Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects

Abstract
:
1. Introduction
2. Syndromic IRD Types
3. Genetic Heterogeneity in Syndromic IRDs
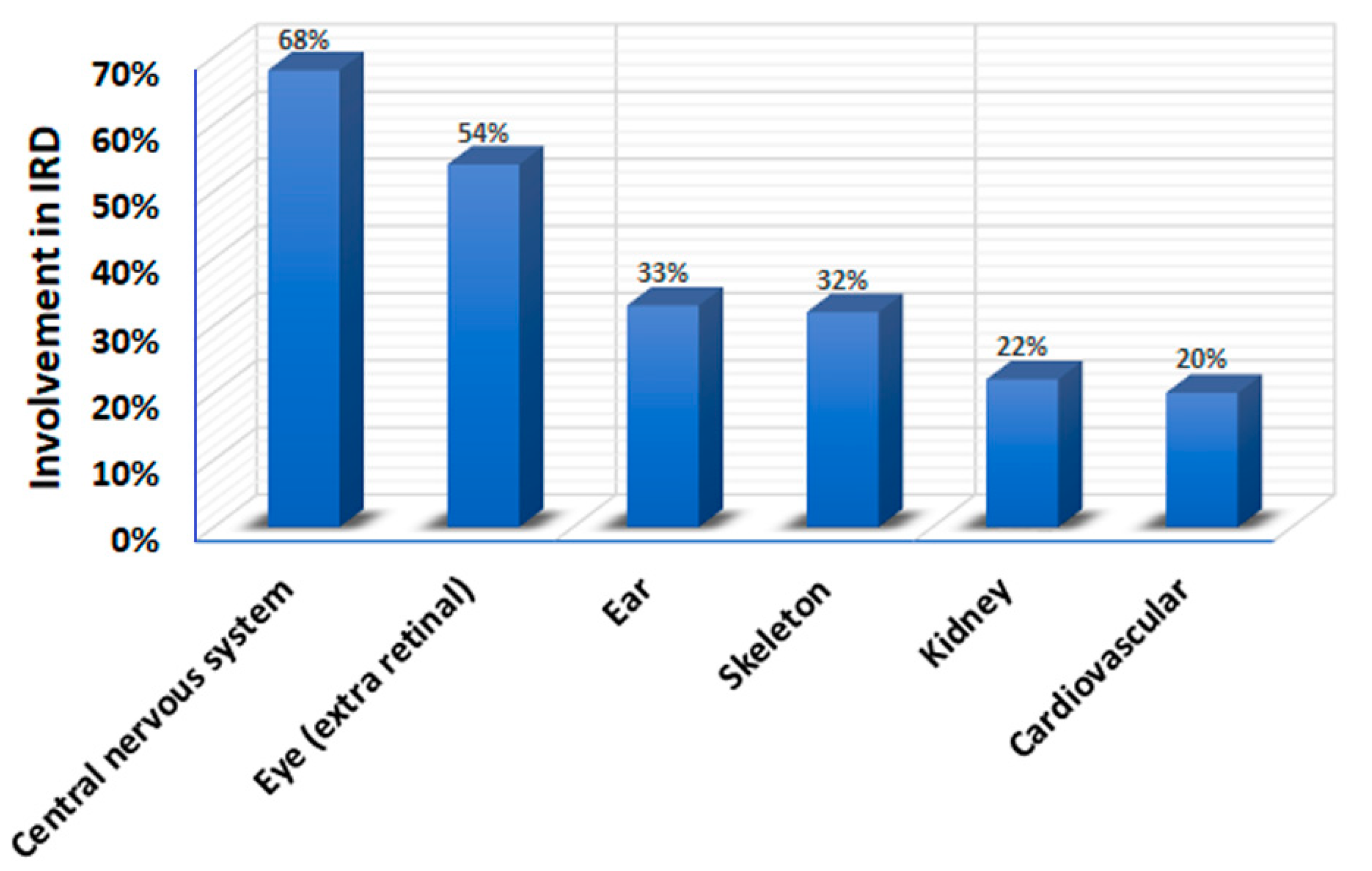
4. Phenotypic Overlap in Syndromic IRDs
4.1. Phenotypic Overlap between Different IRD Syndromes
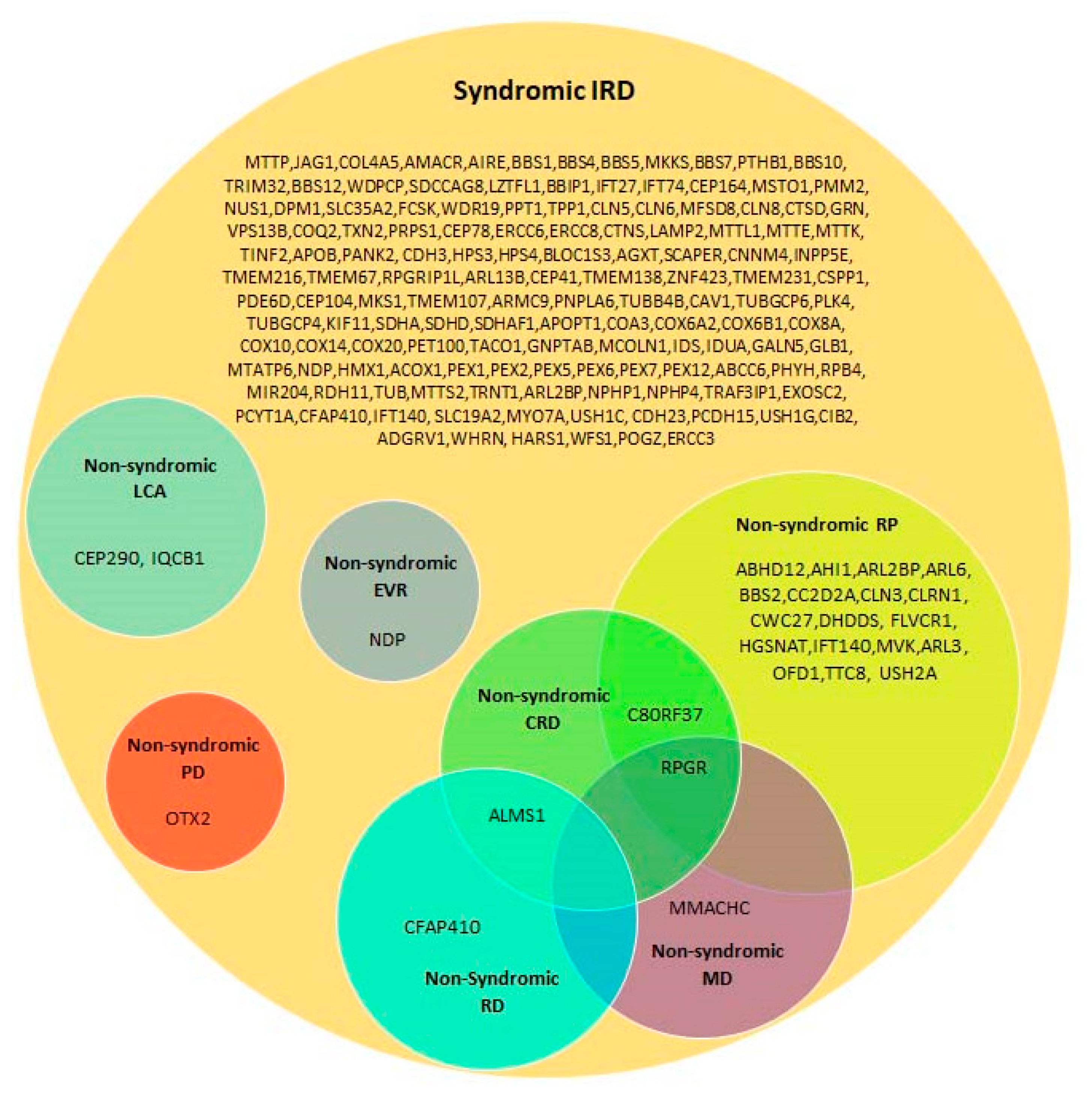
4.2. Syndromic Versus Non-Syndromic IRD Caused by the Same Genes
4.3. Co-Existence of Non-Syndromic IRD and Additional Non-Ocular Diseases
5. Diagnostic Challenges
6. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Duncan, J.L.; Pierce, E.A.; Laster, A.M.; Daiger, S.P.; Birch, D.G.; Ash, J.D.; Iannaccone, A.; Flannery, J.G.; Sahel, J.A.; Zack, D.J.; et al. Inherited Retinal Degenerations: Current Landscape and Knowledge Gaps. Transl. Vis. Sci. Technol. 2018, 7, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Thiadens, A.A.; Phan, T.M.; Zekveld-Vroon, R.C.; Leroy, B.P.; van den Born, L.I.; Hoyng, C.B.; Klaver, C.C.; Roosing, S.; Pott, J.W.; van Schooneveld, M.J.; et al. Clinical course, genetic etiology, and visual outcome in cone and cone-rod dystrophy. Ophthalmology 2012, 119, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Sharma, T. Rod Monochromatism (Achromatopsia). Adv. Exp. Med. Biol. 2018, 1085, 119–123. [Google Scholar]
- Werdich, X.Q.; Place, E.M.; Pierce, E.A. Systemic diseases associated with retinal dystrophies. Semin. Ophthalmol. 2014, 29, 319–328. [Google Scholar] [CrossRef]
- Shamseldin, H.E.; Shaheen, R.; Ewida, N.; Bubshait, D.K.; Alkuraya, H.; Almardawi, E.; Howaidi, A.; Sabr, Y.; Abdalla, E.M.; Alfaifi, A.Y.; et al. The morbid genome of ciliopathies: An update. Genet. Med. 2020, 22, 1051–1060. [Google Scholar] [CrossRef]
- Iwama, K.; Takaori, T.; Fukushima, A.; Tohyama, J.; Ishiyama, A.; Ohba, C.; Mitsuhashi, S.; Miyatake, S.; Takata, A.; Miyake, N.; et al. Novel recessive mutations in MSTO1 cause cerebellar atrophy with pigmentary retinopathy. J. Hum. Genet. 2018, 63, 263–270. [Google Scholar] [CrossRef]
- Ferreira, C.R.; van Karnebeek, C.D.M. Inborn errors of metabolism. Handb. Clin. Neurol. 2019, 162, 449–481. [Google Scholar]
- Freeze, H.H.; Schachter, H.; Kinoshita, T. Genetic Disorders of Glycosylation. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2017; Chapter 45. [Google Scholar]
- Nita, D.A.; Mole, S.E.; Minassian, B.A. Neuronal ceroid lipofuscinoses. Epileptic Disord. 2016, 18, 73–88. [Google Scholar] [CrossRef]
- Muenzer, J. Overview of the mucopolysaccharidoses. Rheumatology (Oxford) 2011, 50 (Suppl. 5), v4–v12. [Google Scholar] [CrossRef] [Green Version]
- Imanaka, T. Biogenesis and Function of Peroxisomes in Human Disease with a Focus on the ABC Transporter. Biol. Pharm. Bull. 2019, 42, 649–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreekumar, V.; Norris, D.P. Cilia and development. Curr. Opin. Genet. Dev. 2019, 56, 15–21. [Google Scholar] [CrossRef] [PubMed]
- May-Simera, H.; Nagel-Wolfrum, K.; Wolfrum, U. Cilia—The sensory antennae in the eye. Prog. Retin. Eye Res. 2017, 60, 144–180. [Google Scholar] [CrossRef]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Bardet-Biedl Syndrome. Adv. Exp. Med. Biol. 2018, 1085, 171–174. [Google Scholar] [PubMed]
- Valente, E.M.; Dallapiccola, B.; Bertini, E. Joubert syndrome and related disorders. Handb. Clin. Neurol. 2013, 113, 1879–1888. [Google Scholar] [PubMed]
- Geleoc, G.G.S.; El-Amraoui, A. Disease mechanisms and gene therapy for Usher syndrome. Hear. Res. 2020, 394, 107932. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Senior-Loken Syndrome. Adv. Exp. Med. Biol. 2018, 1085, 175–178. [Google Scholar] [PubMed]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Alstrom Syndrome. Adv. Exp. Med. Biol. 2018, 1085, 179–180. [Google Scholar] [PubMed]
- Petriman, N.A.; Lorentzen, E. Moving proteins along in the cilium. Elife 2020, 9, e55254. [Google Scholar] [CrossRef]
- Hsu, Y.; Garrison, J.E.; Kim, G.; Schmitz, A.R.; Searby, C.C.; Zhang, Q.; Datta, P.; Nishimura, D.Y.; Seo, S.; Sheffield, V.C. BBSome function is required for both the morphogenesis and maintenance of the photoreceptor outer segment. PLoS Genet. 2017, 13, e1007057. [Google Scholar] [CrossRef]
- Hsu, Y.; Garrison, J.E.; Seo, S.; Sheffield, V.C. The absence of BBSome function decreases synaptogenesis and causes ectopic synapse formation in the retina. Sci. Rep. 2020, 10, 8321. [Google Scholar] [CrossRef]
- Wang, S.F.; Kowal, T.J.; Ning, K.; Koo, E.B.; Wu, A.Y.; Mahajan, V.B.; Sun, Y. Review of Ocular Manifestations of Joubert Syndrome. Genes 2018, 9, 605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Amraoui, A.; Petit, C. The retinal phenotype of Usher syndrome: Pathophysiological insights from animal models. Comptes Rendus Biol. 2014, 337, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.R.; Benson, M.D.; MacDonald, I.M.; Innes, A.M. A diagnostic approach to syndromic retinal dystrophies with intellectual disability. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 538–570. [Google Scholar] [CrossRef] [PubMed]
- Namburi, P.; Ratnapriya, R.; Khateb, S.; Lazar, C.H.; Kinarty, Y.; Obolensky, A.; Erdinest, I.; Marks-Ohana, D.; Pras, E.; Ben-Yosef, T.; et al. Bi-allelic Truncating Mutations in CEP78, Encoding Centrosomal Protein 78, Cause Cone-Rod Degeneration with Sensorineural Hearing Loss. Am. J. Hum. Genet. 2016, 99, 777–784. [Google Scholar] [CrossRef] [Green Version]
- Luscan, R.; Mechaussier, S.; Paul, A.; Tian, G.; Gerard, X.; Defoort-Dellhemmes, S.; Loundon, N.; Audo, I.; Bonnin, S.; LeGargasson, J.F.; et al. Mutations in TUBB4B Cause a Distinctive Sensorineural Disease. Am. J. Hum. Genet. 2017, 101, 1006–1012. [Google Scholar] [CrossRef] [Green Version]
- Sims, K.B. NDP-Related Retinopathies. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2014. [Google Scholar]
- Tsang, S.H.; Sharma, T. Inborn Errors of Metabolism: Refsum Disease. Adv. Exp. Med. Biol. 2018, 1085, 191–192. [Google Scholar]
- Raas-Rothschild, A.; Wanders, R.J.; Mooijer, P.A.; Gootjes, J.; Waterham, H.R.; Gutman, A.; Suzuki, Y.; Shimozawa, N.; Kondo, N.; Eshel, G.; et al. A PEX6-defective peroxisomal biogenesis disorder with severe phenotype in an infant, versus mild phenotype resembling Usher syndrome in the affected parents. Am. J. Hum. Genet. 2002, 70, 1062–1068. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.E.; Poulter, J.A.; Levin, A.V.; Capasso, J.E.; Price, S.; Ben-Yosef, T.; Sharony, R.; Newman, W.G.; Shore, R.C.; Brookes, S.J.; et al. Spectrum of PEX1 and PEX6 variants in Heimler syndrome. Eur. J. Hum. Genet. 2016, 24, 1565–1571. [Google Scholar] [CrossRef]
- Le Quesne Stabej, P.; Saihan, Z.; Rangesh, N.; Steele-Stallard, H.B.; Ambrose, J.; Coffey, A.; Emmerson, J.; Haralambous, E.; Hughes, Y.; Steel, K.P.; et al. Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J. Med. Genet. 2012, 49, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dockery, A.; Stephenson, K.; Keegan, D.; Wynne, N.; Silvestri, G.; Humphries, P.; Kenna, P.F.; Carrigan, M.; Farrar, G.J. Target 5000: Target Capture Sequencing for Inherited Retinal Degenerations. Genes 2017, 8, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharon, D.; Ben-Yosef, T.; Goldenberg-Cohen, N.; Pras, E.; Gradstein, L.; Soudry, S.; Mezer, E.; Zur, D.; Abbasi, A.H.; Zeitz, C.; et al. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum. Mutat. 2019, 41, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Lenassi, E.; Vincent, A.; Li, Z.; Saihan, Z.; Coffey, A.J.; Steele-Stallard, H.B.; Moore, A.T.; Steel, K.P.; Luxon, L.M.; Heon, E.; et al. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur. J. Hum. Genet. 2015, 23, 1318–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierrache, L.H.; Hartel, B.P.; van Wijk, E.; Meester-Smoor, M.A.; Cremers, F.P.; de Baere, E.; de Zaeytijd, J.; van Schooneveld, M.J.; Cremers, C.W.; Dagnelie, G.; et al. Visual Prognosis in USH2A-Associated Retinitis Pigmentosa Is Worse for Patients with Usher Syndrome Type IIa Than for Those with Nonsyndromic Retinitis Pigmentosa. Ophthalmology 2016, 123, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Sengillo, J.D.; Cabral, T.; Schuerch, K.; Duong, J.; Lee, W.; Boudreault, K.; Xu, Y.; Justus, S.; Sparrow, J.R.; Mahajan, V.B.; et al. Electroretinography Reveals Difference in Cone Function between Syndromic and Nonsyndromic USH2A Patients. Sci. Rep. 2017, 7, 11170. [Google Scholar] [CrossRef]
- Nishiguchi, K.M.; Avila-Fernandez, A.; van Huet, R.A.; Corton, M.; Perez-Carro, R.; Martin-Garrido, E.; Lopez-Molina, M.I.; Blanco-Kelly, F.; Hoefsloot, L.H.; van Zelst-Stams, W.A.; et al. Exome sequencing extends the phenotypic spectrum for ABHD12 mutations: From syndromic to nonsyndromic retinal degeneration. Ophthalmology 2014, 121, 1620–1627. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Hull, S.; Roepman, R.; van den Born, L.I.; Oud, M.M.; de Vrieze, E.; Hetterschijt, L.; Letteboer, S.J.F.; van Beersum, S.E.C.; Blokland, E.A.; et al. Missense mutations in the WD40 domain of AHI1 cause non-syndromic retinitis pigmentosa. J. Med. Genet. 2017, 54, 624–632. [Google Scholar] [CrossRef]
- Aldrees, A.; Abdelkader, E.; Al-Habboubi, H.; Alrwebah, H.; Rahbeeni, Z.; Schatz, P. Non-syndromic retinal dystrophy associated with homozygous mutations in the ALMS1 gene. Ophthalmic Genet. 2019, 40, 77–79. [Google Scholar] [CrossRef]
- Audo, I.; El Shamieh, S.; Mejecase, C.; Michiels, C.; Demontant, V.; Antonio, A.; Condroyer, C.; Boyard, F.; Letexier, M.; Saraiva, J.P.; et al. ARL2BP mutations account for 0.1% of autosomal recessive rod-cone dystrophies with the report of a novel splice variant. Clin. Genet. 2017, 92, 109–111. [Google Scholar] [PubMed]
- Davidson, A.E.; Schwarz, N.; Zelinger, L.; Stern-Schneider, G.; Shoemark, A.; Spitzbarth, B.; Gross, M.; Laxer, U.; Sosna, J.; Sergouniotis, P.I.; et al. Mutations in ARL2BP, encoding ADP-ribosylation-factor-like 2 binding protein, cause autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 2013, 93, 321–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtan, J.P.; Teigen, K.; Aukrust, I.; Bragadottir, R.; Houge, G. Dominant ARL3-related retinitis pigmentosa. Ophthalmic Genet. 2019, 40, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Aldahmesh, M.A.; Safieh, L.A.; Alkuraya, H.; Al-Rajhi, A.; Shamseldin, H.; Hashem, M.; Alzahrani, F.; Khan, A.O.; Alqahtani, F.; Rahbeeni, Z.; et al. Molecular characterization of retinitis pigmentosa in Saudi Arabia. Mol. Vis. 2009, 15, 2464–2469. [Google Scholar]
- Shevach, E.; Ali, M.; Mizrahi-Meissonnier, L.; McKibbin, M.; El-Asrag, M.; Watson, C.M.; Inglehearn, C.F.; Ben-Yosef, T.; Blumenfeld, A.; Jalas, C.; et al. Association Between Missense Mutations in the BBS2 Gene and Nonsyndromic Retinitis Pigmentosa. JAMA Ophthalmol. 2015, 133, 312–318. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.O.; Decker, E.; Bachmann, N.; Bolz, H.J.; Bergmann, C. C8orf37 is mutated in Bardet-Biedl syndrome and constitutes a locus allelic to non-syndromic retinal dystrophies. Ophthalmic Genet. 2016, 37, 290–293. [Google Scholar] [CrossRef]
- Mejecase, C.; Hummel, A.; Mohand-Said, S.; Andrieu, C.; El Shamieh, S.; Antonio, A.; Condroyer, C.; Boyard, F.; Foussard, M.; Blanchard, S.; et al. Whole exome sequencing resolves complex phenotype and identifies CC2D2A mutations underlying non-syndromic rod-cone dystrophy. Clin. Genet. 2019, 95, 329–333. [Google Scholar] [CrossRef]
- den Hollander, A.I.; Koenekoop, R.K.; Yzer, S.; Lopez, I.; Arends, M.L.; Voesenek, K.E.; Zonneveld, M.N.; Strom, T.M.; Meitinger, T.; Brunner, H.G.; et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006, 79, 556–561. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.O.; Eisenberger, T.; Nagel-Wolfrum, K.; Wolfrum, U.; Bolz, H.J. C21orf2 is mutated in recessive early-onset retinal dystrophy with macular staphyloma and encodes a protein that localises to the photoreceptor primary cilium. Br. J. Ophthalmol. 2015, 99, 1725–1731. [Google Scholar] [CrossRef]
- Suga, A.; Mizota, A.; Kato, M.; Kuniyoshi, K.; Yoshitake, K.; Sultan, W.; Yamazaki, M.; Shimomura, Y.; Ikeo, K.; Tsunoda, K.; et al. Identification of Novel Mutations in the LRR-Cap Domain of C21orf2 in Japanese Patients With Retinitis Pigmentosa and Cone-Rod Dystrophy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4255–4263. [Google Scholar] [CrossRef] [Green Version]
- Ku, C.A.; Hull, S.; Arno, G.; Vincent, A.; Carss, K.; Kayton, R.; Weeks, D.; Anderson, G.W.; Geraets, R.; Parker, C.; et al. Detailed Clinical Phenotype and Molecular Genetic Findings in CLN3-Associated Isolated Retinal Degeneration. JAMA Ophthalmol. 2017, 135, 749–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.I.; Kersten, F.F.; Azam, M.; Collin, R.W.; Hussain, A.; Shah, S.T.; Keunen, J.E.; Kremer, H.; Cremers, F.P.; Qamar, R.; et al. CLRN1 mutations cause nonsyndromic retinitis pigmentosa. Ophthalmology 2011, 118, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Xie, Y.A.; Abouzeid, H.; Gordon, C.T.; Fiorentino, A.; Sun, Z.; Lehman, A.; Osman, I.S.; Dharmat, R.; Riveiro-Alvarez, R.; et al. Mutations in the Spliceosome Component CWC27 Cause Retinal Degeneration with or without Additional Developmental Anomalies. Am. J. Hum. Genet. 2017, 100, 592–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, B.L.; Zuchner, S.L.; Dallman, J.; Wen, R.; Alfonso, E.C.; Vance, J.M.; Pericak-Vance, M.A. Mutation K42E in dehydrodolichol diphosphate synthase (DHDDS) causes recessive retinitis pigmentosa. Adv. Exp. Med. Biol. 2014, 801, 165–170. [Google Scholar]
- Zelinger, L.; Banin, E.; Obolensky, A.; Mizrahi-Meissonnier, L.; Beryozkin, A.; Bandah-Rozenfeld, D.; Frenkel, S.; Ben-Yosef, T.; Merin, S.; Schwartz, S.B.; et al. A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. Am. J. Hum. Genet. 2011, 88, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Kuehlewein, L.; Schols, L.; Llavona, P.; Grimm, A.; Biskup, S.; Zrenner, E.; Kohl, S. Phenotypic spectrum of autosomal recessive retinitis pigmentosa without posterior column ataxia caused by mutations in the FLVCR1 gene. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 629–638. [Google Scholar] [CrossRef]
- Haer-Wigman, L.; Newman, H.; Leibu, R.; Bax, N.M.; Baris, H.N.; Rizel, L.; Banin, E.; Massarweh, A.; Roosing, S.; Lefeber, D.J.; et al. Non-syndromic retinitis pigmentosa due to mutations in the mucopolysaccharidosis type IIIC gene, heparan-alpha-glucosaminide N-acetyltransferase (HGSNAT). Hum. Mol. Genet. 2015, 24, 3742–3751. [Google Scholar] [CrossRef]
- Xu, M.; Yang, L.; Wang, F.; Li, H.; Wang, X.; Wang, W.; Ge, Z.; Wang, K.; Zhao, L.; Li, H.; et al. Mutations in human IFT140 cause non-syndromic retinal degeneration. Hum. Genet. 2015, 134, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- Stone, E.M.; Cideciyan, A.V.; Aleman, T.S.; Scheetz, T.E.; Sumaroka, A.; Ehlinger, M.A.; Schwartz, S.B.; Fishman, G.A.; Traboulsi, E.I.; Lam, B.L.; et al. Variations in NPHP5 in patients with nonsyndromic leber congenital amaurosis and Senior-Loken syndrome. Arch. Ophthalmol. 2011, 129, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Khan, K.N.; El-Asrag, M.E.; Ku, C.A.; Holder, G.E.; McKibbin, M.; Arno, G.; Poulter, J.A.; Carss, K.; Bommireddy, T.; Bagheri, S.; et al. Specific Alleles of CLN7/MFSD8, a Protein That Localizes to Photoreceptor Synaptic Terminals, Cause a Spectrum of Nonsyndromic Retinal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2906–2914. [Google Scholar] [CrossRef] [Green Version]
- Collison, F.T.; Xie, Y.A.; Gambin, T.; Jhangiani, S.; Muzny, D.; Gibbs, R.; Lupski, J.R.; Fishman, G.A.; Allikmets, R. Whole Exome Sequencing Identifies an Adult-Onset Case of Methylmalonic Aciduria and Homocystinuria Type C (cblC) with Non-Syndromic Bull’s Eye Maculopathy. Ophthalmic Genet. 2015, 36, 270–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siemiatkowska, A.M.; van den Born, L.I.; van Hagen, P.M.; Stoffels, M.; Neveling, K.; Henkes, A.; Kipping-Geertsema, M.; Hoefsloot, L.H.; Hoyng, C.B.; Simon, A.; et al. Mutations in the mevalonate kinase (MVK) gene cause nonsyndromic retinitis pigmentosa. Ophthalmology 2013, 120, 2697–2705. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Battinelli, E.M.; Fielder, A.; Bundey, S.; Sims, K.; Breakefield, X.O.; Craig, I.W. A mutation in the Norrie disease gene (NDP) associated with X-linked familial exudative vitreoretinopathy. Nat. Genet. 1993, 5, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Webb, T.R.; Parfitt, D.A.; Gardner, J.C.; Martinez, A.; Bevilacqua, D.; Davidson, A.E.; Zito, I.; Thiselton, D.L.; Ressa, J.H.; Apergi, M.; et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum. Mol. Genet. 2012, 21, 3647–3654. [Google Scholar] [CrossRef] [Green Version]
- Vincent, A.; Forster, N.; Maynes, J.T.; Paton, T.A.; Billingsley, G.; Roslin, N.M.; Ali, A.; Sutherland, J.; Wright, T.; Westall, C.A.; et al. OTX2 mutations cause autosomal dominant pattern dystrophy of the retinal pigment epithelium. J. Med. Genet. 2014, 51, 797–805. [Google Scholar] [CrossRef]
- Tee, J.J.; Smith, A.J.; Hardcastle, A.J.; Michaelides, M. RPGR-associated retinopathy: Clinical features, molecular genetics, animal models and therapeutic options. Br. J. Ophthalmol. 2016, 100, 1022–1027. [Google Scholar] [CrossRef]
- Riazuddin, S.A.; Iqbal, M.; Wang, Y.; Masuda, T.; Chen, Y.; Bowne, S.; Sullivan, L.S.; Waseem, N.H.; Bhattacharya, S.; Daiger, S.P.; et al. A splice-site mutation in a retina-specific exon of BBS8 causes nonsyndromic retinitis pigmentosa. Am. J. Hum. Genet. 2010, 86, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Rivolta, C.; Sweklo, E.A.; Berson, E.L.; Dryja, T.P. Missense mutation in the USH2A gene: Association with recessive retinitis pigmentosa without hearing loss. Am. J. Hum. Genet. 2000, 66, 1975–1978. [Google Scholar] [CrossRef] [Green Version]
- Ehrenberg, M.; Weiss, S.; Orenstein, N.; Goldenberg-Cohen, N.; Ben-Yosef, T. The co-occurrence of rare non-ocular phenotypes in patients with inherited retinal degenerations. Mol. Vis. 2019, 25, 691–702. [Google Scholar]
- Ku, C.A.; Pennesi, M.E. The new landscape of retinal gene therapy. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 846–859. [Google Scholar] [CrossRef]
- Bach, G.; Webb, M.B.; Bargal, R.; Zeigler, M.; Ekstein, J. The frequency of mucolipidosis type IV in the Ashkenazi Jewish population and the identification of 3 novel MCOLN1 mutations. Hum. Mutat. 2005, 26, 591. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yosef, T.; Ness, S.L.; Madeo, A.C.; Bar-Lev, A.; Wolfman, J.H.; Ahmed, Z.M.; Desnick, R.J.; Willner, J.P.; Avraham, K.B.; Ostrer, H.; et al. A mutation of PCDH15 among Ashkenazi Jews with the type 1 Usher syndrome. N. Engl. J. Med. 2003, 348, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Fedick, A.; Jalas, C.; Abeliovich, D.; Krakinovsky, Y.; Ekstein, J.; Ekstein, A.; Treff, N.R. Carrier frequency of two BBS2 mutations in the Ashkenazi population. Clin. Genet. 2014, 85, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Ness, S.L.; Ben-Yosef, T.; Bar-Lev, A.; Madeo, A.C.; Brewer, C.C.; Avraham, K.B.; Kornreich, R.; Desnick, R.J.; Willner, J.P.; Friedman, T.B.; et al. Genetic homogeneity and phenotypic variability among Ashkenazi Jews with Usher syndrome type III. J. Med. Genet. 2003, 40, 767–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joensuu, T.; Hamalainen, R.; Yuan, B.; Johnson, C.; Tegelberg, S.; Gasparini, P.; Zelante, L.; Pirvola, U.; Pakarinen, L.; Lehesjoki, A.E.; et al. Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am. J. Hum. Genet. 2001, 69, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Kyttala, M.; Tallila, J.; Salonen, R.; Kopra, O.; Kohlschmidt, N.; Paavola-Sakki, P.; Peltonen, L.; Kestila, M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat. Genet. 2006, 38, 155–157. [Google Scholar] [CrossRef]
- Branham, K.; Schlegel, D.; Fahim, A.T.; Jayasundera, K.T. Genetic testing for inherited retinal degenerations: Triumphs and tribulations. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 571–577. [Google Scholar] [CrossRef]
- Mansfield, B.C.; Yerxa, B.R.; Branham, K.H. Implementation of a registry and open access genetic testing program for inherited retinal diseases within a non-profit foundation. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 838–845. [Google Scholar] [CrossRef]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Haer-Wigman, L.; van Zelst-Stams, W.A.; Pfundt, R.; van den Born, L.I.; Klaver, C.C.; Verheij, J.B.; Hoyng, C.B.; Breuning, M.H.; Boon, C.J.; Kievit, A.J.; et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur. J. Hum. Genet. 2017, 25, 591–599. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Syndrome (MIM/Reference) | Gene | Inheritance * | Main Ocular Phenotypes # | Main Extra-Ocular Phenotypes ¶ |
|---|---|---|---|---|
| Abetalipoproteinemia; ABL (#200100) | MTTP | AR | RP | Fat malabsorption, neurodegeneration, acanthocytosis |
| Aicardi Syndrome; AIC (#304050) | Xp22 abnormalities | XLD | Chorioretinopathy, OA, microphthalmia, optic nerve coloboma, cataract | Callosal agenesis, PGR, microcephaly, ID, skeletal anomalies, neoplasia |
| Alagille Syndrome 1; ALGS1 (#118450) | JAG1 | AD | Iris stromal hypoplasia, posterior embryotoxon, microcornea, anomalous optic disc, peripapillary retinal depigmentation, chorioretinopathy | Liver disease, skeletal and renal involvement, characteristic facial features, ID, FTT |
| Alport Syndrome 1; ATS1 (#3010150) | COL4A5 | XLD | Fleck retinopathy, cataract, myopia, corneal abnormalities | HL, renal disease |
| Alstrom Syndrome; ALMS (#203800) | ALMS1 | AR | CRD, MD, cataract | DD, SS, obesity, HL, cardiac, skeletal, hepatic, renal and endocrine involvement |
| Alpha-Methylacyl-CoA Racemase Deficiency; AMACRD (#614307) | AMACR | AR | RP | Neurodegeneration |
| Autoimmune Polyendocrine Syndrome, Type I, with or without Reversible Metaphyseal Dysplasia; APS1 (#240300) | AIRE | AD, AR | RP, keratopathy, keratoconjunctivitis | Multiple autoantibodies, anemia, hepatic, gastrointestinal, dental, skin, hair and endocrine involvement, hypogonadism |
| Bardet–Biedl Syndrome; BBS (#209900, #615981, #600151, #615982, #615983, #605231, #615984, #615985, #615986, #615987, #615988, #615989, #615990, #615991, 615992, #615993, #615994, #615995, #615996, #617119, #617406) [7] | BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, TTC8, PTHB1, BBS10, TRIM32, BBS12, MKS1, CEP290, WDPCP, SDCCAG8, LZTFL1, BBIP1, IFT27, IFT74, C8ORF37, CEP164 | AR | RP, strabismus, cataract | ID, SS, obesity, hypogonadism, renal disease, polydactyly |
| Cerebellar Atrophy with Pigmentary Retinopathy [8] | MSTO1 | AR | RD | Cerebellar atrophy, ID, PGR |
| Congenital Disorder of Glycosylation; CDG (#212065, #617082, #613861, #608799, #300896) | PMM2, NUS1, DHDDS, DPM1, SLC35A2 | AR | RP | FTT, microcephaly, ID, neurodegeneration, cardiac, hepatic, gastrointestinal, renal and hematological involvement |
| Congenital Disorder of Glycosylation with Defective Fucosylation 2; CDGF2 (#618324) | FCSK | AR | MD, OA, strabismus | FTT, ID, hypotonia, neurodegeneration, gastrointestinal anomalies |
| Cranioectodermal Dysplasia 4; CED4 (#614378) | WDR19 | AR | RP | Skeletal anomalies, SS, respiratory, hepatic and renal involvement |
| Ceroid Lipofuscinosis, Neuronal; CLN (#256730, #204500, #204200, #256731, #601780, #610951, #600143, #610127, #614706) | PPT1, TPP1, CLN3, CLN5, CLN6, MFSD8, CLN8, CTSD, GRN | AR | RP, CRD, OA | Microcephaly, ID, neurodegeneration |
| Cohen Syndrome; COH1 (#216550) | VPS13B | AR | RD, OA, strabismus, high myopia | ID, DD, microcephaly, SS, obesity, skeletal, cardiac, hematological and endocrine involvement |
| Coenzyme Q10 Deficiency, Primary, 1; COQ10D1 (#607426) | COQ2 | AR | RP | ID, cerebellar atrophy, HL, cardiac, hepatic, renal and muscular involvement |
| Combined Oxidative Phosphorylation Deficiency 29; COXPD29 (#616811) | TXN2 | AR | RD, OA | Microcephaly, hypotonia, DD, ID, neurodegeneration |
| Charcot–Marie–Tooth Disease, X-linked recessive, 5; CMTX5 (#311070) | PRPS1 | XLR | RP, OA | Peripheral neuropathy, HL |
| Cone–Rod Dystrophy and Hearing Loss 1; CRDHL1 (#617236) | CEP78 | AR | CRD | HL |
| Cockayne Syndrome; CS (#216400, #133540) | ERCC8, ERCC6 | AR | RD, OA, cataract, strabismus | IUGR, PGR, microcephaly, ID, neurodegeneration, HL, renal, skeletal and skin involvement |
| Cystinosis, Nephropathic; CTNS (#219800, #219900) | CTNS | AR | RD, corneal crystals | Renal disease, neurodegeneration, skeletal and endocrine anomalies |
| Danon Disease (#300257) | LAMP2 | XLD | RD | Cardiac disease, myopathy, ID |
| Diabetes and Deafness, Maternally Inherited; MIDD (#520000) | MTTL1, MTTE, MTTK, mitochondrial DNA rearrangements | Mi | RD, MD, ophthalmoplegia | HL, cardiac and neurological anomalies, diabetes mellitus |
| Dyskeratosis Congenita, Autosomal Dominant 3; DKCA3 (#613990) | TINF2 | AD | RD, blockage of lacrimal ducts | IUGR, SS, microcephaly, ID, HL, respiratory, skin, skeletal and hematological involvement, neoplasia |
| Hypobetalipoproteinemia, Familial, 1; FHBL1 (#615558) | APOB | AR | RP | Fat malabsorption, neurodegeneration, acanthocytosis |
| Hypobetalipoproteinemia, Acanthocytosis, Retinitis Pigmentosa and Pallidal Degeneration; HARP (#607236) | PANK2 | AR | RP | Fat malabsorption, neurodegeneration, acanthocytosis |
| Hypotrichosis, Congenital, with Juvenile Macular Dystrophy; HJMD (#601553) | CDH3 | AR | MD | Hypotrichosis |
| Hermansky–Pudlak Syndrome; HPS (#614072, #614073, #614077) | HPS3, HPS4, BLOC1S3 | AR | Hypopigmentation of retina and choroid, foveal hypoplasia, nystagmus, iris transillumination | Skin and hair hypopigmentation, bleeding diathesis |
| Hyper-IgD Syndrome; HIDS (#260920) | MVK | AR | RP | Hematological anomalies, gastrointestinal and skeletal involvement, periodic fever |
| Hyperoxaluria, Primary, Type I; HP1 (#259900) | AGXT | AR | RD, OA | Renal disease, dental, cardiovascular and skin involvement, peripheral neuropathy |
| Intellectual Developmental Disorder and Retinitis Pigmentosa; IDDRP (#618195) | SCAPER | AR | RP, MD, cataract | ID, skeletal abnormalities, male sterility |
| Jalili Syndrome (#217080) | CNNM4 | AR | CRD | Amelogenesis imperfecta |
| Joubert Syndrome; JBTS (#213300, #608091, #608629, #610188, #610688, #611560, #612291, #612285, #614464, #614465, #614844, #614970, #615636, #615665, #616781, #617121, #617562, #617622, #618161, #300804) | INPP5E, TMEM216, AHI1, CEP290, TMEM67, RPGRIP1L, ARL13B, CC2D2A, CEP41, TMEM138, ZNF423, TMEM231, CSPP1, PDE6D, CEP104, MKS1, TMEM107, ARMC9, ARL3 | AR | RD, chorioretinal coloboma, optic nerve coloboma, microphthalmia, oculomotor apraxia, esotropia, ptosis | Brain structural anomalies, FTT, macrocephaly, ID, neurodegeneration, genitourinary, hepatic, respiratory and skeletal involvement |
| OFD1 | XLR | |||
| Kearns–Sayre Syndrome; KSS (#530000) | Mitochondrial DNA deletions | Mi | RD, ophthalmoplegia | SS, microcephaly, neurodegeneration, cardiac, renal and endocrine involvement |
| Laurence–Moon Syndrome; LNMS (#245800) | PNPLA6 | AR | Chorioretinal degeneration | ID, neurodegeneration, genitourinary abnormalities |
| Leber Congenital Amaurosis with Early-Onset Deafness; LCAEOD (#617879) | TUBB4B | AD | LCA | HL |
| Lipodystrophy, familial partial, type7; FPLD7 (#606721) | CAV1 | AD | RD, cataract | Lack of facial fat, orthostatic hypotension, neurological and skin involvement |
| Methylmalonic Aciduria and Homocystinuria, cblC type; MAHCC (#277400) | MMACHC | AR | RP, CRD | FTT, microcephaly, ID, neurodegeneration, renal and hematological involvement |
| Mevalonic Aciduria; MEVA (#610377) | MVK | AR | RP, OA, cataract | FTT, DD, neurodegeneration, spleen, hepatic, skeletal, skin and hematological involvement |
| Microcephaly and Chorioretinopathy, autosomal recessive; MCCRP (#251270, #616171, #616335) | TUBGCP6, PLK4, TUBGCP4 | AR | Chorioretinopathy, OA, microphthalmia, microcornea, cataract | IUGR, microcephaly, brain structural anomalies, DD, ID, neurodegeneration, SS |
| Microcephaly with or without Chorioretinopathy, Lymphedema or Mental Retardation; MCLMR (#152950) | KIF11 | AD | Chorioretinopathy, myopia, hypermetropia, corneal opacity, microcornea, microphthalmia, cataract | Microcephaly, ID, neurodegeneration, lymphedema |
| Microphthalmia, Syndromic 5; MCOPS5 (#610125) | OTX2 | AD | RD, microphthalmia, anophthalmia, optic nerve hypoplasia or agenesis, microcornea, cataract | Brain structural anomalies, hypotonia, pituitary dysfunction, DD, SS, cleft palate, abnormal genitalia, joint laxity |
| Mitochondrial Complex II Deficiency (#252011) | SDHA, SDHD, SDHAF1 | AR | RD, OA, ptosis, ophthalmoplegia | SS, cardiac, skeletal, muscular and neurological involvement |
| Mitochondrial Complex IV Deficiency (#220110) | APOPT1, COA3, COX6A2, COX6B1, COX8A, COX10, COX14, COX20, PET100, TACO1 | AR | RD, OA, ptosis | FTT, brain structural anomalies, ID, HL, cardiac, respiratory, hepatic, renal and muscular involvement |
| Mucolipidosis III alpha/beta; MLIII A/B (#252600) | GNPTAB | AR | RD, corneal clouding | Neurodegeneration, ID, SS, coarse facies, skeletal, cardiac and skin involvement |
| Mucolipidosis IV; ML4 (#252650) | MCOLN1 | AR | RD, OA, corneal disease, strabismus | Microcephaly, ID, neurodegeneration |
| Mucopolysaccharidosis; MPS (#309900, #252930, #607014, #253000, #253010) | IDS | XLR | RP, ptosis, corneal clouding | Neurodegeneration, ID, SS, coarse facies, HL, skeletal, cardiac, respiratory, hepatic, gastrointestinal and skin involvement |
| HGSNAT, IDUA, GALN5, GLB1 | AR | |||
| Nephronophthisis 15; NPHP15 (#614845) | CEP164 | AR | LCA | Renal disease |
| Neurodegeneration with Brain Iron Accumulation 1; NBIA1 (#234200) | PANK2 | AR | RD, OA, eyelid apraxia | Neurodegeneration, gastrointestinal, skeletal, skin and muscular involvement |
| Neuropathy, Ataxia and Retinitis Pigmentosa; NARP (#551500) | MTATP6 | Mi | RP | Neurodegeneration, ataxia |
| Norrie Disease; ND (#310600) | NDP | XLR | Retinal dysgenesis, retinal dysplasia, OA, microphthalmia, vitreous atrophy, corneal opacities, iris atrophy, cataract | HL, ID, neurodegeneration |
| Oculoauricular Syndrome; OCACS (#612109) | HMX1 | AR | RP, microphthalmia, microcornea, cataract, microphakia, sclerocornea, increased intraocular pressure | External ear abnormalities |
| Orofaciodigital Syndrome XVI; OFD16 (#617563) | TMEM107 | AR | RD, oculomotor apraxia, ptosis | Facial anomalies, breathing abnormalities, polydactyly, hypotonia, ID, neurological anomalies |
| Oliver–McFarlane Syndrome; OMCS (#275400) | PNPLA6 | AR | Chorioretinopathy, OA | SS, ID, neurodegeneration, obesity, male external genitalia abnormalities, endocrine anomalies |
| Peroxisomal Acyl-CoA Oxidase Deficiency (#264470) | ACOX1 | AR | RD, OA, strabismus | Neurodegeneration, ID, HL, liver disease |
| Peroxisome Biogenesis Disorder; PBD (#214100, #614866, #601539, #234580, #614879, #266510) | PEX1, PEX2, PEX5, PEX6, PEX7, PEX12 | AR | RD, OA, corneal clouding, cataract | FTT, neurodegeneration, ID, HL, dental, cardiac, hepatic, genitourinary and skeletal involvement |
| Posterior Column Ataxia with Retinitis Pigmentosa; AXPC1 (#609033) | FLVCR1 | AR | RP, OA | Posterior column ataxia, neurodegeneration, gastrointestinal and skeletal involvement |
| Polyneuropathy, Hearing Loss, Ataxia, Retinitis Pigmentosa and Cataract; PHARC (#612674) | ABHD12 | AR | RP, OA, cataract | Ataxia, neurodegeneration, HL |
| Pseudoxanthoma Elasticum; PXE (#264800) | ABCC6 | AR | RD, MD, choroidal neovascularization | Skin lesions, cardiovascular disease, gastrointestinal and genitourinary involvement |
| Refsum Disease, classic (#266500) | PHYH | AR | RP | Neurodegeneration, ataxia, HL, anosmia, cardiac, skeletal and skin involvement |
| Retinal Dystrophy, Iris Coloboma and Comedogenic Acne Syndrome; RDCCAS (#615147) | RPB4 | AR | RD, coloboma of the iris, displacement of the pupil, microcornea, cataract | Comedogenic acne |
| Retinal Dystrophy and Iris Coloboma with or without Cataract; RDICC (#616722) | MIR204 | AD | RD, coloboma of the iris, congenital cataract | |
| Retinal Dystrophy, Juvenile Cataracts and Short Stature Syndrome; RDJCSS (#616108) | RDH11 | AR | RD, juvenile cataracts | SS, DD, ID, dental anomalies |
| Retinal Dystrophy and Obesity; RDOB (#616188) | TUB | AR | RD | Obesity |
| Revesz Syndrome (#268130) | TINF2 | AD | RD | IUGR, brain structural anomalies, neurodegeneration, ID, aplastic anemia, skin, hair and nail abnormalities |
| Retinitis Pigmentosa–Deafness Syndrome (#500004) | MTTS2 | Mi | RP | HL |
| Retinitis Pigmentosa and Erythrocytic Microcytosis; RPEM (#616959) | TRNT1 | AR | RP | Erythrocytic microcytosis and additional hematologic abnormalities |
| Retinitis Pigmentosa, Hypopituitarism, Nephronophtisis and mild Skeletal Dysplasia; RHYNS (#602152) | TMEM67 | AR | RP | Hypopituitarism, renal disease, skeletal anomalies, HL |
| Retinitis Pigmentosa 82 with or without Situs Inversus; RP82 (#615434) | ARL2BP | AR | RP | Situs inversus, male infertility |
| Retinitis Pigmentosa with or without Skeletal Anomalies; RPSKA (#250410) | CWC27 | AR | RP | SS, skeletal anomalies, ID |
| Retinitis Pigmentosa, X-linked and Sinorespiratory Infections, with or without Deafness (#300455) | RPGR | XL | RP | Recurrent respiratory infections, HL |
| Senior–Løken Syndrome; SLSN (#266900, #606996, #609254, #610189, #613615, #616307, #616629) | NPHP1, NPHP4, IQCB1, CEP290, SDCCAG8, WDR19, TRAF3IP1 | AR | RP, LCA | Renal disease |
| Short Stature, Hearing Loss, Retinitis Pigmentosa and Distinctive Facies; SHRF (#617763) | EXOSC2 | AR | RP, corneal dystrophy, glaucoma, strabismus | SS, facial anomalies, HL, neurodegeneration, DD, ID |
| Sideroblastic Anemia with B-cell Immunodeficiency, Periodic Fevers and Developmental Delay; SIFD (#616084) | TRNT1 | AR | RP | Sideroblastic anemia, immunodeficiency, growth retardation, DD, periodic fever, HL, neurological, cardiac and renal involvement |
| Spondylometaphyseal Dysplasia with Cone–Rod Dystrophy; SMDCRD (#608940) | PCYT1A | AR | CRD | Skeletal anomalies, PGR |
| Spondylometaphyseal Dysplasia, Axial; SMDAX (#602271) | CFAP410 | AR | RP, CRD, OA | Skeletal anomalies, respiratory disease, reduced sperm motility |
| Short-Rib Thoracic Dysplasia 9 with or without Polydactyly; SRTD9 (#266920) | IFT140 | AR | RP | Skeletal anomalies, renal disease, ID |
| Thiamine-Responsive Megaloblastic Anemia Syndrome; TRMA (#249270) | SLC19A2 | AR | OA, RD | Megaloblastic anemia, diabetes mellitus, HL |
| Usher Syndrome; USH (#276900, #276904, #601067, #602083, #606943, #614869, #276901, #605472, #611383, #276902, #614504) | MYO7A, USH1C, CDH23, PCDH15, USH1G, CIB2, USH2A, ADGRV1, WHRN, CLRN1, HARS1 | AR | RP | HL, vestibular dysfunction |
| Wolfram Syndrome 1, WFS1 (#222300) | WFS1 | AR | OA, RD | Diabetes mellitus, diabetes insipidus, HL, neurodegeneration, genitourinary and neurologic involvement |
| White–Sutton Syndrome, WHSUS (#616364) | POGZ | AD | RP, OA, cortical blindness | DD, characteristic facial features, hypotonia, HL, joint laxity, gastrointestinal anomalies |
| Xeroderma Pigmentosum, group B; XPB (#610651) | ERCC3 | AR | RD, OA, micropathalmia | Neoplasia, skin anomalies, SS, microcephaly, HL, ID, brain structural anomalies, neurodegeneration |
| Gene | Syndromic IRD | Non-Syndromic IRD (MIM) | Reference |
|---|---|---|---|
| ABHD12 | PHARC | arRP | [40] |
| AHI1 | JBTS3 | arRP | [41] |
| ALMS1 | ALMS | arCRD, arEORD | [42] |
| ARL2BP | RP with situs inversus | arRP (#615434) | [43,44] |
| ARL3 | JBTS35 | adRP (#618173) | [45] |
| ARL6 | BBS3 | arRP (#613575) | [46] |
| BBS2 | BBS2 | arRP (#616562) | [47] |
| C8ORF37 | BBS21 | arCRD, arRP (#614500) | [48] |
| CC2D2A | JBTS9, MKS6 | arRP | [49] |
| CEP290 | BBS14, JBTS5, MKS4, SLSN6 | arLCA (#611755) | [50] |
| CFAP410 | SMDAX | arRD with or without macular staphyloma (#617547) | [51,52] |
| CLN3 | CLN3 | arRP | [53] |
| CLRN1 | USH3A | arRP (#614180) | [54] |
| CWC27 | RPSKA | arRP (#250410) | [55] |
| DHDDS | CDG1BB | arRP (#613861) | [56,57] |
| FLVCR1 | PCARP | arRP | [58] |
| HGSNAT | MPS3C | arRP (#616544) | [59] |
| IFT140 | SRTD9 with/without polydactyly | arRP (#617781) | [60] |
| IQCB1 | SLSN5 | arLCA | [61] |
| MFSD8 | CLN7 | arMD (#616170), arRD | [62] |
| MMACHC | MAHCC | arMD | [63] |
| MVK | HIDS, MEVA | arRP | [64] |
| NDP | ND | XL EVR (#305390) | [65] |
| OFD1 | JBTS10 | XL RP (#300424) | [66] |
| OTX2 | RD with pituitary dysfunction | adPD (#610125) | [67] |
| RPGR | RP, sinorespiratory infections and deafness | XL CRD (#304020), XL MD (#300834), XL RP (#300029) | [68] |
| TTC8 | BBS8 | arRP (#613464) | [69] |
| USH2A | USH2A | arRP (#613809) | [70] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tatour, Y.; Ben-Yosef, T. Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects. Diagnostics 2020, 10, 779. https://doi.org/10.3390/diagnostics10100779
Tatour Y, Ben-Yosef T. Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects. Diagnostics. 2020; 10(10):779. https://doi.org/10.3390/diagnostics10100779
Chicago/Turabian StyleTatour, Yasmin, and Tamar Ben-Yosef. 2020. "Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects" Diagnostics 10, no. 10: 779. https://doi.org/10.3390/diagnostics10100779
APA StyleTatour, Y., & Ben-Yosef, T. (2020). Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects. Diagnostics, 10(10), 779. https://doi.org/10.3390/diagnostics10100779