C.; Cannon, G.; Ng, S.W.; Popkin, B. Ultra-processed products are becoming dominant in the global food system. Obes. Rev. 2013, 2 (Suppl. S14), 21–28. [Google Scholar] [CrossRef]
- Spreadbury, I. Comparison with ancestral diets suggests dense acellular carbohydrates promote an inflammatory microbiota, and may be the primary dietary cause of leptin resistance and obesity. Diabetes Metab. Syndr. Obes. 2012, 5, 175–189. [Google Scholar] [CrossRef]
- Zinocker, M.K.; Lindseth, I.A. The Western Diet-Microbiome-Host Interaction and Its Role in Metabolic Disease. Nutrients 2018, 10, 365. [Google Scholar] [CrossRef]
- Mendonca, R.D.; Pimenta, A.M.; Gea, A.; de la Fuente-Arrillaga, C.; Martinez-Gonzalez, M.A.; Lopes, A.C.; Bes-Rastrollo, M. Ultraprocessed food consumption and risk of overweight and obesity: The University of Navarra Follow-Up (SUN) cohort study. Am. J. Clin. Nutr. 2016, 104, 1433–1440. [Google Scholar] [CrossRef]
- Monteiro, C.A.; Moubarac, J.C.; Levy, R.B.; Canella, D.S.; Louzada, M.; Cannon, G. Household availability of ultra-processed foods and obesity in nineteen European countries. Public Health Nutr. 2018, 21, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Canella, D.S.; Levy, R.B.; Martins, A.P.; Claro, R.M.; Moubarac, J.C.; Baraldi, L.G.; Cannon, G.; Monteiro, C.A. Ultra-processed food products and obesity in Brazilian households (2008–2009). PLoS ONE 2014, 9, e92752. [Google Scholar] [CrossRef]
- De Paula, G.C.; Simoes, R.F.; Garcia-Serrano, A.M.; Duarte, J.M.N. High-fat and High-sucrose Diet-induced Hypothalamic Inflammation Shows Sex Specific Features in Mice. Neurochem. Res. 2024, 49, 3356–3366. [Google Scholar] [CrossRef] [PubMed]
- Buckley, L.A.; Kulhanek, D.R.; Bruder, A.; Gisslen, T.; Paulsen, M.E. Inflammation as a Sex-Specific Mediator in the Relationship between Maternal and Offspring Obesity in C57Bl/6J Mice. Biology 2024, 13, 399. [Google Scholar] [CrossRef]
- Mohr, A.A.; Garcia-Serrano, A.M.; Vieira, J.P.; Skoug, C.; Davidsson, H.; Duarte, J.M. A glucose-stimulated BOLD fMRI study of hypothalamic dysfunction in mice fed a high-fat and high-sucrose diet. J. Cereb. Blood Flow. Metab. 2021, 41, 1734–1743. [Google Scholar] [CrossRef]
- Slomp, M.; Koekkoek, L.L.; Mutersbaugh, M.; Linville, I.; Luquet, S.H.; la Fleur, S.E. Free-choice high-fat diet consumption reduces lateral hypothalamic GABAergic activity, without disturbing neural response to sucrose drinking in mice. Front. Neurosci. 2023, 17, 1219569. [Google Scholar] [CrossRef]
- Perusse, L.; Rankinen, T.; Zuberi, A.; Chagnon, Y.C.; Weisnagel, S.J.; Argyropoulos, G.; Walts, B.; Snyder, E.E.; Bouchard, C. The human obesity gene map: The 2004 update. Obes. Res. 2005, 13, 381–490. [Google Scholar] [CrossRef]
- McPherson, R. Genetic contributors to obesity. Can. J. Cardiol. 2007, 23 (Suppl. A), 23A–27A. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Chaffin, M.; Wade, K.H.; Zahid, S.; Brancale, J.; Xia, R.; Distefano, M.; Senol-Cosar, O.; Haas, M.E.; Bick, A.; et al. Polygenic Prediction of Weight and Obesity Trajectories from Birth to Adulthood. Cell 2019, 177, 587–596.e9. [Google Scholar] [CrossRef]
- Maes, H.H.; Neale, M.C.; Eaves, L.J. Genetic and environmental factors in relative body weight and human adiposity. Behav. Genet. 1997, 27, 325–351. [Google Scholar] [CrossRef]
- Rohde, K.; Keller, M.; la Cour Poulsen, L.; Bluher, M.; Kovacs, P.; Bottcher, Y. Genetics and epigenetics in obesity. Metabolism 2019, 92, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Sierra, A.; Ramos-Lopez, O.; Riezu-Boj, J.I.; Milagro, F.I.; Martinez, J.A. Diet, Gut Microbiota, and Obesity: Links with Host Genetics and Epigenetics and Potential Applications. Adv. Nutr. 2019, 10, S17–S30. [Google Scholar] [CrossRef]
- Morris, M.J.; Monteggia, L.M. Role of DNA methylation and the DNA methyltransferases in learning and memory. Dialogues Clin. Neurosci. 2014, 16, 359–371. [Google Scholar] [CrossRef]
- Sweatt, J.D. The emerging field of neuroepigenetics. Neuron 2013, 80, 624–632. [Google Scholar] [CrossRef]
- Hamilton, P.J.; Nestler, E.J. Epigenetics and addiction. Curr. Opin. Neurobiol. 2019, 59, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Pena, C.J.; Nestler, E.J. Progress in Epigenetics of Depression. Prog. Mol. Biol. Transl. Sci. 2018, 157, 41–66. [Google Scholar] [PubMed]
- Krasilnikova, J.; Lauberte, L.; Stoyanova, E.; Abadjieva, D.; Chervenkov, M.; Mori, M.; De Paolis, E.; Mladenova, V.; Telysheva, G.; Botta, B. Oregonin from Alnus incana bark affects DNA methyltransferases expression and mitochondrial DNA copies in mouse embryonic fibroblasts. J. Enzym. Inhib. Med. Chem. 2018, 33, 1055–1063. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Kim, S.; Kaang, B.K. Epigenetic regulation and chromatin remodeling in learning and memory. Exp. Mol. Med. 2017, 49, e281. [Google Scholar] [CrossRef]
- Kooistra, S.M.; Helin, K. Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [Google Scholar] [CrossRef]
- Nelson, C.J.; Santos-Rosa, H.; Kouzarides, T. Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell 2006, 126, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Jo, C.; Park, S.; Oh, S.; Choi, J.; Kim, E.K.; Youn, H.D.; Cho, E.J. Histone acylation marks respond to metabolic perturbations and enable cellular adaptation. Exp. Mol. Med. 2020, 52, 2005–2019. [Google Scholar] [CrossRef]
- Liu, J.; Shangguan, Y.; Tang, D.; Dai, Y. Histone succinylation and its function on the nucleosome. J. Cell. Mol. Med. 2021, 25, 7101–7109. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef]
- Elgazzaz, M.; Berdasco, C.; Garai, J.; Baddoo, M.; Lu, S.; Daoud, H.; Zabaleta, J.; Mauvais-Jarvis, F.; Lazartigues, E. Maternal Western diet programs cardiometabolic dysfunction and hypothalamic inflammation via epigenetic mechanisms predominantly in the male offspring. Mol. Metab. 2024, 80, 101864. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.M.; Dias-Rocha, C.P.; Reis-Gomes, C.F.; Wang, H.; Atella, G.C.; Cordeiro, A.; Pazos-Moura, C.C.; Joss-Moore, L.; Trevenzoli, I.H. Maternal high-fat diet impairs leptin signaling and up-regulates type-1 cannabinoid receptor with sex-specific epigenetic changes in the hypothalamus of newborn rats. Psychoneuroendocrinology 2019, 103, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Gali Ramamoorthy, T.; Allen, T.J.; Davies, A.; Harno, E.; Sefton, C.; Murgatroyd, C.; White, A. Maternal overnutrition programs epigenetic changes in the regulatory regions of hypothalamic Pomc in the offspring of rats. Int. J. Obes. 2018, 42, 1431–1444. [Google Scholar] [CrossRef]
- Kuehnen, P.; Mischke, M.; Wiegand, S.; Sers, C.; Horsthemke, B.; Lau, S.; Keil, T.; Lee, Y.A.; Grueters, A.; Krude, H. An Alu element-associated hypermethylation variant of the POMC gene is associated with childhood obesity. PLoS Genet. 2012, 8, e1002543. [Google Scholar] [CrossRef]
- Kuhnen, P.; Handke, D.; Waterland, R.A.; Hennig, B.J.; Silver, M.; Fulford, A.J.; Dominguez-Salas, P.; Moore, S.E.; Prentice, A.M.; Spranger, J.; et al. Interindividual Variation in DNA Methylation at a Putative POMC Metastable Epiallele Is Associated with Obesity. Cell Metab. 2016, 24, 502–509. [Google Scholar] [CrossRef]
- Cross, S.H.; Meehan, R.R.; Nan, X.; Bird, A. A component of the transcriptional repressor MeCP1 shares a motif with DNA methyltransferase and HRX proteins. Nat. Genet. 1997, 16, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Campoy, F.J.; Bird, A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 1997, 88, 471–481. [Google Scholar] [CrossRef]
- Frayre, J.; Frayre, P.; Wong, I.; Mithani, A.; Bishop, S.; Mani, C.; Ponce-Rubio, K.; Virk, R.; Morris, M.J.; Na, E.S. Perinatal exposure to high fat diet alters expression of MeCP2 in the hypothalamus. Behav. Brain Res. 2021, 415, 113518. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xiao, X.; Zhang, Q.; Yu, M.; Xu, J.; Wang, Z.; Qi, C.; Wang, T. Maternal and post-weaning high-fat, high-sucrose diet modulates glucose homeostasis and hypothalamic POMC promoter methylation in mouse offspring. Metab. Brain Dis. 2015, 30, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhang, L.; Liu, J.; Li, Y.; Zhang, J. Long-Term Effects of Maternal Low-Protein Diet and Post-weaning High-Fat Feeding on Glucose Metabolism and Hypothalamic POMC Promoter Methylation in Offspring Mice. Front. Nutr. 2021, 8, 657848. [Google Scholar] [CrossRef]
- Padilla, S.L.; Reef, D.; Zeltser, L.M. Defining POMC neurons using transgenic reagents: Impact of transient Pomc expression in diverse immature neuronal populations. Endocrinology 2012, 153, 1219–1231. [Google Scholar] [CrossRef]
- Wang, X.; Lacza, Z.; Sun, Y.E.; Han, W. Leptin resistance and obesity in mice with deletion of methyl-CpG-binding protein 2 (MeCP2) in hypothalamic pro-opiomelanocortin (POMC) neurons. Diabetologia 2014, 57, 236–245. [Google Scholar] [CrossRef]
- Frayre, P.; Ponce-Rubio, K.; Frayre, J.; Medrano, J.; Na, E.S. POMC-specific knockdown of MeCP2 leads to adverse phenotypes in mice chronically exposed to high fat diet. Behav. Brain Res. 2024, 461, 114863. [Google Scholar] [CrossRef]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef]
- Morris, M.J.; Monteggia, L.M. Unique functional roles for class I and class II histone deacetylases in central nervous system development and function. Int. J. Dev. Neurosci. 2013, 31, 370–381. [Google Scholar] [CrossRef]
- Funato, H.; Oda, S.; Yokofujita, J.; Igarashi, H.; Kuroda, M. Fasting and high-fat diet alter histone deacetylase expression in the medial hypothalamus. PLoS ONE 2011, 6, e18950. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Marin de Evsikova, C.; Bian, K.; Achille, A.; Telles, E.; Pei, H.; Seto, E. Programming and Regulation of Metabolic Homeostasis by HDAC11. EBioMedicine 2018, 33, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Cakir, I.; Hadley, C.K.; Pan, P.L.; Bagchi, R.A.; Ghamari-Langroudi, M.; Porter, D.T.; Wang, Q.; Litt, M.J.; Jana, S.; Hagen, S.; et al. Histone deacetylase 6 inhibition restores leptin sensitivity and reduces obesity. Nat. Metab. 2022, 4, 44–59. [Google Scholar] [CrossRef]
- Nillni, E.A. The metabolic sensor Sirt1 and the hypothalamus: Interplay between peptide hormones and pro-hormone convertases. Mol. Cell. Endocrinol. 2016, 438, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, M.O.; Antunes, C.; Geliang, G.; Liu, Z.W.; Borok, E.; Nie, Y.; Xu, A.W.; Souza, D.O.; Gao, Q.; Diano, S.; et al. Agrp neurons mediate Sirt1’s action on the melanocortin system and energy balance: Roles for Sirt1 in neuronal firing and synaptic plasticity. J. Neurosci. 2010, 30, 11815–11825. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stancakova, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar] [CrossRef]
- Rapps, K.; Kisliouk, T.; Marco, A.; Weller, A.; Meiri, N. Dieting reverses histone methylation and hypothalamic AgRP regulation in obese rats. Front. Endocrinol. 2023, 14, 1121829. [Google Scholar] [CrossRef]
- McFadden, T.; Carucci, I.; Farrell, K.; Fletchall, E.; Jarome, T.J. Hypothalamic DNA 5-hydroxymethylation levels are altered by diet-induced weight gain during the development of obesity in a sex-specific manner. Brain Res. 2023, 1817, 148478. [Google Scholar] [CrossRef]
- Li, J.; Long, J.; Zhang, Q.; Shen, H.; Guo, A.Y.; Ma, Z.; Zhang, G. Hypothalamic long noncoding RNA AK044061 is involved in the development of dietary obesity in mice. Int. J. Obes. 2021, 45, 2638–2647. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef]
- Shi, X.; Wang, X.; Li, Q.; Su, M.; Chew, E.; Wong, E.T.; Lacza, Z.; Radda, G.K.; Tergaonkar, V.; Han, W. Nuclear factor kappaB (NF-kappaB) suppresses food intake and energy expenditure in mice by directly activating the Pomc promoter. Diabetologia 2013, 56, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.; Ghosh, S. Regulation of the NF-kappaB-Mediated Transcription of Inflammatory Genes. Front. Immunol. 2014, 5, 71. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wan, B.; Zhang, H.; Zhang, L.; Zhang, R.; Li, L.; Zhang, Y.; Hu, C. Histone lactylation mediated by Fam172a in POMC neurons regulates energy balance. Nat. Commun. 2024, 15, 10111. [Google Scholar] [CrossRef] [PubMed]
- van Baak, M.A.; Mariman, E.C.M. Mechanisms of weight regain after weight loss—The role of adipose tissue. Nat. Rev. Endocrinol. 2019, 15, 274–287. [Google Scholar] [CrossRef]
- Hall, K.D.; Kahan, S. Maintenance of Lost Weight and Long-Term Management of Obesity. Med. Clin. N. Am. 2018, 102, 183–197. [Google Scholar] [CrossRef]
- Magro, D.O.; Geloneze, B.; Delfini, R.; Pareja, B.C.; Callejas, F.; Pareja, J.C. Long-term weight regain after gastric bypass: A 5-year prospective study. Obes. Surg. 2008, 18, 648–651. [Google Scholar] [CrossRef]
- Lauti, M.; Kularatna, M.; Hill, A.G.; MacCormick, A.D. Weight Regain Following Sleeve Gastrectomy-a Systematic Review. Obes. Surg. 2016, 26, 1326–1334. [Google Scholar] [CrossRef]
- Hinte, L.C.; Castellano-Castillo, D.; Ghosh, A.; Melrose, K.; Gasser, E.; Noe, F.; Massier, L.; Dong, H.; Sun, W.; Hoffmann, A.; et al. Adipose tissue retains an epigenetic memory of obesity after weight loss. Nature 2024, 636, 457–465. [Google Scholar] [CrossRef]
- Liang, X.; Yang, Q.; Zhang, L.; Maricelli, J.W.; Rodgers, B.D.; Zhu, M.J.; Du, M. Maternal high-fat diet during lactation impairs thermogenic function of brown adipose tissue in offspring mice. Sci. Rep. 2016, 6, 34345. [Google Scholar] [CrossRef]
- Yi, D.; Nguyen, H.P.; Sul, H.S. Epigenetic dynamics of the thermogenic gene program of adipocytes. Biochem. J. 2020, 477, 1137–1148. [Google Scholar] [CrossRef]
- Librizzi, M.; Naselli, F.; Abruscato, G.; Luparello, C.; Caradonna, F. Parathyroid Hormone Related Protein (PTHrP)-Associated Molecular Signatures in Tissue Differentiation and Non-Tumoral Diseases. Biology 2023, 12, 950. [Google Scholar] [CrossRef]
- Liu, X.; Chen, Y.; Zhao, L.; Tian, Q.; deAvila, J.M.; Zhu, M.J.; Du, M. Dietary succinate supplementation to maternal mice improves fetal brown adipose tissue development and thermogenesis of female offspring. J. Nutr. Biochem. 2022, 100, 108908. [Google Scholar] [CrossRef] [PubMed]
- Brons, C.; Jacobsen, S.; Nilsson, E.; Ronn, T.; Jensen, C.B.; Storgaard, H.; Poulsen, P.; Groop, L.; Ling, C.; Astrup, A.; et al. Deoxyribonucleic acid methylation and gene expression of PPARGC1A in human muscle is influenced by high-fat overfeeding in a birth-weight-dependent manner. J. Clin. Endocrinol. Metab. 2010, 95, 3048–3056. [Google Scholar] [CrossRef] [PubMed]
- Gillberg, L.; Perfilyev, A.; Brons, C.; Thomasen, M.; Grunnet, L.G.; Volkov, P.; Rosqvist, F.; Iggman, D.; Dahlman, I.; Riserus, U.; et al. Adipose tissue transcriptomics and epigenomics in low birthweight men and controls: Role of high-fat overfeeding. Diabetologia 2016, 59, 799–812. [Google Scholar] [CrossRef]
- Jacobsen, S.C.; Brons, C.; Bork-Jensen, J.; Ribel-Madsen, R.; Yang, B.; Lara, E.; Hall, E.; Calvanese, V.; Nilsson, E.; Jorgensen, S.W.; et al. Effects of short-term high-fat overfeeding on genome-wide DNA methylation in the skeletal muscle of healthy young men. Diabetologia 2012, 55, 3341–3349. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.C.; Gillberg, L.; Bork-Jensen, J.; Ribel-Madsen, R.; Lara, E.; Calvanese, V.; Ling, C.; Fernandez, A.F.; Fraga, M.F.; Poulsen, P.; et al. Young men with low birthweight exhibit decreased plasticity of genome-wide muscle DNA methylation by high-fat overfeeding. Diabetologia 2014, 57, 1154–1158. [Google Scholar] [CrossRef]
- Ronn, T.; Volkov, P.; Gillberg, L.; Kokosar, M.; Perfilyev, A.; Jacobsen, A.L.; Jorgensen, S.W.; Brons, C.; Jansson, P.A.; Eriksson, K.F.; et al. Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum. Mol. Genet. 2015, 24, 3792–3813. [Google Scholar] [CrossRef]
- Dahlman, I.; Sinha, I.; Gao, H.; Brodin, D.; Thorell, A.; Ryden, M.; Andersson, D.P.; Henriksson, J.; Perfilyev, A.; Ling, C.; et al. The fat cell epigenetic signature in post-obese women is characterized by global hypomethylation and differential DNA methylation of adipogenesis genes. Int. J. Obes. 2015, 39, 910–919. [Google Scholar] [CrossRef]
- Shanaki, M.; Omidifar, A.; Shabani, P.; Toolabi, K. Association between HDACs and pro-inflammatory cytokine gene expressions in obesity. Arch. Physiol. Biochem. 2022, 128, 880–886. [Google Scholar] [CrossRef]
- Lechner, L.; Opitz, R.; Silver, M.J.; Krabusch, P.M.; Prentice, A.M.; Field, M.S.; Stachelscheid, H.; Leitao, E.; Schroder, C.; Fernandez Vallone, V.; et al. Early-set POMC methylation variability is accompanied by increased risk for obesity and is addressable by MC4R agonist treatment. Sci. Transl. Med. 2023, 15, eadg1659. [Google Scholar] [CrossRef]
- Tatton-Brown, K.; Zachariou, A.; Loveday, C.; Renwick, A.; Mahamdallie, S.; Aksglaede, L.; Baralle, D.; Barge-Schaapveld, D.; Blyth, M.; Bouma, M.; et al. The Tatton-Brown-Rahman Syndrome: A clinical study of 55 individuals with de novo constitutive DNMT3A variants. Wellcome Open Res. 2018, 3, 46. [Google Scholar] [CrossRef] [PubMed]
- Xin, B.; Cruz Marino, T.; Szekely, J.; Leblanc, J.; Cechner, K.; Sency, V.; Wensel, C.; Barabas, M.; Therriault, V.; Wang, H. Novel DNMT3A germline mutations are associated with inherited Tatton-Brown-Rahman syndrome. Clin. Genet. 2017, 91, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Tovy, A.; Reyes, J.M.; Zhang, L.; Huang, Y.H.; Rosas, C.; Daquinag, A.C.; Guzman, A.; Ramabadran, R.; Chen, C.W.; Gu, T.; et al. Constitutive loss of DNMT3A causes morbid obesity through misregulation of adipogenesis. Elife 2022, 11, e72359. [Google Scholar] [CrossRef]
- Kohno, D.; Lee, S.; Harper, M.J.; Kim, K.W.; Sone, H.; Sasaki, T.; Kitamura, T.; Fan, G.; Elmquist, J.K. Dnmt3a in Sim1 neurons is necessary for normal energy homeostasis. J. Neurosci. 2014, 34, 15288–15296. [Google Scholar] [CrossRef] [PubMed]
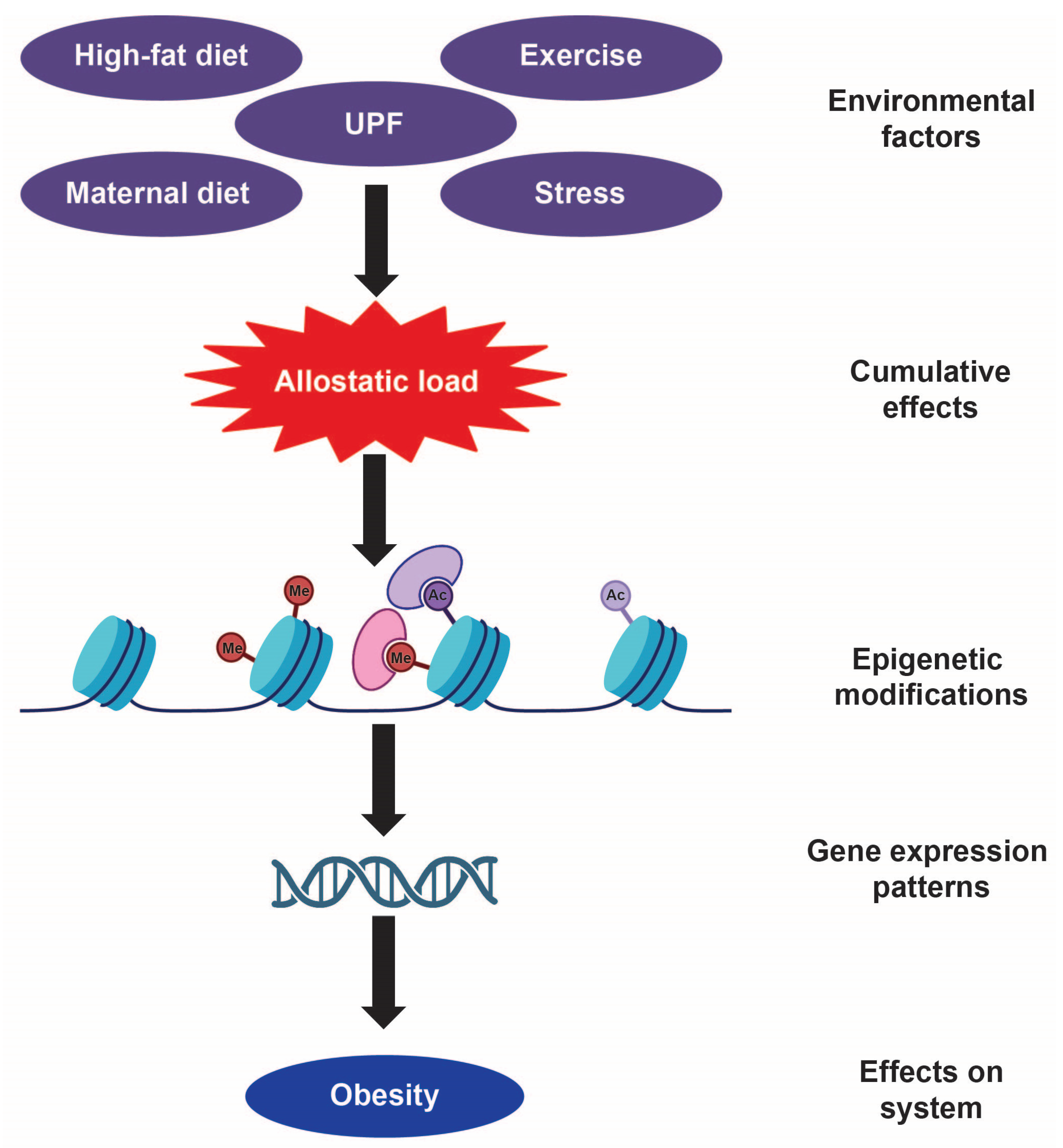
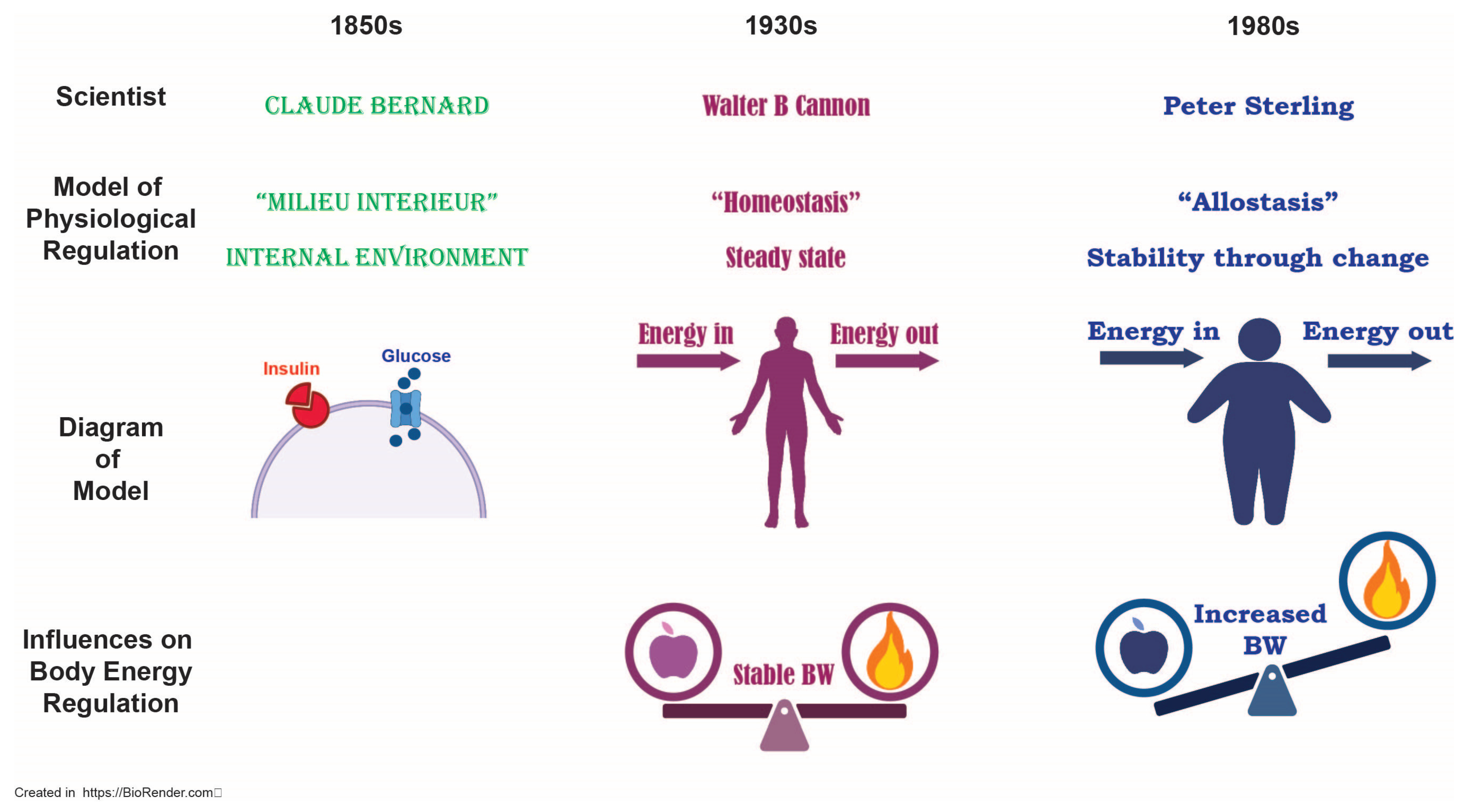
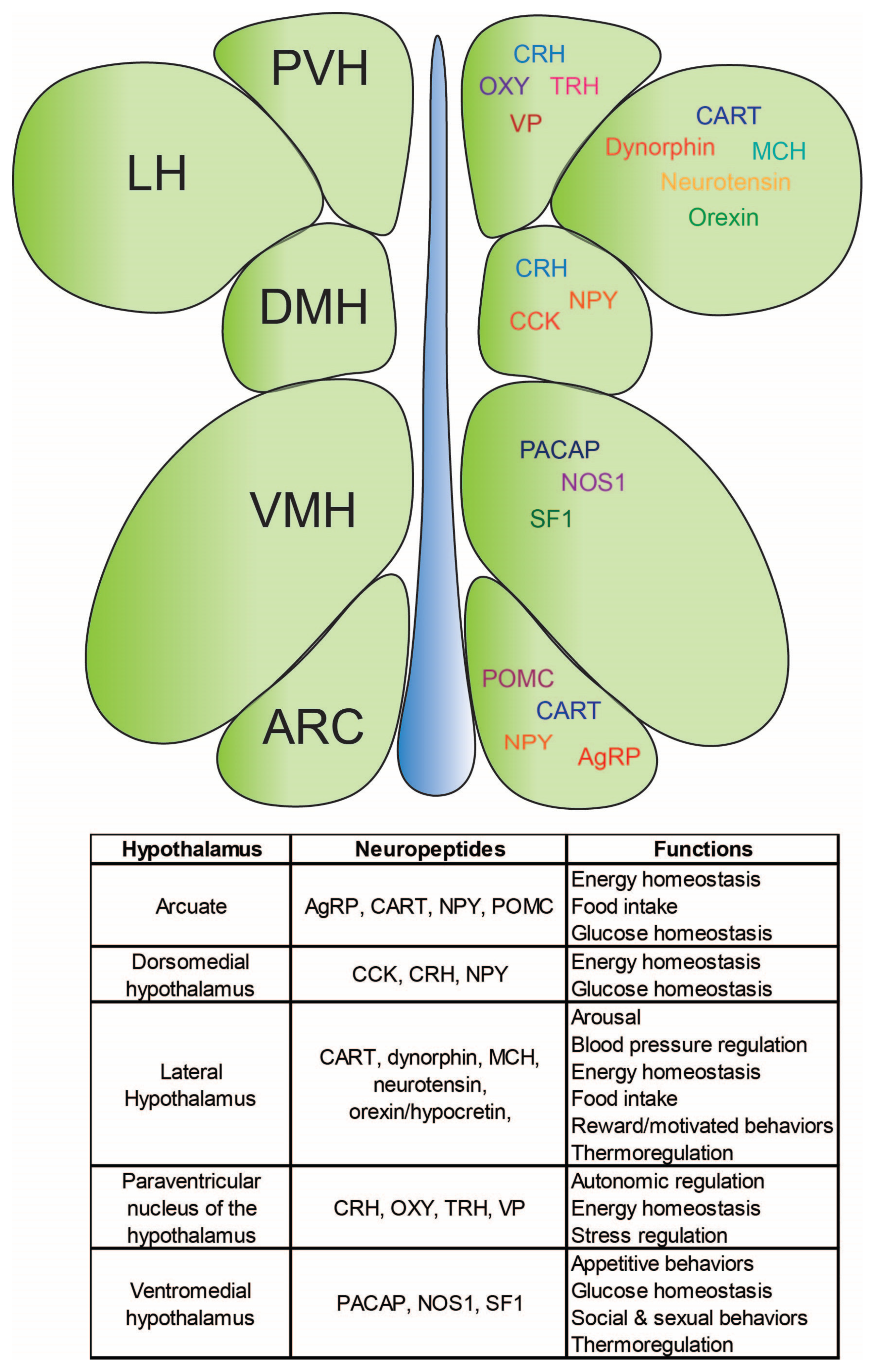
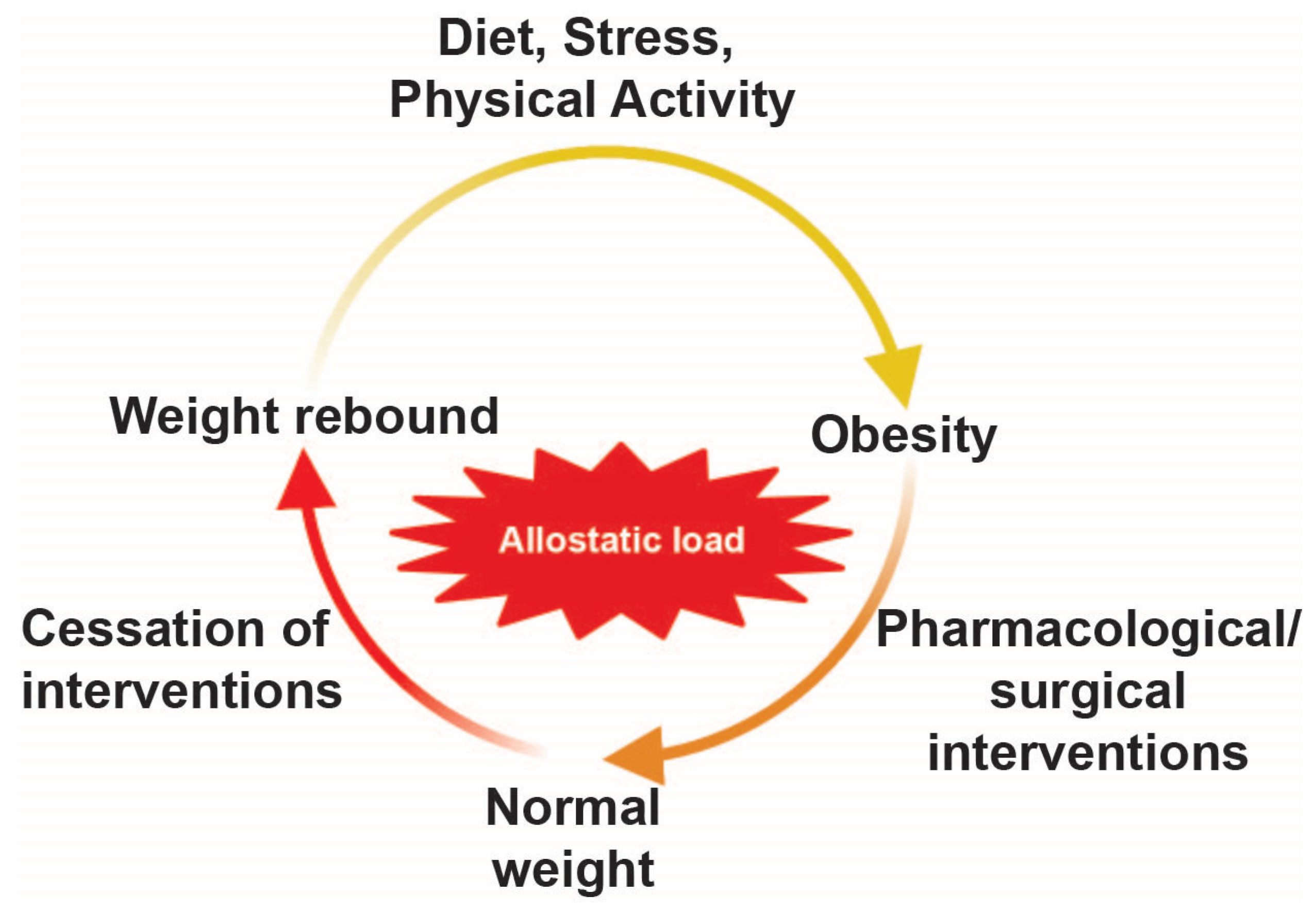
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Na, E.S. Epigenetic Mechanisms of Obesity: Insights from Transgenic Animal Models. Life 2025, 15, 653. https://doi.org/10.3390/life15040653
Na ES. Epigenetic Mechanisms of Obesity: Insights from Transgenic Animal Models. Life. 2025; 15(4):653. https://doi.org/10.3390/life15040653
Chicago/Turabian StyleNa, Elisa S. 2025. "Epigenetic Mechanisms of Obesity: Insights from Transgenic Animal Models" Life 15, no. 4: 653. https://doi.org/10.3390/life15040653
APA StyleNa, E. S. (2025). Epigenetic Mechanisms of Obesity: Insights from Transgenic Animal Models. Life, 15(4), 653. https://doi.org/10.3390/life15040653