Advantages and Limitations of 16S rRNA Next-Generation Sequencing for Pathogen Identification in the Diagnostic Microbiology Laboratory: Perspectives from a Middle-Income Country

 , , , , , ,
, , , , , ,  and
and 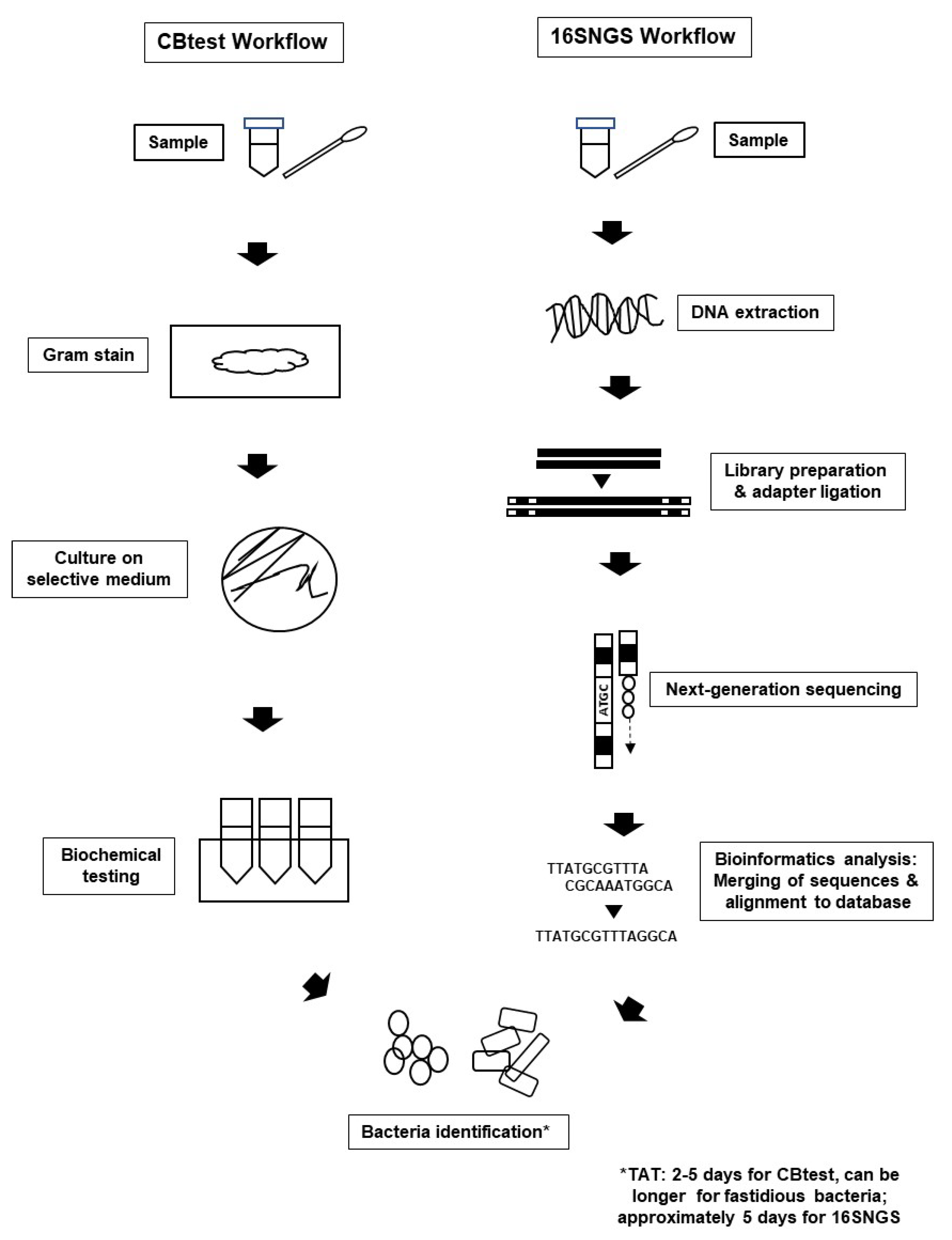{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Culture and Biochemical Testing (CBtest) for Bacterial Pathogen Identification
1.2. Limitations of CBtest
1.3. 16S rRNA Next-Generation Sequencing (16SNGS): An Alternative to CBtest
1.4. Diagnostic Microbiology in Malaysia, a Middle-Income Country
2. 16SNGS: Platforms, Workflow and Bioinformatics Analysis
2.1. NGS Technology
2.2. 16SNGS Workflow and Bioinformatics Analysis
3. Limitations and Challenges in Implementing 16SNGS for Pathogen Identification in Diagnostic Microbiology Laboratories of Middle-Income Countries
3.1. Low Taxonomical Resolution in 16SNGS Sequencing Reads
3.2. Bioinformatics Analysis of Results
3.3. Costly Laboratory Set-Up, Maintenance, Staff Training and Reagent Procurement
3.4. Lack of Sample Trail for Repeat Testing and Antibiotic Susceptibility Testing (AST)
3.5. Lack of Workflow Standardization and Validation
4. Advantages of 16SNGS for Bacterial Pathogen Detection
4.1. Identification of Unculturable and Fastidious Bacteria
4.2. Shorter and Predictable Turn-Around-Time with Streamlined Identification Protocol
4.3. Accuracy of Results
4.4. Data Portability and Technology Transition Readiness
5. Future Considerations
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Washington, J.A. Principles of Diagnosis. In Medical Microbiology; Baron, S., Ed.; University of Texas Medical Branch: Galveston, TX, USA, 1996. [Google Scholar]
- Sayed, S.; Cherniak, W.; Lawler, M.; Tan, S.Y.; El Sadr, W.; Wolf, N.; Silkensen, S.; Brand, N.; Looi, L.M.; Pai, S.A.; et al. Improving pathology and laboratory medicine in low-income and middle-income countries: Roadmap to solutions. Lancet 2018, 391, 1939–1952. [Google Scholar] [CrossRef]
- Jegathesan, M.; De Witt, G.F. Organisation of laboratory services in Malaysia. Malays. J. Pathol. 1982, 5, 1–5. [Google Scholar]
- Clarridge, J.E. Impact of 16S rRNA Gene Sequence Analysis for Identification of Bacteria on Clinical Microbiology and Infectious Diseases Impact of 16S rRNA Gene Sequence Analysis for Identification of Bacteria on Clinical Microbiology and Infectious Diseases. Clin. Microbiol. Rev. 2004, 17, 840–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagier, J.C.; Edouard, S.; Pagnier, I.; Mediannikov, O.; Drancourt, M.; Raoult, D. Current and past strategies for bacterial culture in clinical microbiology. Clin. Microbiol. Rev. 2015, 28, 208–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, V.K. The Medical Microbiological Laboratory Services in Malaysia. Malays. J. Pathol. 1982, 5, 15–18. [Google Scholar] [PubMed]
- Schroeder, L.F.; Guarner, J.; Amukele, T.K. Essential Diagnostics for the Use of World Health Organization Essential Medicines. Clin. Chem. 2018, 64, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Nkengasong, J.N.; Yao, K.; Onyebujoh, P. Laboratory medicine in low-income and middle-income countries: Progress and challenges. Lancet 2018, 391, 1873–1875. [Google Scholar] [CrossRef] [Green Version]
- Petti, C.A.; Polage, C.R.; Schreckenberger, P. The role of 16S rRNA gene sequencing in identification of microorganisms misidentified by conventional methods. J. Clin. Microbiol. 2005, 43, 6123–6125. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, R.; Karaoz, U.; Volegova, M.; MacKichan, J.; Kato-Maeda, M.; Miller, S.; Nadarajan, R.; Brodie, E.L.; Lynch, S.V. Use of 16S rRNA gene for identification of a broad range of clinically relevant bacterial pathogens. PLoS ONE 2015, 10, e0117617. [Google Scholar] [CrossRef]
- Fournier, P.E.; Dubourg, G.; Raoult, D. Clinical detection and characterization of bacterial pathogens in the genomics era. Genome Med. 2014, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S. Improving Diagnostic and Laboratory Capacity Helps in Control of Infection: An Indian Perspective. Curr. Treat. Options Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Sastry, S.; Masroor, N.; Bearman, G.; Hajjeh, R.; Holmes, A.; Memish, Z.; Lassmann, B.; Pittet, D.; Macnab, F.; Kamau, R.; et al. The 17th International Congress on Infectious Diseases Workshop on Developing Infection Prevention and Control Resources for Low to Middle Income Countries. Int. J. Infect. Dis. 2017, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sune, D.; Rydberg, H.; Augustinsson, Å.N.; Serrander, L.; Jungeström, M.B. Optimization of 16S rRNA gene analysis for use in the diagnostic clinical microbiology service. J. Microbiol. Methods 2020, 170, 105854. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N.; Alland, D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, L.A.; Kurosawa, K.; Hoogestraat, D.R.; SenGupta, D.J.; Candra, F.; Doyle, M.; Thielges, S.; Land, T.A.; Rosenthal, C.A.; Hoffman, N.G.; et al. Clinical next generation sequencing outperforms standard microbiological culture for characterizing polymicrobial samples. Clin. Chem. 2016, 62, 1465–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, J.C.; Mccallum, N.; Sintchenko, V.; Howden, B.P. Whole genome sequencing in clinical and public health microbiology. Pathology 2015, 47, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.L.; Fleming, K.A.; Kuti, M.A.; Looi, L.M.; Lago, N.; Ru, K. Access to pathology and laboratory medicine services: A crucial gap. Lancet 2018, 391, 1927–1938. [Google Scholar] [CrossRef]
- Horton, S.; Sullivan, R.; Flanigan, J.; Fleming, K.A.; Kuti, M.A.; Looi, L.M.; Pai, S.A.; Lawler, M. Delivering modern, high-quality, affordable pathology and laboratory medicine to low-income and middle-income countries: A call to action. Lancet 2018, 391, 1953–1964. [Google Scholar] [CrossRef]
- Rosli, F. Exit the middle income trap. New Straits Times, 3 April 2019. [Google Scholar]
- International Monetary Fund World Economic Outlook (October 2019). Available online: https://www.imf.org/external/datamapper/NGDPD@WEO/THA/MYS/SGP/PHL/IDN (accessed on 6 April 2020).
- Al-Darraji, H.A.A.; Razak, H.A.; Ng, K.P.; Altice, F.L.; Kamarulzaman, A. The Diagnostic Performance of a Single GeneXpert MTB/RIF Assay in an Intensified Tuberculosis Case Finding Survey among HIV-Infected Prisoners in Malaysia. PLoS ONE 2013, 8, e73717. [Google Scholar] [CrossRef]
- Escobar-Zepeda, A.; Vera-Ponce de León, A.; Sanchez-Flores, A. The Road to Metagenomics: From Microbiology to DNA Sequencing Technologies and Bioinformatics. Front. Genet. 2015, 6, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buermans, H.P.J.; Den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1932–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.; Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5448. [Google Scholar] [CrossRef] [PubMed]
- Valouev, A.; Ichikawa, J.; Tonthat, T.; Stuart, J.; Ranade, S.; Peckham, H.; Zeng, K.; Malek, J.A.; Costa, G.; McKernan, K.; et al. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res. 2008, 18, 1051–1063. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-F.; Chen, S.-C.; Chiang, Y.-S.; Chen, T.-H.; Chiu, K.-P. Palindromic sequence impedes sequencing-by-ligation mechanism. BMC Syst. Biol. 2012, 6 (Suppl. S2), S10. [Google Scholar] [CrossRef] [Green Version]
- Porreca, G.J. Genome sequencing on nanoballs. Nat. Biotechnol. 2010, 28, 43–44. [Google Scholar] [CrossRef]
- Patch, A.-M.; Nones, K.; Kazakoff, S.H.; Newell, F.; Wood, S.; Leonard, C.; Holmes, O.; Xu, Q.; Addala, V.; Creaney, J.; et al. Germline and somatic variant identification using BGISEQ-500 and HiSeq X Ten whole genome sequencing. PLoS ONE 2018, 13, e0190264. [Google Scholar] [CrossRef]
- Mak, S.S.T.; Gopalakrishnan, S.; Carøe, C.; Geng, C.; Liu, S.; Sinding, M.-H.S.; Kuderna, L.F.K.; Zhang, W.; Fu, S.; Vieira, F.G.; et al. Comparative performance of the BGISEQ-500 vs Illumina HiSeq2500 sequencing platforms for palaeogenomic sequencing. Gigascience 2017, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.-Y.; Chen, M.-X.; Ye, N.-H.; Qiao, W.-M.; Gao, B.; Law, W.-K.; Tian, Y.; Zhang, D.; Zhang, D.; Liu, T.-Y.; et al. Comparative performance of the BGISEQ-500 and Illumina HiSeq4000 sequencing platforms for transcriptome analysis in plants. Plant. Methods 2018, 14, 69. [Google Scholar] [CrossRef] [Green Version]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Kai, S.; Matsuo, Y.; Nakagawa, S.; Kryukov, K.; Matsukawa, S.; Tanaka, H.; Iwai, T.; Imanishi, T.; Hirota, K. Rapid bacterial identification by direct PCR amplification of 16S rRNA genes using the MinIONTM nanopore sequencer. FEBS Open Bio 2019, 9, 548–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D.; Jenior, M.L.; Koumpouras, C.C.; Westcott, S.L.; Highlander, S.K. Sequencing 16S rRNA gene fragments using the PacBio SMRT DNA sequencing system. PeerJ 2016, 4, e1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambardar, S.; Gupta, R.; Trakroo, D.; Lal, R.; Vakhlu, J. High Throughput Sequencing: An Overview of Sequencing Chemistry. Indian J. Microbiol. 2016, 56, 394–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besser, J.; Carleton, H.A.; Gerner-Smidt, P.; Lindsey, R.L.; Trees, E. Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clin. Microbiol. Infect. 2018, 24, 335–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef] [PubMed]
- Alekseyev, Y.O.; Fazeli, R.; Yang, S.; Basran, R.; Maher, T.; Miller, N.S.; Remick, D. A Next-Generation Sequencing Primer-How Does It Work and What Can It Do? Acad. Pathol. 2018, 5, 2374289518766521. [Google Scholar] [CrossRef] [Green Version]
- Handelsman, J.; Rondon, M.; Brady, S.; Clardy, J.; Goodman, R.; Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular Biological access to the chemistry of unknown soil microbes: A new frontier for natural products. Chem. Biol. 1998, 5, R245–R249. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Pachter, L. Bioinformatics for Whole-Genome Shotgun Sequencing of Microbial Communities. PLOS Comput. Biol. 2005, 1, e24. [Google Scholar] [CrossRef] [Green Version]
- Deurenberg, R.H.; Bathoorn, E.; Chlebowicz, M.A.; Couto, N.; Ferdous, M.; García-Cobos, S.; Kooistra-Smid, A.M.D.; Raangs, E.C.; Rosema, S.; Veloo, A.C.M.; et al. Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 2017, 243, 16–24. [Google Scholar] [CrossRef]
- Watanabe, N.; Kryukov, K.; Nakagawa, S.; Takeuchi, J.S.; Takeshita, M.; Kirimura, Y.; Mitsuhashi, S.; Ishihara, T.; Aoki, H.; Inokuchi, S.; et al. Detection of pathogenic bacteria in the blood from sepsis patients using 16S rRNA gene amplicon sequencing analysis. PLoS ONE 2018, 13, e0202049. [Google Scholar] [CrossRef]
- Sabat, A.J.; Van Zanten, E.; Akkerboom, V.; Wisselink, G.; Van Slochteren, K.; De Boer, R.F.; Hendrix, R.; Friedrich, A.W.; Rossen, J.W.A.; Kooistra-Smid, A.M.D. (Mirjam) Targeted next-generation sequencing of the 16S-23S rRNA region for culture-independent bacterial identification–increased discrimination of closely related species. Sci. Rep. 2017, 7, 3434. [Google Scholar] [CrossRef] [PubMed]
- Bartram, A.K.; Lynch, M.D.J.; Stearns, J.C.; Moreno-Hagelsieb, G.; Neufeld, J.D. Generation of Multimillion-Sequence 16S rRNA Gene Libraries from Complex Microbial Communities by Assembling Paired-End Illumina Reads. Appl. Environ. Microbiol. 2011, 77, 3846–3852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, G.S.; Youens-Clark, K.; Slepian, M.J.; Wolk, D.M.; Oshiro, M.M.; Metzger, G.S.; Dhingra, D.; Cranmer, L.D.; Hurwitz, B.L. 16S rRNA gene sequencing on a benchtop sequencer: Accuracy for identification of clinically important bacteria. J. Appl. Microbiol. 2017, 123, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Bukin, Y.S.; Galachyants, Y.P.; Morozov, I.V.; Bukin, S.V.; Zakharenko, A.S.; Zemskaya, T.I. The effect of 16S rRNA region choice on bacterial community metabarcoding results. Sci. Data 2019, 6, 190007. [Google Scholar] [CrossRef] [Green Version]
- Fouhy, F.; Clooney, A.G.; Stanton, C.; Claesson, M.J.; Cotter, P.D. 16S rRNA gene sequencing of mock microbial populations- impact of DNA extraction method, primer choice and sequencing platform. BMC Microbiol. 2016, 16, 123. [Google Scholar] [CrossRef]
- Yang, B.; Wang, Y.; Qian, P.Y. Sensitivity and correlation of hypervariable regions in 16S rRNA genes in phylogenetic analysis. BMC Bioinform. 2016, 17, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Inc., Illumina. Part # 15044223 Rev. B. In 16S Metagenomic Sequencing Library Preparation; Illumina Inc.: San Diego, CA, USA, 2013. [Google Scholar]
- Thermo Fisher Scientific. MAN0010799. In Ion. 16S Metanomics Kit; Thermo Fisher Scientific: Waltham, MA, USA, 2020. [Google Scholar]
- Pichler, M.; Coskun, Ö.K.; Ortega-Arbulú, A.-S.; Conci, N.; Wörheide, G.; Vargas, S.; Orsi, W.D. A 16S rRNA gene sequencing and analysis protocol for the Illumina MiniSeq platform. Microbiologyopen 2018, 7, e00611. [Google Scholar] [CrossRef]
- Tan, S.C.; Yiap, B.C. DNA, RNA, and protein extraction: The past and the present. J. Biomed. Biotechnol. 2009, 2009, 574398. [Google Scholar] [CrossRef] [Green Version]
- Hess, J.F.; Kohl, T.A.; Kotrová, M.; Rönsch, K.; Paprotka, T.; Mohr, V.; Hutzenlaub, T.; Brüggemann, M.; Zengerle, R.; Niemann, S.; et al. Library preparation for next generation sequencing: A review of automation strategies. Biotechnol. Adv. 2020, 41, 107537. [Google Scholar] [CrossRef]
- Kim, H.; Jebrail, M.J.; Sinha, A.; Bent, Z.W.; Solberg, O.D.; Williams, K.P.; Langevin, S.A.; Renzi, R.F.; Van De Vreugde, J.L.; Meagher, R.J.; et al. A Microfluidic DNA Library Preparation Platform for Next-Generation Sequencing. PLoS ONE 2013, 8, e68988. [Google Scholar] [CrossRef]
- Tan, S.J.; Phan, H.; Gerry, B.M.; Kuhn, A.; Hong, L.Z.; Min Ong, Y.; Poon, P.S.Y.; Unger, M.A.; Jones, R.C.; Quake, S.R.; et al. A Microfluidic Device for Preparing Next Generation DNA Sequencing Libraries and for Automating Other Laboratory Protocols That Require One or More Column Chromatography Steps. PLoS ONE 2013, 8, e64084. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.F.; Kotrová, M.; Calabrese, S.; Darzentas, N.; Hutzenlaub, T.; Zengerle, R.; Brüggemann, M.; Paust, N. Automation of Amplicon-Based Library Preparation for Next-Generation Sequencing by Centrifugal Microfluidics. Anal. Chem. 2020. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.-L.; Weng, J.-C.; Hsiao, C.-C.; Chou, M.-T.; Tseng, C.-W.; Hung, J.-H. PEAT: An intelligent and efficient paired-end sequencing adapter trimming algorithm. BMC Bioinform. 2015, 16 (Suppl. S1), S2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef]
- Nguyen, N.-P.; Warnow, T.; Pop, M.; White, B. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. NPJ Biofilms Microbiomes 2016, 2, 16004. [Google Scholar] [CrossRef] [Green Version]
- Bharti, R.; Grimm, D.G. Current challenges and best-practice protocols for microbiome analysis. Brief. Bioinform. 2019. [Google Scholar] [CrossRef] [Green Version]
- Nearing, J.T.; Douglas, G.M.; Comeau, A.M.; Langille, M.G.I. Denoising the Denoisers: An independent evaluation of microbiome sequence error-correction approaches. PeerJ 2018, 6, e5364. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 2016, 81257. [Google Scholar] [CrossRef] [Green Version]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 2020, 15, e0227434. [Google Scholar] [CrossRef] [Green Version]
- Federhen, S. The NCBI Taxonomy database. Nucleic Acids Res. 2011, 40, D136–D143. [Google Scholar] [CrossRef] [Green Version]
- Balvočiūtė, M.; Huson, D.H. SILVA, RDP, Greengenes, NCBI and OTT—How do these taxonomies compare? BMC Genom. 2017, 18, 114. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Glöckner, F.O.; Yilmaz, P.; Quast, C.; Gerken, J.; Beccati, A.; Ciuprina, A.; Bruns, G.; Yarza, P.; Peplies, J.; Westram, R.; et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 2017, 261, 169–176. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-C.; Won, S. Evaluation of 16S rRNA Databases for Taxonomic Assignments Using Mock Community. Genom. Inform. 2018, 16, e24. [Google Scholar] [CrossRef] [Green Version]
- Eisen, J.A. Environmental shotgun sequencing: Its potential and challenges for studying the hidden world of microbes. PLoS Biol. 2007, 5, e82. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, I.; Tap, J.; Roudot-Thoraval, F.; Roperch, J.P.; Letulle, S.; Langella, P.; Corthier, G.; Tran Van Nhieu, J.; Furet, J.P. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS ONE 2011, 6, e16393. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.W.-C.; Gan, H.-M.; Lee, Y.-P.; Leow, A.H.-R.; Azmi, A.N.; Francois, F.; Perez-Perez, G.I.; Loke, M.-F.; Goh, K.-L.; Vadivelu, J. Helicobacter pylori Eradication Causes Perturbation of the Human Gut Microbiome in Young Adults. PLoS ONE 2016, 11, e0151893. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.C.; Yap, P.S.X.; Chong, C.W.; Cheok, Y.Y.; Lee, C.Y.Q.; Tan, G.M.Y.; Sulaiman, S.; Hassan, J.; Sabet, N.S.; Looi, C.Y.; et al. Diversity of endocervical microbiota associated with genital Chlamydia trachomatis infection and infertility among women visiting obstetrics and gynecology clinics in Malaysia. PLoS ONE 2019, 14, e0224658. [Google Scholar] [CrossRef] [PubMed]
- Nurul, A.N.A.; Muhammad, D.-D.; Okomoda, V.T.; Nur, A.A.B. 16S rRNA-Based metagenomic analysis of microbial communities associated with wild Labroides dimidiatus from Karah Island, Terengganu, Malaysia. Biotechnol. Rep. 2019, 21, e00303. [Google Scholar] [CrossRef]
- Khalid, N.A.; Rajandas, H.; Parimannan, S.; Croft, L.J.; Loke, S.; Chong, C.S.; Bruce, N.C.; Yahya, A. Insights into microbial community structure and diversity in oil palm waste compost. 3 Biotech. 2019, 9, 364. [Google Scholar] [CrossRef]
- Nygaard, A.B.; Tunsjø, H.S.; Meisal, R.; Charnock, C. A preliminary study on the potential of Nanopore MinION and Illumina MiSeq 16S rRNA gene sequencing to characterize building-dust microbiomes. Sci. Rep. 2020, 10, 3209. [Google Scholar] [CrossRef] [Green Version]
- Pearman, W.S.; Freed, N.E.; Silander, O.K. Testing the advantages and disadvantages of short- and long- read eukaryotic metagenomics using simulated reads. BMC Bioinform. 2020, 21, 220. [Google Scholar] [CrossRef]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L.; et al. Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef] [Green Version]
- McLean, K.; Rosenthal, C.A.; Sengupta, D.; Owens, J.; Cookson, B.T.; Hoffman, N.G.; Salipante, S.J. Improved Species-Level Clinical Identification of Enterobacteriaceae through Broad-Range dnaJ PCR and Sequencing. J. Clin. Microbiol. 2019, 57. [Google Scholar] [CrossRef]
- Mahenthiralingam, E.; Bischof, J.; Byrne, S.K.; Radomski, C.; Davies, J.E.; Av-Gay, Y.; Vandamme, P. DNA-Based diagnostic approaches for identification of Burkholderia cepacia complex, Burkholderia vietnamiensis, Burkholderia multivorans, Burkholderia stabilis, and Burkholderia cepacia genomovars I and III. J. Clin. Microbiol. 2000, 38, 3165–3173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Jang, H.; Kim, C.; Chung, B.; Chang, C.L.; Park, S.K.; Song, S. Detection and identification of mycobacteria by amplification of the internal transcribed spacer regions with genus- and species-specific PCR primers. J. Clin. Microbiol. 2000, 38, 4080–4085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tortoli, E. Impact of genotypic studies on mycobacterial taxonomy: The new mycobacteria of the 1990s. Clin. Microbiol. Rev. 2003, 16, 319–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeaiter, Z.; Liang, Z.; Raoult, D. Genetic classification and differentiation of Bartonella species based on comparison of partial ftsZ gene sequences. J. Clin. Microbiol. 2002, 40, 3641–3647. [Google Scholar] [CrossRef] [Green Version]
- Fournier, P.-E.; Dumler, J.S.; Greub, G.; Zhang, J.; Wu, Y.; Raoult, D. Gene sequence-based criteria for identification of new rickettsia isolates and description of Rickettsia heilongjiangensis sp. nov. J. Clin. Microbiol. 2003, 41, 5456–5465. [Google Scholar] [CrossRef] [Green Version]
- Church, D.L.; Cerutti, L.; Gürtler, A.; Griener, T.; Zelazny, A.; Emler, S. Performance and Application of 16S rRNA Gene Cycle Sequencing for Routine Identification of Bacteria in the Clinical Microbiology Laboratory. Clin. Microbiol. Rev. 2020, 33, e00053-19. [Google Scholar] [CrossRef]
- Fleming, K.A.; Naidoo, M.; Wilson, M.; Flanigan, J.; Horton, S.; Kuti, M.; Looi, L.M.; Price, C.; Ru, K.; Ghafur, A.; et al. An Essential Pathology Package for Low- and Middle-Income Countries. Am. J. Clin. Pathol. 2017, 147, 15–32. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Sirisena, N.D.; Dissanayake, V.H.W. Strategies for Genomic Medicine Education in Low- and Middle-Income Countries. Front. Genet. 2019, 10, 944. [Google Scholar] [CrossRef] [Green Version]
- Chow, K. Comparison of Bioinformatics Industry between Malaysia and India: An Overview. Int. J. Bus. Soc. Sci. 2011, 2, 83–92. [Google Scholar]
- Sherry, N.L.; Porter, J.L.; Seemann, T.; Watkins, A.; Stinear, T.P.; Howden, B.P. Outbreak investigation using high-throughput genome sequencing within a diagnostic microbiology laboratory. J. Clin. Microbiol. 2013, 51, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwinn, M.; MacCannell, D.; Armstrong, G.L. Next-Generation Sequencing of Infectious Pathogens. JAMA 2019, 321, 893–894. [Google Scholar] [CrossRef] [Green Version]
- College of Pathologists, Academy of Medicine of Malaysia. Guidelines on Minimum Qualification, Training and Experience of Professional Personnel Working in a Pathology Laboratory. Malays. J. Pathol. 2005, 27, 57–62. [Google Scholar]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection. Annu. Rev. Pathol. 2019, 14, 319–338. [Google Scholar] [CrossRef]
- Angiuoli, S.V.; White, J.R.; Matalka, M.; White, O.; Fricke, W.F. Resources and Costs for Microbial Sequence Analysis Evaluated Using Virtual Machines and Cloud Computing. PLoS ONE 2011, 6, e26624. [Google Scholar] [CrossRef] [Green Version]
- Alividza, V.; Mariano, V.; Ahmad, R.; Charani, E.; Rawson, T.M.; Holmes, A.H.; Castro-Sánchez, E. Investigating the impact of poverty on colonization and infection with drug-resistant organisms in humans: A systematic review. Infect. Dis. Poverty 2018, 7, 76. [Google Scholar] [CrossRef]
- Simner, P.J.; Miller, S.; Carroll, K.C. Understanding the Promises and Hurdles of Metagenomic Next-Generation Sequencing as a Diagnostic Tool for Infectious Diseases. Clin. Infect. Dis. 2017, 66, 778–788. [Google Scholar] [CrossRef] [Green Version]
- Schlaberg, R.; Chiu, C.Y.; Miller, S.; Procop, G.W.; Weinstock, G. Validation of metagenomic next-generation sequencing tests for universal pathogen detection. Arch. Pathol. Lab. Med. 2017, 141, 776–786. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.Y.; Miller, S.A. Clinical metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Abel, H.J.; Merker, J.D.; Bodner, J.B.; Zhao, Q.; Voelkerding, K.V.; Pfeifer, J.D. A Model Study of In Silico Proficiency Testing for Clinical Next-Generation Sequencing. Arch. Pathol. Lab. Med. 2016, 140, 1085–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zengler, K.; Toledo, G.; Rappé, M.; Elkins, J.; Mathur, E.J.; Short, J.M.; Keller, M. Cultivating the uncultured. Proc. Natl. Acad. Sci. USA 2002, 99, 15681–15686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wade, W. Unculturable bacteria—The uncharacterized organisms that cause oral infections. J. R. Soc. Med. 2002, 95, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Lagier, J.-C.; Hugon, P.; Khelaifia, S.; Fournier, P.-E.; La Scola, B.; Raoult, D. The Rebirth of Culture in Microbiology through the Example of Culturomics To Study Human Gut Microbiota. Clin. Microbiol. Rev. 2015, 28, 237–264. [Google Scholar] [CrossRef] [Green Version]
- Coburn, B.; Morris, A.M.; Tomlinson, G.; Detsky, A.S. Does this adult patient with suspected bacteremia require blood cultures? JAMA 2012, 308, 502–511. [Google Scholar] [CrossRef]
- Baron, E.J.; Scott, J.D.; Tompkins, L.S. Prolonged incubation and extensive subculturing do not increase recovery of clinically significant microorganisms from standard automated blood cultures. Clin. Infect. Dis. 2005, 41, 1677–1680. [Google Scholar] [CrossRef] [Green Version]
- Scheer, C.S.; Fuchs, C.; Grundling, M.; Vollmer, M.; Bast, J.; Bohnert, J.A.; Zimmermann, K.; Hahnenkamp, K.; Rehberg, S.; Kuhn, S.-O. Impact of antibiotic administration on blood culture positivity at the beginning of sepsis: A prospective clinical cohort study. Clin. Microbiol. Infect. 2019, 25, 326–331. [Google Scholar] [CrossRef] [Green Version]
- Phua, J.; Ngerng, W.J.; See, K.C.; Tay, C.K.; Kiong, T.; Lim, H.F.; Chew, M.Y.; Yip, H.S.; Tan, A.; Khalizah, H.J.; et al. Characteristics and outcomes of culture-negative versus culture-positive severe sepsis. Crit. Care 2013, 17, R202. [Google Scholar] [CrossRef] [Green Version]
- Raoult, D.; Roux, V. Rickettsioses as paradigms of new or emerging infectious diseases. Clin. Microbiol. Rev. 1997, 10, 694–719. [Google Scholar] [CrossRef]
- Gouriet, F.; Fenollar, F.; Patrice, J.Y.; Drancourt, M.; Raoult, D. Use of shell-vial cell culture assay for isolation of bacteria from clinical specimens: 13 Years of experience. J. Clin. Microbiol. 2005, 43, 4993–5002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Lahiri, R.; Scollard, D.M.; Pena, M.; Williams, D.L.; Adams, L.B.; Figarola, J.; Truman, R.W. The armadillo: A model for the neuropathy of leprosy and potentially other neurodegenerative diseases. Dis. Model. Mech. 2013, 6, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbour, A.G.; Hayes, S.F. Biology of Borrelia species. Microbiol. Rev. 1986, 50, 381–400. [Google Scholar] [CrossRef] [PubMed]
- Lim, V.K.; Cheong, Y.M. Bacteriology turnaround time in seven Malaysian general hospitals. Malays. J. Pathol. 1992, 14, 41–43. [Google Scholar] [PubMed]
- Tabak, Y.P.; Vankeepuram, L.; Ye, G.; Jeffers, K.; Gupta, V.; Murray, P.R. Blood Culture Turnaround Time in U.S. Acute Care Hospitals and Implications for Laboratory Process Optimization. J. Clin. Microbiol. 2018, 56, e00500-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, C.; Fitzgibbon, M.; Powell, J.; Barron, D.; O’Mahony, J.; Power, L.; O’Connell, N.; Dunne, C. A commentary on the role of molecular technology and automation in clinical diagnostics. Bioengineered 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arena, F.; Argentieri, M.; Bernaschi, P.; Fortina, G.; Kroumova, V.; Manso, E.; Montanera, P.G.; Nicoletti, P.; Pecile, P.; Rassu, M.; et al. Real life turnaround time of blood cultures in the clinical microbiology laboratory: Results of the first Italian survey, May 2015. Microbiol. Med. 2016, 31. [Google Scholar] [CrossRef] [Green Version]
- Van Horn, K.G.; Audette, C.D.; Tucker, K.A.; Sebeck, D. Comparison of 3 swab transport systems for direct release and recovery of aerobic and anaerobic bacteria. Diagn. Microbiol. Infect. Dis. 2008, 62, 471–473. [Google Scholar] [CrossRef]
- El Khechine, A.; Henry, M.; Raoult, D.; Drancourt, M. Detection of Mycobacterium tuberculosis complex organisms in the stools of patients with pulmonary tuberculosis. Microbiology 2009, 155, 2384–2389. [Google Scholar] [CrossRef] [Green Version]
- Chapin, K.C.; Lauderdale, T.-L. Reagents, stains, and media: Bacteriology. In Manual of Clinical Microbiology; Murray, P.R., Baron, E.J., Jorgensen, J.H., Landry, M.L., Pfaller, M.A., Eds.; ASM Press: Washington, DC, USA, 2007; pp. 334–364. [Google Scholar]
- Chapin, K.C. Principles of Stains and Media. In Manual of Clinical Microbiology; Murray, P.R., Baron, E.J., Jorgensen, J.H., Landry, M.L., Pfaller, M.A., Eds.; ASM Press: Washington, DC, USA, 2007; pp. 182–191. [Google Scholar]
- Raoult, D.; La Scola, B.; Enea, M.; Fournier, P.E.; Roux, V.; Fenollar, F.; Galvao, M.A.; De Lamballerie, X. A flea-associated Rickettsia pathogenic for humans. Emerg. Infect. Dis. 2001, 7, 73–81. [Google Scholar] [CrossRef]
- Fitzgerald, C.; Nachamkin, I. Campylobacter and Arcobacter. In Manual of Clinical Microbiology; Murray, P.R., Baron, E.J., Jorgensen, J.H., Landry, M.L., Pfaller, M., Eds.; ASM Press: Washington, DC, USA, 2007; pp. 933–946. [Google Scholar]
- Ghodbane, R.; Raoult, D.; Drancourt, M. Dramatic reduction of culture time of Mycobacterium tuberculosis. Sci. Rep. 2014, 4, 4236. [Google Scholar] [CrossRef] [Green Version]
- Teng, F.; Darveekaran Nair, S.S.; Zhu, P.; Li, S.; Huang, S.; Li, X.; Xu, J.; Yang, F. Impact of DNA extraction method and targeted 16S-rRNA hypervariable region on oral microbiota profiling. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ducarmon, Q.R.; Hornung, B.V.H.; Geelen, A.R.; Kuijper, E.J.; Zwittink, R.D. Toward Standards in Clinical Microbiota Studies: Comparison of Three DNA Extraction Methods and Two Bioinformatic Pipelines. mSystems 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salipante, S.J.; Sengupta, D.J.; Rosenthal, C.; Costa, G.; Spangler, J.; Sims, E.H.; Jacobs, M.A.; Miller, S.I.; Hoogestraat, D.R.; Cookson, B.T.; et al. Rapid 16S rRNA next-generation sequencing of polymicrobial clinical samples for diagnosis of complex bacterial infections. PLoS ONE 2013, 8, e65226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagier, J.-C.; Bilen, M.; Cadoret, F.; Drancourt, M.; Fournier, P.-E.; La Scola, B.; Raoult, D. Naming microorganisms: The contribution of the IHU Méditerranée Infection, Marseille, France. New Microbes New Infect. 2018, 26, S89–S95. [Google Scholar] [CrossRef]
- Munson, E.; Carroll, K.C. What′s in a Name? New Bacterial Species and Changes to Taxonomic Status from 2012 through 2015. J. Clin. Microbiol. 2017, 55, 24–42. [Google Scholar] [CrossRef] [Green Version]
- Looi, L.-M. The Pathology Laboratory Act 2007 explained. Malays. J. Pathol. 2008, 30, 1–10. [Google Scholar]
- Audu, R.A.; Sylvester-Ikondu, U.; Onwuamah, C.K.; Salu, O.B.; Ige, F.A.; Meshack, E.; Aniedobe, M.; Amoo, O.S.; Okwuraiwe, A.P.; Okhiku, F.; et al. Experience of quality management system in a clinical laboratory in Nigeria. Afr. J. Lab. Med. 2012, 1, 18. [Google Scholar] [CrossRef] [Green Version]
- Kenya Accreditation Service Medical Laboratories. Available online: https://kenas.go.ke/our-services/medical-laboratories/ (accessed on 8 April 2020).
- Woodcock, S.; Fine, G.; McClure, K.; Unger, B.; Rizzo-Price, P. The role of standards and training in preparing for accreditation. Am. J. Clin. Pathol. 2010, 134, 388–392. [Google Scholar] [CrossRef] [Green Version]
- Ohuabunwa, E.C.; Sun, J.; Jean Jubanyik, K.; Wallis, L.A. Electronic Medical Records in low to middle income countries: The case of Khayelitsha Hospital, South Africa. Afr. J. Emerg. Med. Rev. 2016, 6, 38–43. [Google Scholar] [CrossRef] [Green Version]
- Narattharaksa, K.; Speece, M.; Newton, C.; Bulyalert, D. Key success factors behind electronic medical record adoption in Thailand. J. Health Organ. Manag. 2016, 30, 985–1008. [Google Scholar] [CrossRef] [Green Version]
- Ismail, N.; Abdullah, N.H. Developing Electronic Medical Records (EMR) Framework for Malaysia’s Public Hospitals. In 2011 IEEE Colloquium on Humanities, Science and Engineering; IEEE: Piscataway, NJ, USA, 2011. [Google Scholar]
- Willis, S.J.; Cocoros, N.M.; Randall, L.M.; Ochoa, A.M.; Haney, G.; Hsu, K.K.; DeMaria, A.J.; Klompas, M. Electronic Health Record Use in Public Health Infectious Disease Surveillance, USA, 2018–2019. Curr. Infect. Dis. Rep. 2019, 21, 32. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.-S.; Lee, W.-S.; Chen, G.-B.; Liu, C.-T. Improving the work efficiency of healthcare-associated infection surveillance using electronic medical records. Comput. Methods Programs Biomed. 2014, 117, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Sarkar, S.; Lin, Z.S.; McKenney, S.; Konry, T. Ultrafast Parallelized Microfluidic Platform for Antimicrobial Susceptibility Testing of Gram Positive and Negative Bacteria. Anal. Chem. 2019, 91, 6242–6249. [Google Scholar] [CrossRef]
- Wistrand-Yuen, P.; Malmberg, C.; Fatsis-Kavalopoulos, N.; Lübke, M.; Tängdén, T.; Kreuger, J. A Multiplex Fluidic Chip for Rapid Phenotypic Antibiotic Susceptibility Testing. MBio 2020, 11, e03109-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boers, S.A.; Hiltemann, S.D.; Stubbs, A.P.; Jansen, R.; Hays, J.P. Development and evaluation of a culture-free microbiota profiling platform (MYcrobiota) for clinical diagnostics. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1081–1089. [Google Scholar] [CrossRef] [Green Version]
- Rizal, N.M.; Neoh, H.R.R.; Periyasamy, P.; Hanafiah, A.; Samat, M.A.; Tan, T.; Wong, K.; Nathan, S.; Chieng, S.; Saw, S.; et al. Culture and Biochemical Testing versus 16S rRNA Gene Sequencing for Pathogen Identification in the Clinical Microbiology Laboratory: Preliminary Findings on Cost Comparison and Breaking the Bioinformatics Barrier. In Proceedings of the Abstracts of the 4th International Interscience Conference of Infection and Chemotherapy and 12th International Symposium on Antimicrobial Agents and Resistance (ICIC & ISAAR 2019), Gyeongju, Korea, 26–28 September 2019; The Korean Society of Infectious Diseases; The Korean Society for Chemotherapy; The Korean Society for AIDS: Seocho-gu, Seoul, Korea, 2019; Volume 51, p. S94. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhamad Rizal, N.S.; Neoh, H.-m.; Ramli, R.; A/L K Periyasamy, P.@.R.; Hanafiah, A.; Abdul Samat, M.N.; Tan, T.L.; Wong, K.K.; Nathan, S.; Chieng, S.; et al. Advantages and Limitations of 16S rRNA Next-Generation Sequencing for Pathogen Identification in the Diagnostic Microbiology Laboratory: Perspectives from a Middle-Income Country. Diagnostics 2020, 10, 816. https://doi.org/10.3390/diagnostics10100816
Muhamad Rizal NS, Neoh H-m, Ramli R, A/L K Periyasamy P@R, Hanafiah A, Abdul Samat MN, Tan TL, Wong KK, Nathan S, Chieng S, et al. Advantages and Limitations of 16S rRNA Next-Generation Sequencing for Pathogen Identification in the Diagnostic Microbiology Laboratory: Perspectives from a Middle-Income Country. Diagnostics. 2020; 10(10):816. https://doi.org/10.3390/diagnostics10100816
Chicago/Turabian StyleMuhamad Rizal, Nurnabila Syafiqah, Hui-min Neoh, Ramliza Ramli, Petrick @ Ramesh A/L K Periyasamy, Alfizah Hanafiah, Muttaqillah Najihan Abdul Samat, Toh Leong Tan, Kon Ken Wong, Sheila Nathan, Sylvia Chieng, and et al. 2020. "Advantages and Limitations of 16S rRNA Next-Generation Sequencing for Pathogen Identification in the Diagnostic Microbiology Laboratory: Perspectives from a Middle-Income Country" Diagnostics 10, no. 10: 816. https://doi.org/10.3390/diagnostics10100816
APA StyleMuhamad Rizal, N. S., Neoh, H.-m., Ramli, R., A/L K Periyasamy, P. @. R., Hanafiah, A., Abdul Samat, M. N., Tan, T. L., Wong, K. K., Nathan, S., Chieng, S., Saw, S. H., & Khor, B. Y. (2020). Advantages and Limitations of 16S rRNA Next-Generation Sequencing for Pathogen Identification in the Diagnostic Microbiology Laboratory: Perspectives from a Middle-Income Country. Diagnostics, 10(10), 816. https://doi.org/10.3390/diagnostics10100816