
Beyond a Single Marker: An Update on the Comprehensive Evaluation of Right Ventricular Dysfunction in Pulmonary Thromboembolism


 , ,
, , 

Abstract
:1. Introduction
2. Pathophysiological Overview and Risk Assessment of Right Ventricular Dysfunction in Pulmonary Thromboembolism
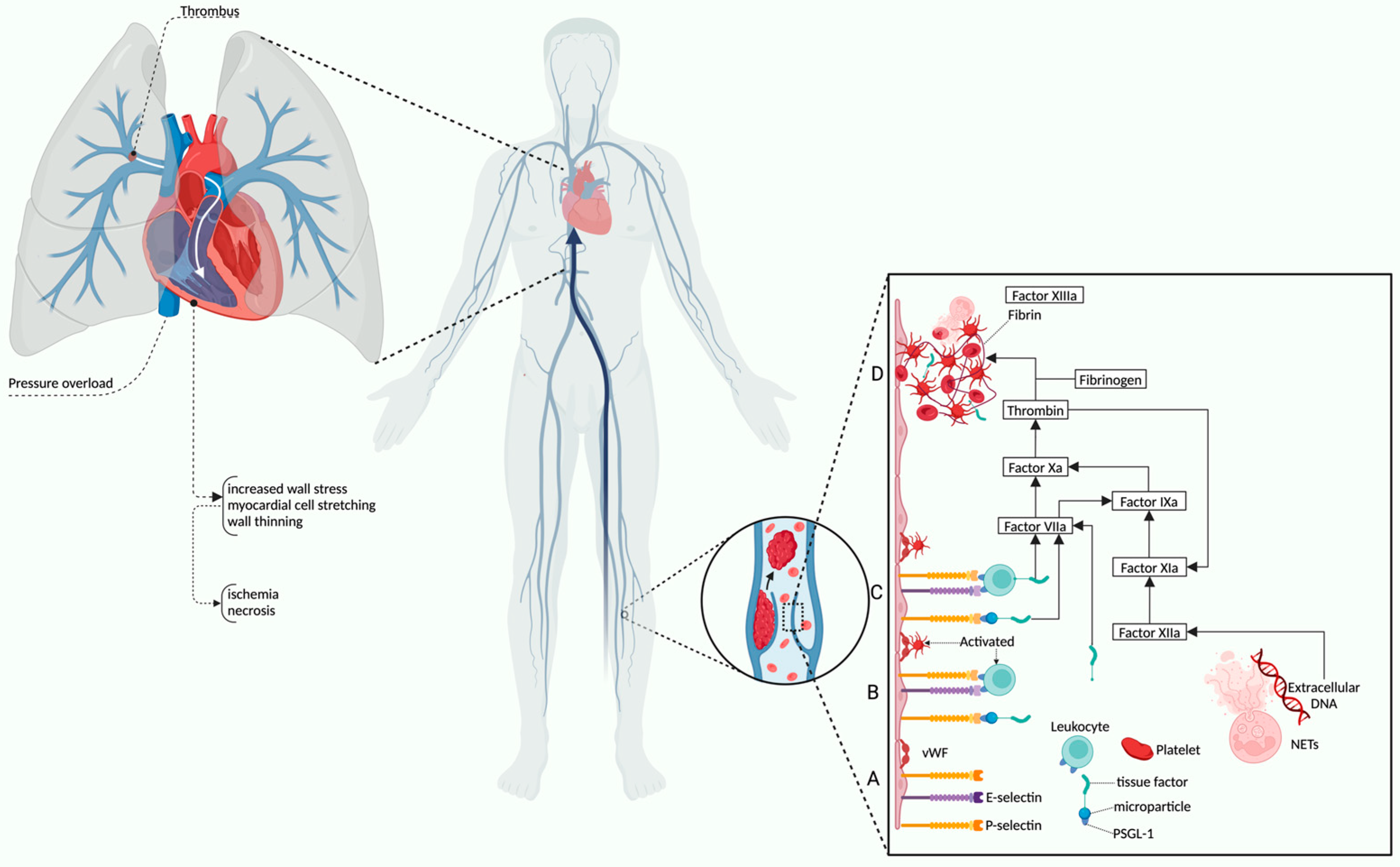
2.1. Physiology and Pathophysiological Mechanisms of RV Dysfunction in PE
2.2. Clinical Assessment and Risk Stratification of RV Dysfunction
3. Biomarkers Correlated with Right Ventricular Dysfunction in Pulmonary Thromboembolism
3.1. High-Sensitivity Cardiac Troponin I (hs-cTnI)
3.2. N-Terminal Pro-B-Type Natriuretic Peptide (NT-proBNP)
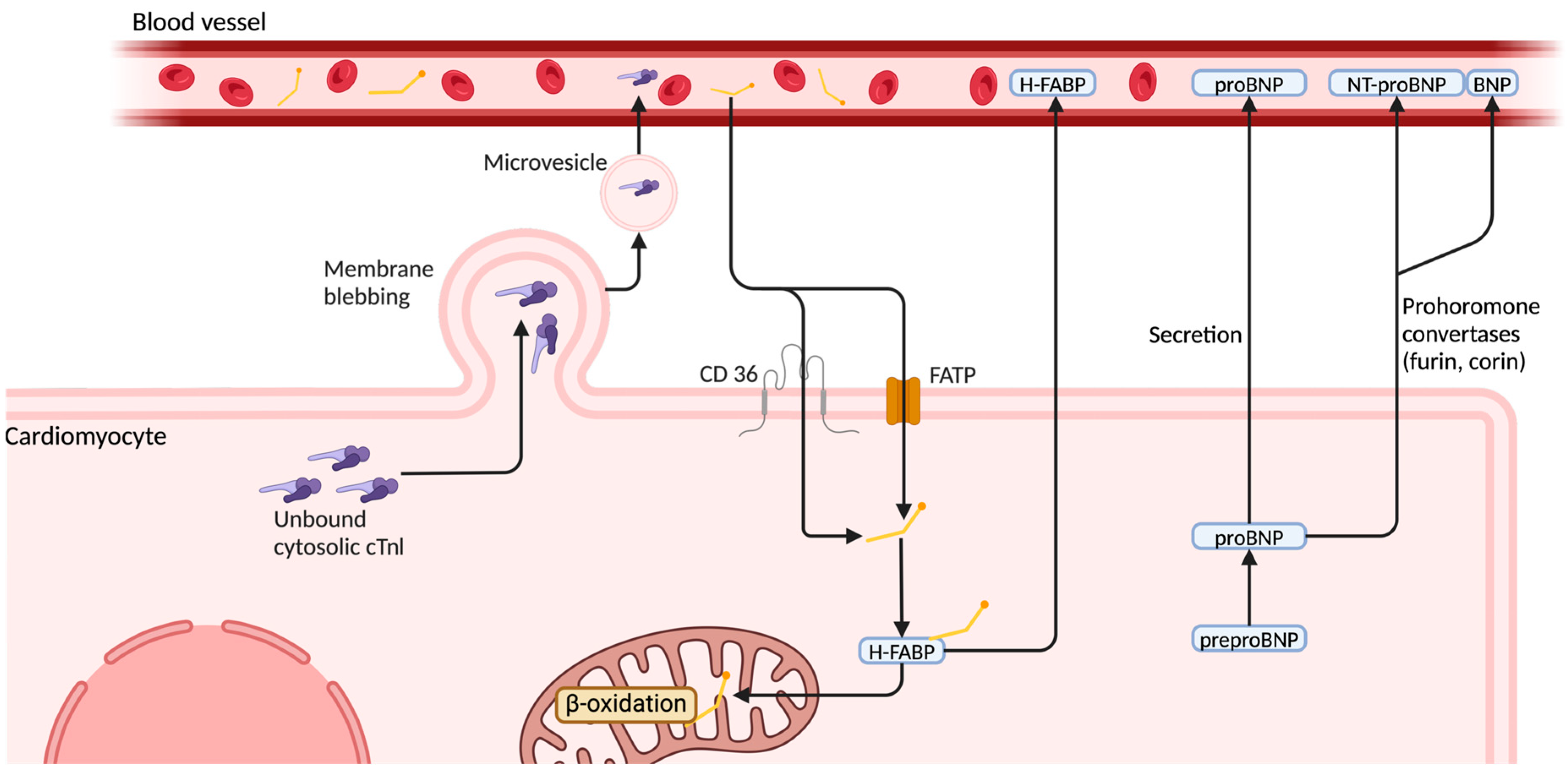
3.3. Heart-Type Fatty Acid-Binding Protein (H-FABP)
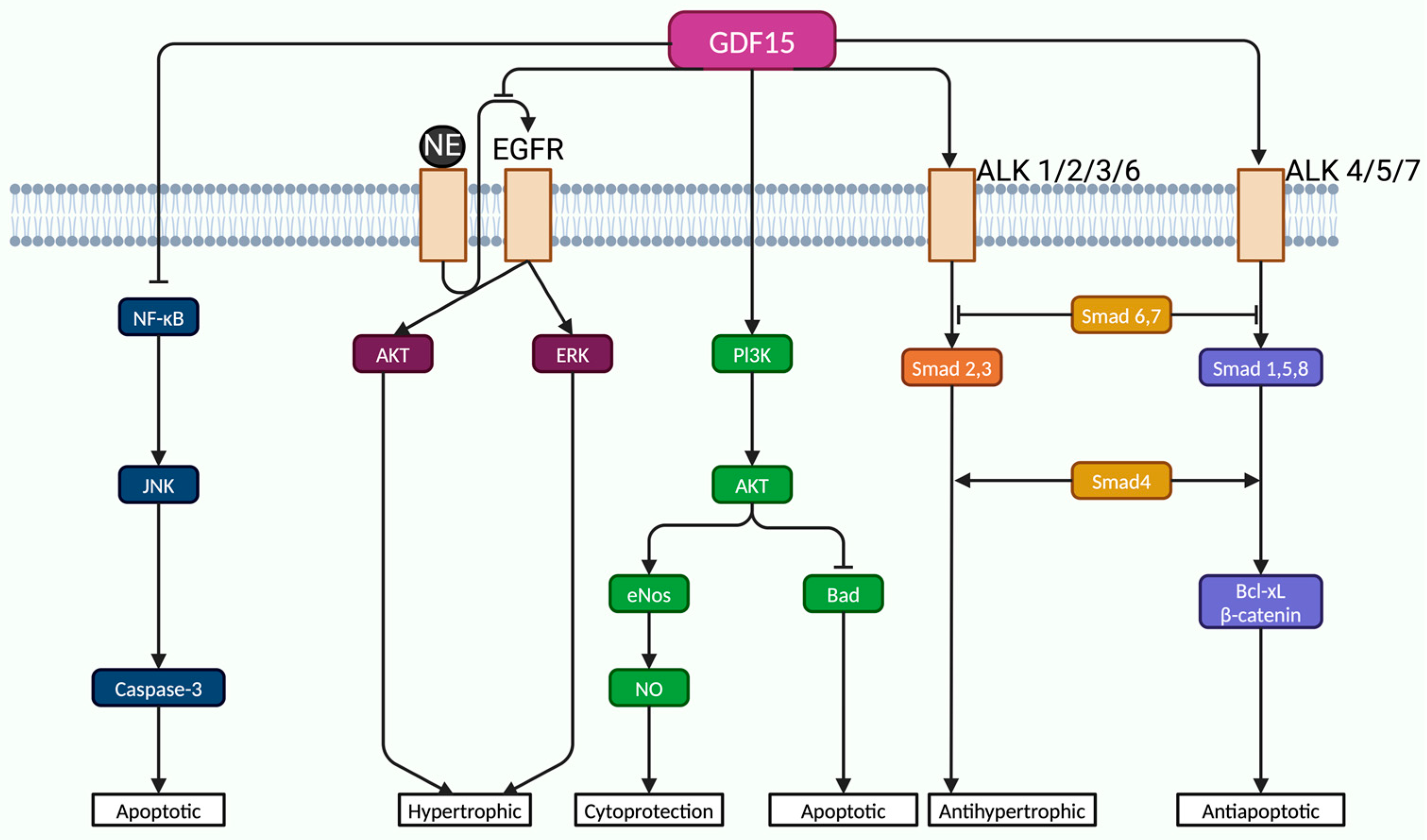
3.4. Growth Differentiation Factor 15 (GDF-15)
3.5. Clinical Utility of Biomarkers Across Risk Stratification Subgroups in PE
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tritschler, T.; Kraaijpoel, N.; Girard, P.; Büller, H.R.; Langlois, N.; Righini, M.; Schulman, S.; Segers, A.; Le Gal, G. Definition of Pulmonary Embolism-related Death and Classification of the Cause of Death in Venous Thromboembolism Studies: Communication from the SSC of the ISTH. J. Thromb. Haemost. 2020, 18, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Piazza, G. Advanced Management of Intermediate-and High-Risk Pulmonary Embolism. J. Am. Coll. Cardiol. 2020, 76, 2117–2127. [Google Scholar] [CrossRef]
- Lutsey, P.L.; Zakai, N.A. Epidemiology and Prevention of Venous Thromboembolism. Nat. Rev. Cardiol. 2023, 20, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Duffett, L.; Castellucci, L.A.; Forgie, M.A. Pulmonary Embolism: Update on Management and Controversies. BMJ 2020, 370, m2177. [Google Scholar] [CrossRef] [PubMed]
- Freund, Y.; Cohen-Aubart, F.; Bloom, B. Acute Pulmonary Embolism: A Review. JAMA 2022, 328, 1336. [Google Scholar] [CrossRef]
- Sanz, J.; Sánchez-Quintana, D.; Bossone, E.; Bogaard, H.J.; Naeije, R. Anatomy, Function, and Dysfunction of the Right Ventricle. J. Am. Coll. Cardiol. 2019, 73, 1463–1482. [Google Scholar] [CrossRef]
- Lyhne, M.D.; Kline, J.A.; Nielsen-Kudsk, J.E.; Andersen, A. Pulmonary Vasodilation in Acute Pulmonary Embolism—a Systematic Review. Pulm. Circ. 2020, 10, 1–16. [Google Scholar] [CrossRef]
- Bertoletti, L.; Montani, D.; Humbert, M. Right Ventricle Dysfunction in Patients with Acute Pulmonary Embolism Supposedly at Low Risk for Death: When Evidence-Based Medicine Rescues Clinical Practice. Eur. Heart J. 2021, 42, 3200–3202. [Google Scholar] [CrossRef]
- Elshahaat, H.A.; Zayed, N.E.; Ateya, M.A.; Safwat, M.; El Hawary, A.T.; Abozaid, M.N. Role of Serum Biomarkers in Predicting Management Strategies for Acute Pulmonary Embolism. Heliyon 2023, 9, e21068. [Google Scholar] [CrossRef]
- Yang, L.; Li, B.; Chen, H.; Belfeki, N.; Monchi, M.; Moini, C. The Role of Troponin in the Diagnosis and Treatment of Acute Pulmonary Embolism: Mechanisms of Elevation, Prognostic Evaluation, and Clinical Decision-Making. Cureus 2024, 16, e67922. [Google Scholar] [CrossRef]
- Inampudi, C.; Tedford, R.J.; Hemnes, A.R.; Hansmann, G.; Bogaard, H.-J.; Koestenberger, M.; Lang, I.M.; Brittain, E.L. Treatment of Right Ventricular Dysfunction and Heart Failure in Pulmonary Arterial Hypertension. Cardiovasc. Diagn. Ther. 2020, 10, 1659–1674. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Liang, S.; He, Z.; Jin, Y.; Lang, Z.; Liu, H.; Wang, Y.; Li, S. The Prognostic Value of the Serum Levels of Brain Natriuretic Peptide, Troponin I, and D-Dimer, in Addition to the Neutrophil-to-Lymphocyte Ratio, for the Disease Evaluation of Patients with Acute Pulmonary Embolism. IJGM 2021, 14, 303–308. [Google Scholar] [CrossRef]
- Konstantinides, S.V.; Meyer, G.; Becattini, C.; Bueno, H.; Geersing, G.-J.; Harjola, V.-P.; Huisman, M.V.; Humbert, M.; Jennings, C.S.; Jiménez, D.; et al. 2019 ESC Guidelines for the Diagnosis and Management of Acute Pulmonary Embolism Developed in Collaboration with the European Respiratory Society (ERS): The Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). Eur. Respir. J. 2019, 54, 1901647. [Google Scholar] [CrossRef] [PubMed]
- Bryce, Y.C.; Perez-Johnston, R.; Bryce, E.B.; Homayoon, B.; Santos-Martin, E.G. Pathophysiology of Right Ventricular Failure in Acute Pulmonary Embolism and Chronic Thromboembolic Pulmonary Hypertension: A Pictorial Essay for the Interventional Radiologist. Insights Imaging 2019, 10, 18. [Google Scholar] [CrossRef]
- Chen, Y.L.; Wright, C.; Pietropaoli, A.P.; Elbadawi, A.; Delehanty, J.; Barrus, B.; Gosev, I.; Trawick, D.; Patel, D.; Cameron, S.J. Right Ventricular Dysfunction Is Superior and Sufficient for Risk Stratification by a Pulmonary Embolism Response Team. J. Thromb. Thrombolysis 2020, 49, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, M.; Huber, L.C.; Winnik, S.; Mikulicic, F.; Guidetti, F.; Frank, M.; Flammer, A.J.; Ruschitzka, F. Right Ventricular Failure: Pathophysiology, Diagnosis and Treatment. Card. Fail. Rev. 2019, 5, 140–146. [Google Scholar] [CrossRef]
- Bianco, F.; Bucciarelli, V.; Ammirati, E.; Occhi, L.; Musca, F.; Tonti, G.; Frigerio, M.; Gallina, S. Assessment of Right Ventricular Function in Advanced Heart Failure with Nonischemic Dilated Cardiomyopathy: Insights of Right Ventricular Elastance. J. Cardiovasc. Med. 2020, 21, 134–143. [Google Scholar] [CrossRef]
- Mirambeaux, R.; Le Mao, R.; Muriel, A.; Pintado, B.; Pérez, A.; Velasco, D.; Lobo, J.L.; Barrios, D.; Morillo, R.; Bikdeli, B.; et al. Implications of Abnormal Troponin Levels with Normal Right Ventricular Function in Normotensive Patients with Acute Pulmonary Embolism. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620967760. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Celermajer, D.S.; D’Alto, M.; Haddad, F.; Hassoun, P.M.; Prins, K.W.; Naeije, R.; Vonk Noordegraaf, A. Pathophysiology of the Right Ventricle and Its Pulmonary Vascular Interaction. Eur. Respir. J. 2024, 64, 2401321. [Google Scholar] [CrossRef]
- Wang, J.M.H.; Rai, R.; Carrasco, M.; Sam-Odusina, T.; Salandy, S.; Gielecki, J.; Zurada, A.; Loukas, M. An Anatomical Review of the Right Ventricle. Transl. Res. Anat. 2019, 17, 100049. [Google Scholar] [CrossRef]
- Martinez Licha, C.R.; McCurdy, C.M.; Maldonado, S.M.; Lee, L.S. Current Management of Acute Pulmonary Embolism. ATCS 2020, 26, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, P.; Tian, H.; Zhen, K.; McCabe, C.; Zhao, L.; Zhai, Z. Right Ventricle Remodeling in Chronic Thromboembolic Pulmonary Hypertension. J. Transl. Intern. Med. 2022, 10, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Buckberg, G.; Hoffman, J.I.E. Right Ventricular Architecture Responsible for Mechanical Performance: Unifying Role of Ventricular Septum. J. Thorac. Cardiovasc. Surg. 2014, 148, 3166–3171.e4. [Google Scholar] [CrossRef] [PubMed]
- Peracaula, M.; Sebastian, L.; Francisco, I.; Vilaplana, M.B.; Rodríguez-Chiaradía, D.A.; Tura-Ceide, O. Decoding Pulmonary Embolism: Pathophysiology, Diagnosis, and Treatment. Biomedicines 2024, 12, 1936. [Google Scholar] [CrossRef]
- Alerhand, S.; Sundaram, T.; Gottlieb, M. What Are the Echocardiographic Findings of Acute Right Ventricular Strain That Suggest Pulmonary Embolism? Anaesth. Crit. Care Pain. Med. 2021, 40, 100852. [Google Scholar] [CrossRef]
- Escopy, S.; Chaikof, E.L. Targeting the P-Selectin/PSGL-1 Pathway: Discovery of Disease-Modifying Therapeutics for Disorders of Thromboinflammation. Blood Vessel. Thromb. Hemost. 2024, 1, 100015. [Google Scholar] [CrossRef]
- Thålin, C.; Hisada, Y.; Lundström, S.; Mackman, N.; Wallén, H. Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. ATVB 2019, 39, 1724–1738. [Google Scholar] [CrossRef]
- Reddy, S.; Bernstein, D. Molecular Mechanisms of Right Ventricular Failure. Circulation 2015, 132, 1734–1742. [Google Scholar] [CrossRef]
- Greyson, C.R. Pathophysiology of Right Ventricular Failure. Crit. Care Med. 2008, 36, S57–S65. [Google Scholar] [CrossRef]
- Vamsidhar, A.; Rajasekhar, D.; Vanajakshamma, V.; Lakshmi, A.Y.; Latheef, K.; Siva Sankara, C.; Obul Reddy, G. Comparison of PESI, Echocardiogram, CTPA, and NT-proBNP as Risk Stratification Tools in Patients with Acute Pulmonary Embolism. Indian. Heart J. 2017, 69, 68–74. [Google Scholar] [CrossRef]
- Yang, P.; Li, H.; Zhang, J.; Xu, X. Research Progress on Biomarkers of Pulmonary Embolism. Clin. Respir. J. 2021, 15, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Machanahalli Balakrishna, A.; Reddi, V.; Belford, P.M.; Alvarez, M.; Jaber, W.A.; Zhao, D.X.; Vallabhajosyula, S. Intermediate-Risk Pulmonary Embolism: A Review of Contemporary Diagnosis, Risk Stratification and Management. Medicina 2022, 58, 1186. [Google Scholar] [CrossRef] [PubMed]
- Bikdeli, B.; Muriel, A.; Rodríguez, C.; González, S.; Briceño, W.; Mehdipoor, G.; Piazza, G.; Ballaz, A.; Lippi, G.; Yusen, R.D.; et al. High-Sensitivity vs Conventional Troponin Cutoffs for Risk Stratification in Patients with Acute Pulmonary Embolism. JAMA Cardiol. 2024, 9, 64. [Google Scholar] [CrossRef]
- Houston, B.A.; Brittain, E.L.; Tedford, R.J. Right Ventricular Failure. N. Engl. J. Med. 2023, 388, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, N.M.; Mullin, C.; Poor, H.D. Biomarkers and Right Ventricular Dysfunction. Crit. Care Clin. 2020, 36, 141–153. [Google Scholar] [CrossRef]
- Briceño, W.; Díaz, G.; Castillo, A.; Jara, I.; Yong, E.; Lago, L.; Dam Lyhne, M.; Monreal, M.; Bikdeli, B.; Jimenez, D. Time to Resolution of Right Ventricle Dysfunction in Patients with Acute Pulmonary Embolism. Arch. De. Bronconeumol. 2024, 60, 448–450. [Google Scholar] [CrossRef]
- Thandavarayan, R.A.; Chitturi, K.R.; Guha, A. Pathophysiology of Acute and Chronic Right Heart Failure. Cardiol. Clin. 2020, 38, 149–160. [Google Scholar] [CrossRef]
- Hobohm, L.; Becattini, C.; Konstantinides, S.V.; Casazza, F.; Lankeit, M. Validation of a Fast Prognostic Score for Risk Stratification of Normotensive Patients with Acute Pulmonary Embolism. Clin. Res. Cardiol. 2020, 109, 1008–1017. [Google Scholar] [CrossRef]
- Sagcan, G.; Dogan, Z.; Uzun, H.; Cuhadaroglu, C.; Okumus, G.; Arseven, O. Impact of Promising Biomarkers on Severity and Outcome of Acute Pulmonary Embolism. IJGM 2023, 16, 3301–3309. [Google Scholar] [CrossRef]
- Ajah, O.N. Pulmonary Embolism and Right Ventricular Dysfunction: Mechanism and Management. Cureus 2024, 16, e70561. [Google Scholar] [CrossRef]
- Berglund, F.; Piña, P.; Herrera, C.J. Right Ventricle in Heart Failure with Preserved Ejection Fraction. Heart 2020, 106, 1798–1804. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, P.; Chauhan, A.; Goyal, P.; Singh, A. H-FABP: A Beacon of Hope for Prediabetic Heart Disease. J. Fam. Med. Prim. Care 2020, 9, 3421. [Google Scholar] [CrossRef]
- Elias, A.; Mallett, S.; Daoud-Elias, M.; Poggi, J.-N.; Clarke, M. Prognostic Models in Acute Pulmonary Embolism: A Systematic Review and Meta-Analysis. BMJ Open 2016, 6, e010324. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shao, X.; Zhang, Y.; Zhang, Z.; Tao, X.; Zhai, Z.; Wang, C. Assessment of the Bova Score for Risk Stratification of Acute Normotensive Pulmonary Embolism: A Systematic Review and Meta-Analysis. Thromb. Res. 2020, 193, 99–106. [Google Scholar] [CrossRef]
- Barco, S.; Mahmoudpour, S.H.; Planquette, B.; Sanchez, O.; Konstantinides, S.V.; Meyer, G. Prognostic Value of Right Ventricular Dysfunction or Elevated Cardiac Biomarkers in Patients with Low-Risk Pulmonary Embolism: A Systematic Review and Meta-Analysis. Eur. Heart J. 2019, 40, 902–910. [Google Scholar] [CrossRef]
- Smolarek, D.; Gruchała, M.; Sobiczewski, W. Echocardiographic Evaluation of Right Ventricular Systolic Function: The Traditional and Innovative Approach. Cardiol. J. 2017, 24, 563–572. [Google Scholar] [CrossRef]
- Hahn, R.T.; Lerakis, S.; Delgado, V.; Addetia, K.; Burkhoff, D.; Muraru, D.; Pinney, S.; Friedberg, M.K. Multimodality Imaging of Right Heart Function. J. Am. Coll. Cardiol. 2023, 81, 1954–1973. [Google Scholar] [CrossRef]
- Muraru, D.; Haugaa, K.; Donal, E.; Stankovic, I.; Voigt, J.U.; Petersen, S.E.; Popescu, B.A.; Marwick, T. Right Ventricular Longitudinal Strain in the Clinical Routine: A State-of-the-Art Review. Eur. Heart J. Cardiovasc. Imaging 2022, 23, 898–912. [Google Scholar] [CrossRef]
- Jones, N.; Burns, A.T.; Prior, D.L. Echocardiographic Assessment of the Right Ventricle–State of the Art. Heart Lung Circ. 2019, 28, 1339–1350. [Google Scholar] [CrossRef]
- Dutta, T.; Aronow, W.S. Echocardiographic Evaluation of the Right Ventricle: Clinical Implications. Clin. Cardiol. 2017, 40, 542–548. [Google Scholar] [CrossRef]
- The Steering Committee. Single-Bolus Tenecteplase plus Heparin Compared with Heparin Alone for Normotensive Patients with Acute Pulmonary Embolism Who Have Evidence of Right Ventricular Dysfunction and Myocardial Injury: Rationale and Design of the Pulmonary Embolism Thrombolysis (PEITHO) Trial. Am. Heart J. 2012, 163, 33–38.e1. [Google Scholar] [CrossRef]
- Wang, D.; Fan, G.; Zhang, X.; Xi, L.; Chen, Y.; Li, A.; Zhai, Z. Prevalence of Long-Term Right Ventricular Dysfunction after Acute Pulmonary Embolism: A Systematic Review and Meta-Analysis. eClinicalMedicine 2023, 62, 102153. [Google Scholar] [CrossRef] [PubMed]
- Janisset, L.; Castan, M.; Poenou, G.; Lachand, R.; Mismetti, P.; Viallon, A.; Bertoletti, L. Cardiac Biomarkers in Patients with Acute Pulmonary Embolism. Medicina 2022, 58, 541. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.; Melot, J.; Krinock, M.D.; Kumar, A.; Nadar, S.K.; Lip, G.Y.H. Heart-Type Fatty Acid-Binding Protein: An Overlooked Cardiac Biomarker. Ann. Med. 2020, 52, 444–461. [Google Scholar] [CrossRef]
- Klok, F.A.; Ageno, W.; Ay, C.; Bäck, M.; Barco, S.; Bertoletti, L.; Becattini, C.; Carlsen, J.; Delcroix, M.; Van Es, N.; et al. Optimal Follow-up after Acute Pulmonary Embolism: A Position Paper of the European Society of Cardiology Working Group on Pulmonary Circulation and Right Ventricular Function, in Collaboration with the European Society of Cardiology Working Group on Atherosclerosis and Vascular Biology, Endorsed by the European Respiratory Society. Eur. Heart J. 2022, 43, 183–189. [Google Scholar] [CrossRef]
- Marti, C. Risk Stratification in Patients with Acute Pulmonary Embolisms. Privat-Docent Thesis, Université de Genève, Geneva, Switzerland, 2020. [Google Scholar] [CrossRef]
- El-Menyar, A.; Sathian, B.; Al-Thani, H. Elevated Serum Cardiac Troponin and Mortality in Acute Pulmonary Embolism: Systematic Review and Meta-Analysis. Respir. Med. 2019, 157, 26–35. [Google Scholar] [CrossRef]
- Millington, S.J.; Aissaoui, N.; Bowcock, E.; Brodie, D.; Burns, K.E.A.; Douflé, G.; Haddad, F.; Lahm, T.; Piazza, G.; Sanchez, O.; et al. High and Intermediate Risk Pulmonary Embolism in the ICU. Intensive Care Med. 2024, 50, 195–208. [Google Scholar] [CrossRef]
- Becattini, C.; Maraziti, G.; Vinson, D.R.; Ng, A.C.C.; Den Exter, P.L.; Côté, B.; Vanni, S.; Doukky, R.; Khemasuwan, D.; Weekes, A.J.; et al. Right Ventricle Assessment in Patients with Pulmonary Embolism at Low Risk for Death Based on Clinical Models: An Individual Patient Data Meta-Analysis. Eur. Heart J. 2021, 42, 3190–3199. [Google Scholar] [CrossRef]
- Padang, R.; Chandrashekar, N.; Indrabhinduwat, M.; Scott, C.G.; Luis, S.A.; Chandrasekaran, K.; Michelena, H.I.; Nkomo, V.T.; Pislaru, S.V.; Pellikka, P.A.; et al. Aetiology and Outcomes of Severe Right Ventricular Dysfunction. Eur. Heart J. 2020, 41, 1273–1282. [Google Scholar] [CrossRef]
- Boeddinghaus, J.; Nestelberger, T.; Koechlin, L.; Wussler, D.; Lopez-Ayala, P.; Walter, J.E.; Troester, V.; Ratmann, P.D.; Seidel, F.; Zimmermann, T.; et al. Early Diagnosis of Myocardial Infarction with Point-of-Care High-Sensitivity Cardiac Troponin I. J. Am. Coll. Cardiol. 2020, 75, 1111–1124. [Google Scholar] [CrossRef]
- Turetz, M.; Sideris, A.; Friedman, O.; Triphathi, N.; Horowitz, J. Epidemiology, Pathophysiology, and Natural History of Pulmonary Embolism. Semin. Interv. Radiol. 2018, 35, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rayas, J.M.; Hernandez-Hernandez, J.A.; Lopez-Sanchez, R.D.C.; Rayas-Gomez, A.L.; Gonzalez-Yanez, J.M. Secreção Não Clássica: Um Possível Mecanismo Para Explicar as Elevações Da Troponina Cardíaca Na Ausência de Infarto Agudo Do Miocárdio. Arq. Bras. De. Cardiol. 2022, 118, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Punukollu, G.; Khan, I.A.; Gowda, R.M.; Lakhanpal, G.; Vasavada, B.C.; Sacchi, T.J. Cardiac Troponin I Release in Acute Pulmonary Embolism in Relation to the Duration of Symptoms. Int. J. Cardiol. 2005, 99, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Keller, K.; Beule, J.; Schulz, A.; Coldewey, M.; Dippold, W.; Balzer, J.O. Cardiac Troponin I for Predicting Right Ventricular Dysfunction and Intermediate Risk in Patients with Normotensive Pulmonary Embolism. Neth. Heart J. 2015, 23, 55–61. [Google Scholar] [CrossRef]
- Daquarti, G.; March Vecchio, N.; Mitrione, C.S.; Furmento, J.; Ametrano, M.C.; Dominguez Pace, M.P.; Costabel, J.P. High-Sensitivity Troponin and Right Ventricular Function in Acute Pulmonary Embolism. Am. J. Emerg. Med. 2016, 34, 1579–1582. [Google Scholar] [CrossRef]
- Cotugno, M.; Orgaz-Molina, J.; Rosa-Salazar, V.; Guirado-Torrecillas, L.; García-Pérez, B. Right Ventricular Dysfunction in Acute Pulmonary Embolism: NT-proBNP vs. Troponin, T. Med. Clínica (Engl. Ed.) 2017, 148, 339–344. [Google Scholar] [CrossRef]
- Sonne-Holm, E.; Winther-Jensen, M.; Bang, L.E.; Køber, L.; Fosbøl, E.; Carlsen, J.; Kjaergaard, J. Troponin Dependent 30-Day Mortality in Patients with Acute Pulmonary Embolism. J. Thromb. Thrombolysis 2023, 56, 485–494. [Google Scholar] [CrossRef]
- Neumann, J.T.; Twerenbold, R.; Ojeda, F.; Sörensen, N.A.; Chapman, A.R.; Shah, A.S.V.; Anand, A.; Boeddinghaus, J.; Nestelberger, T.; Badertscher, P.; et al. Application of High-Sensitivity Troponin in Suspected Myocardial Infarction. N. Engl. J. Med. 2019, 380, 2529–2540. [Google Scholar] [CrossRef]
- Matthews, T.M.; Peters, G.A.; Wang, G.; Horick, N.; Chang, K.E.; Harshbarger, S.; Prucnal, C.; Birrenkott, D.A.; Stannek, K.; Lee, E.S.; et al. The optimal cutoff values of high-sensitivity troponin and NT-proBNP for the risk-stratification of patients with acute pulmonary embolism. Clin Chem. 2024, hvae212. [Google Scholar] [CrossRef]
- Chauin, A. The Main Causes and Mechanisms of Increase in Cardiac Troponin Concentrations Other Than Acute Myocardial Infarction (Part 1): Physical Exertion, Inflammatory Heart Disease, Pulmonary Embolism, Renal Failure, Sepsis. VHRM 2021, 17, 601–617. [Google Scholar] [CrossRef]
- Ebner, M.; Guddat, N.; Keller, K.; Merten, M.C.; Lerchbaumer, M.H.; Hasenfuß, G.; Konstantinides, S.V.; Lankeit, M. High-Sensitivity Troponin I for Risk Stratification in Normotensive Pulmonary Embolism. ERJ Open Res. 2020, 6, 00625–02020. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Kim, K.H.; Yoon, H.J.; Hong, Y.J.; Kim, J.H.; Ahn, Y.; Jeong, M.H.; Cho, J.G.; Park, J.C.; Kang, J.C. Usefulness of Cardiac Biomarkers in the Prediction of Right Ventricular Dysfunction before Echocardiography in Acute Pulmonary Embolism. J. Cardiol. 2012, 60, 508–513. [Google Scholar] [CrossRef]
- Amorim, S.; Dias, P.; Rodrigues, R.; Araújo, V.; Macedo, F.; Maciel, M.J.; Gonçalves, F.R. Troponin I as a Marker of Right Ventricular Dysfunction and Severity of Pulmonary Embolism. Rev. Port. Cardiol. 2006, 25, 181–186. [Google Scholar] [PubMed]
- Martens, E.S.L.; Huisman, M.V.; Klok, F.A. Diagnostic Management of Acute Pulmonary Embolism in COVID-19 and Other Special Patient Populations. Diagnostics 2022, 12, 1350. [Google Scholar] [CrossRef]
- Mirna, M.; Rohm, I.; Jirak, P.; Wernly, B.; Bäz, L.; Paar, V.; Kretzschmar, D.; Hoppe, U.C.; Schulze, P.C.; Lichtenauer, M.; et al. Analysis of Novel Cardiovascular Biomarkers in Patients with Pulmonary Hypertension (PH). Heart Lung Circ. 2020, 29, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Surov, A.; Akritidou, M.; Bach, A.G.; Bailis, N.; Lerche, M.; Meyer, H.J.; Pech, M.; Wienke, A. A New Index for the Prediction of 30-Day Mortality in Patients with Pulmonary Embolism: The Pulmonary Embolism Mortality Score (PEMS). Angiology 2021, 72, 787–793. [Google Scholar] [CrossRef]
- Mavromanoli, A.C.; Barco, S.; Ageno, W.; Bouvaist, H.; Brodmann, M.; Cuccia, C.; Couturaud, F.; Dellas, C.; Dimopoulos, K.; Duerschmied, D.; et al. Recovery of Right Ventricular Function after Intermediate-Risk Pulmonary Embolism: Results from the Multicentre Pulmonary Embolism International Trial (PEITHO)-2. Clin. Res. Cardiol. 2023, 112, 1372–1381. [Google Scholar] [CrossRef]
- Wong, Y.-K.; Tse, H.-F. Circulating Biomarkers for Cardiovascular Disease Risk Prediction in Patients with Cardiovascular Disease. Front. Cardiovasc. Med. 2021, 8, 713191. [Google Scholar] [CrossRef]
- Goetze, J.P.; Bruneau, B.G.; Ramos, H.R.; Ogawa, T.; De Bold, M.K.; De Bold, A.J. Cardiac Natriuretic Peptides. Nat. Rev. Cardiol. 2020, 17, 698–717. [Google Scholar] [CrossRef]
- Konstam, M.A.; Kiernan, M.S.; Bernstein, D.; Bozkurt, B.; Jacob, M.; Kapur, N.K.; Kociol, R.D.; Lewis, E.F.; Mehra, M.R.; Pagani, F.D.; et al. Evaluation and Management of Right-Sided Heart Failure: A Scientific Statement from the American Heart Association. Circulation 2018, 137, e578–e622. [Google Scholar] [CrossRef]
- Borkowski, P.; Singh, N.; Borkowska, N.; Mangeshkar, S.; Nazarenko, N. Integrating Cardiac Biomarkers and Electrocardiogram in Pulmonary Embolism Prognosis. Cureus 2024, 16, e53505. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, L.; Han, J.; Song, L.; Geng, H.; Liu, Y. Joint Analysis of D-Dimer, N-Terminal pro b-Type Natriuretic Peptide, and Cardiac Troponin I on Predicting Acute Pulmonary Embolism Relapse and Mortality. Sci. Rep. 2021, 11, 14909. [Google Scholar] [CrossRef]
- Obokata, M.; Reddy, Y.N.V.; Borlaug, B.A. Diastolic Dysfunction and Heart Failure with Preserved Ejection Fraction. JACC Cardiovasc. Imaging 2020, 13, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Bamba, H.; Inban, P.; Chandrasekaran, S.H.; Priyatha, V.; John, J.; Prajjwal, P. The Prognostic Significance of Pro-BNP and Heart Failure in Acute Pulmonary Embolism: A Systematic Review. Dis. Mon. 2024, 70, 101783. [Google Scholar] [CrossRef]
- Bhave, A.; Dumra, H.; Bansal, S. Management of Acute Pulmonary Embolism: A Review. J. Assoc. Physicians India 2024, 72, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Pruszczyk, P.; Skowrońska, M.; Ciurzyński, M.; Kurnicka, K.; Lankei, M.; Konstantinides, S. Assessment of Pulmonary Embolism Severity and the Risk of Early Death. Pol. Arch. Intern. Med. 2021, 131, 16134. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Tian, X.; Liu, X.-W.; Liu, Y.-Z.; Gao, B.-L.; Li, C.-Y. Markers of Right Ventricular Dysfunction Predict 30-Day Adverse Prognosis of Pulmonary Embolism on Pulmonary Computed Tomographic Angiography. Medicine 2023, 102, e34304. [Google Scholar] [CrossRef]
- Vieillard-Baron, A.; Naeije, R.; Haddad, F.; Bogaard, H.J.; Bull, T.M.; Fletcher, N.; Lahm, T.; Magder, S.; Orde, S.; Schmidt, G.; et al. Diagnostic Workup, Etiologies and Management of Acute Right Ventricle Failure: A State-of-the-Art Paper. Intensive Care Med. 2018, 44, 774–790. [Google Scholar] [CrossRef]
- Altuğ, T.; Şentürk, A.; Alişik, M.; BiÇer, C. Clinical Value of Heart Type Fatty Acid Binding Protein (H-FABP) in Acute Pulmonary Thromboembolism. J. Health Sci. Med. 2022, 5, 861–867. [Google Scholar] [CrossRef]
- Elmoughazy, E.M.; Ahmed, M.S. Role of Heart Type Fatty Acid Binding Protein in Early Detection of Different Cardiac Diseases. J. Cardiovasc. Dis. Res. 2021, 12, 2417–2423. [Google Scholar]
- Li, X.; Bi, X. Integrated Control of Fatty Acid Metabolism in Heart Failure. Metabolites 2023, 13, 615. [Google Scholar] [CrossRef] [PubMed]
- Porres-Aguilar, M.; Rosovsky, R.P.; Rivera-Lebron, B.N.; Kaatz, S.; Mukherjee, D.; Anaya-Ayala, J.E.; Jimenez, D.; Jerjes-Sánchez, C. Pulmonary Embolism Response Teams: Changing the Paradigm in the Care for Acute Pulmonary Embolism. J. Thromb. Haemost. 2022, 20, 2457–2464. [Google Scholar] [CrossRef] [PubMed]
- Kaczyñska, A.; Pelsers, M.M.A.L.; Bochowicz, A.; Kostrubiec, M.; Glatz, J.F.C.; Pruszczyk, P. Plasma Heart-Type Fatty Acid Binding Protein Is Superior to Troponin and Myoglobin for Rapid Risk Stratification in Acute Pulmonary Embolism. Clin. Chim. Acta 2006, 371, 117–123. [Google Scholar] [CrossRef]
- Dellas, C.; Puls, M.; Lankeit, M.; Schäfer, K.; Cuny, M.; Berner, M.; Hasenfuss, G.; Konstantinides, S. Elevated Heart-Type Fatty Acid-Binding Protein Levels on Admission Predict an Adverse Outcome in Normotensive Patients with Acute Pulmonary Embolism. J. Am. Coll. Cardiol. 2010, 55, 2150–2157. [Google Scholar] [CrossRef] [PubMed]
- Podlipaeva, A.A.; Mullova, I.S.; Pavlova, T.V.; Ushakova, E.V.; Duplyakov, D.V. Novel Biological Markers for the Diagnosis and Prediction of Mortality Risk in Patients with Pulmonary Embolism. Russ. J. Cardiol. 2021, 25, 4202. [Google Scholar] [CrossRef]
- Bajaj, A.; Rathor, P.; Sehgal, V.; Shetty, A.; Kabak, B.; Hosur, S. Risk Stratification in Acute Pulmonary Embolism with Heart-Type Fatty Acid–Binding Protein: A Meta-Analysis. J. Crit. Care 2015, 30, 1151.e1–1151.e7. [Google Scholar] [CrossRef]
- Geenen, L.W.; Baggen, V.J.M.; Kauling, R.M.; Koudstaal, T.; Boomars, K.A.; Boersma, E.; Roos-Hesselink, J.W.; Van Den Bosch, A.E. Growth Differentiation Factor-15 as Candidate Predictor for Mortality in Adults with Pulmonary Hypertension. Heart 2020, 106, 467–473. [Google Scholar] [CrossRef]
- Wesseling, M.; De Poel, J.H.C.; De Jager, S.C.A. Growth Differentiation Factor 15 in Adverse Cardiac Remodelling: From Biomarker to Causal Player. ESC Heart Fail. 2020, 7, 1488–1501. [Google Scholar] [CrossRef]
- Ago, T.; Sadoshima, J. GDF15, a Cardioprotective TGF-β Superfamily Protein. Circ. Res. 2006, 98, 294–297. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-Dependent and Smad-Independent Pathways in TGF-β Family Signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Xu, J.; Kimball, T.R.; Lorenz, J.N.; Brown, D.A.; Bauskin, A.R.; Klevitsky, R.; Hewett, T.E.; Breit, S.N.; Molkentin, J.D. GDF15/MIC-1 Functions as a Protective and Antihypertrophic Factor Released from the Myocardium in Association With SMAD Protein Activation. Circ. Res. 2006, 98, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Ho, F.M.; Lin, W.W.; Chen, B.C.; Chao, C.M.; Yang, C.-R.; Lin, L.Y.; Lai, C.C.; Liu, S.H.; Liau, C.S. High Glucose-Induced Apoptosis in Human Vascular Endothelial Cells Is Mediated through NF-κB and c-Jun NH2-Terminal Kinase Pathway and Prevented by PI3K/Akt/eNOS Pathway. Cell Signal. 2006, 18, 391–399. [Google Scholar] [CrossRef]
- Lankeit, M.; Kempf, T.; Dellas, C.; Cuny, M.; Tapken, H.; Peter, T.; Olschewski, M.; Konstantinides, S.; Wollert, K.C. Growth Differentiation Factor-15 for Prognostic Assessment of Patients with Acute Pulmonary Embolism. Am. J. Respir. Crit. Care Med. 2008, 177, 1018–1025. [Google Scholar] [CrossRef]
- Jenab, Y.; Pourjafari, M.; Sotoudeh, M.; Lotfi-tokaldany, M.; Etesamifard, N.; Shirani, S.; Jalali, A.; Nozari, Y.; Poorhosseini, H. Comparing the Effect of Cardiac Biomarkers on the Outcome of Normotensive Patients with Acute Pulmonary Embolism. Monaldi Arch. Chest Dis. 2017, 87, 767. [Google Scholar] [CrossRef]
- Widimský, J. Diagnosis and Treatment of Acute Pulmonary Embolism. Cor Vasa 2013, 55, e497–e509. [Google Scholar] [CrossRef]
- Duran, L.; Kayhan, S.; Guzel, A.; Ince, M.; Kati, C.; Akdemir, H.; Yavuz, Y.; Zengin, H.; Okuyucu, A.; Murat, N. The Prognostic Values of GDF-15 in Comparison with NT-proBNP in Patients with Normotensive Acute Pulmonary Embolism. Clin. Lab. 2014, 60, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Becattini, C.; Agnelli, G.; Lankeit, M.; Masotti, L.; Pruszczyk, P.; Casazza, F.; Vanni, S.; Nitti, C.; Kamphuisen, P.; Vedovati, M.C.; et al. Acute Pulmonary Embolism: Mortality Prediction by the 2014 European Society of Cardiology Risk Stratification Model. Eur. Respir. J. 2016, 48, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Kucher, N.; Goldhaber, S.Z. Cardiac Biomarkers for Risk Stratification of Patients with Acute Pulmonary Embolism. Circulation 2003, 108, 2191–2194. [Google Scholar] [CrossRef]
- Becattini, C.; Vedovati, M.C.; Agnelli, G. Prognostic Value of Troponins in Acute Pulmonary Embolism: A Meta-Analysis. Circulation 2007, 116, 427–433. [Google Scholar] [CrossRef]
- Lankeit, M.; Friesen, D.; Aschoff, J.; Dellas, C.; Hasenfuss, G.; Katus, H.; Konstantinides, S.; Giannitsis, E. Highly Sensitive Troponin T Assay in Normotensive Patients with Acute Pulmonary Embolism. Eur. Heart J. 2010, 31, 1836–1844. [Google Scholar] [CrossRef]
- Meyer, G.; Vicaut, E.; Danays, T.; Agnelli, G.; Becattini, C.; Beyer-Westendorf, J.; Bluhmki, E.; Bouvaist, H.; Brenner, B.; Couturaud, F.; et al. Fibrinolysis for Patients with Intermediate-Risk Pulmonary Embolism. N. Engl. J. Med. 2014, 370, 1402–1411. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, M.; Arab, M.M.; Zamani Moghadam, H.; Jalal Yazdi, M.; Rayat Doost, E.; Foroughian, M. Risk Stratification of Pulmonary Thromboembolism Using Brain Natriuretic Peptide and Troponin I; a Brief Report: Diagnostic Value of Serum Troponin and Natriuretic Peptide Levels in Pulmonary Thromboembolism. Arch. Acad. Emerg. Med. 2022, 10, e8. [Google Scholar] [CrossRef] [PubMed]
- Dzudovic, B.; Matijasevic, J.; Salinger, S.; Obradovic, S. Increased B-Type Natriuretic Peptide Is a Better Predictor of Hospital Mortality Then Increased Troponin in Patients with Acute Pulmonary Embolism. Eur. Heart J. 2022, 43, ehac544.1887. [Google Scholar] [CrossRef]
- Hobohm, L.; Hellenkamp, K.; Hasenfuß, G.; Münzel, T.; Konstantinides, S.; Lankeit, M. Comparison of Risk Assessment Strategies for Not-High-Risk Pulmonary Embolism. Eur. Respir. J. 2016, 47, 1170–1178. [Google Scholar] [CrossRef]
- Dellas, C.; Tschepe, M.; Seeber, V.; Zwiener, I.; Kuhnert, K.; Schäfer, K.; Hasenfuß, G.; Konstantinides, S.; Lankeit, M. A Novel H-FABP Assay and a Fast Prognostic Score for Risk Assessment of Normotensive Pulmonary Embolism. Thromb. Haemost. 2014, 112, 996–1003. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Refs. | Biomarker | Mechanism | Kinetics | Clinical Utility | Limitations |
|---|---|---|---|---|---|
| [35,37,57,65,68,73,74] | Hs-cTnI | myocardial injury due to RV ischemia from pressure overload | - peaks in ~10 h - detectable for ~40 h | - early marker of myocardial stress - correlates with RV dysfunction severity - predicts mortality and complications | - low specificity in PE or RV injury - elevated in ACS, sepsis, CKD |
| [55,67,81,85,86,87,89] | NT-proBNP | ventricular wall stretch from acute RV pressure overload | - increases within hours - persists longer than troponin | - strong prognostic marker - useful threshold: >600 pg/mL - predicts ICU need and 30-day mortality | - elevated in heart failure, renal dysfunction |
| [54,94,95,97] | H-FABP | early release due to ischemia and cardiomyocyte stress (before necrosis) | - appears in 1.5–2 h - peaks in 6–8 h - clears in 24–36 h | - sensitive to early RV ischemia - strong predictor of short-term mortality - higher specificity for ischemia than D-dimer or BNP in some cases | - short half-life - less established in clinical use |
| [39,96,98,104,107] | GDF-15 | stress-induced cytokine upregulated in response to pressure, ischemia, inflammation | - elevates under sustained cardiac or systemic stress | - predicts early- and long-term mortality - high NPV at thresholds <1200 ng/L - useful in normotensive PE | - non-specific; - elevated in cancer, CKD, HF - not yet standard in clinical algorithms |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cucută, S.; Badescu, M.C.; Duca, Ș.-T.; Chetran, A.; Cepoi, M.-R.; Ponor, C.-G.; Bobu, A.M.; Serban, I.-L.; Costache-Enache, I.-I. Beyond a Single Marker: An Update on the Comprehensive Evaluation of Right Ventricular Dysfunction in Pulmonary Thromboembolism. Life 2025, 15, 665. https://doi.org/10.3390/life15040665
Cucută S, Badescu MC, Duca Ș-T, Chetran A, Cepoi M-R, Ponor C-G, Bobu AM, Serban I-L, Costache-Enache I-I. Beyond a Single Marker: An Update on the Comprehensive Evaluation of Right Ventricular Dysfunction in Pulmonary Thromboembolism. Life. 2025; 15(4):665. https://doi.org/10.3390/life15040665
Chicago/Turabian StyleCucută, Sandu, Minerva Codruta Badescu, Ștefania-Teodora Duca, Adriana Chetran, Maria-Ruxandra Cepoi, Cosmina-Georgiana Ponor, Amelian Madalin Bobu, Ionela-Lacramioara Serban, and Irina-Iuliana Costache-Enache. 2025. "Beyond a Single Marker: An Update on the Comprehensive Evaluation of Right Ventricular Dysfunction in Pulmonary Thromboembolism" Life 15, no. 4: 665. https://doi.org/10.3390/life15040665
APA StyleCucută, S., Badescu, M. C., Duca, Ș.-T., Chetran, A., Cepoi, M.-R., Ponor, C.-G., Bobu, A. M., Serban, I.-L., & Costache-Enache, I.-I. (2025). Beyond a Single Marker: An Update on the Comprehensive Evaluation of Right Ventricular Dysfunction in Pulmonary Thromboembolism. Life, 15(4), 665. https://doi.org/10.3390/life15040665