Identification of a De Novo Xq26.2 Microduplication Encompassing FIRRE Gene in a Child with Intellectual Disability

 , ,
, , 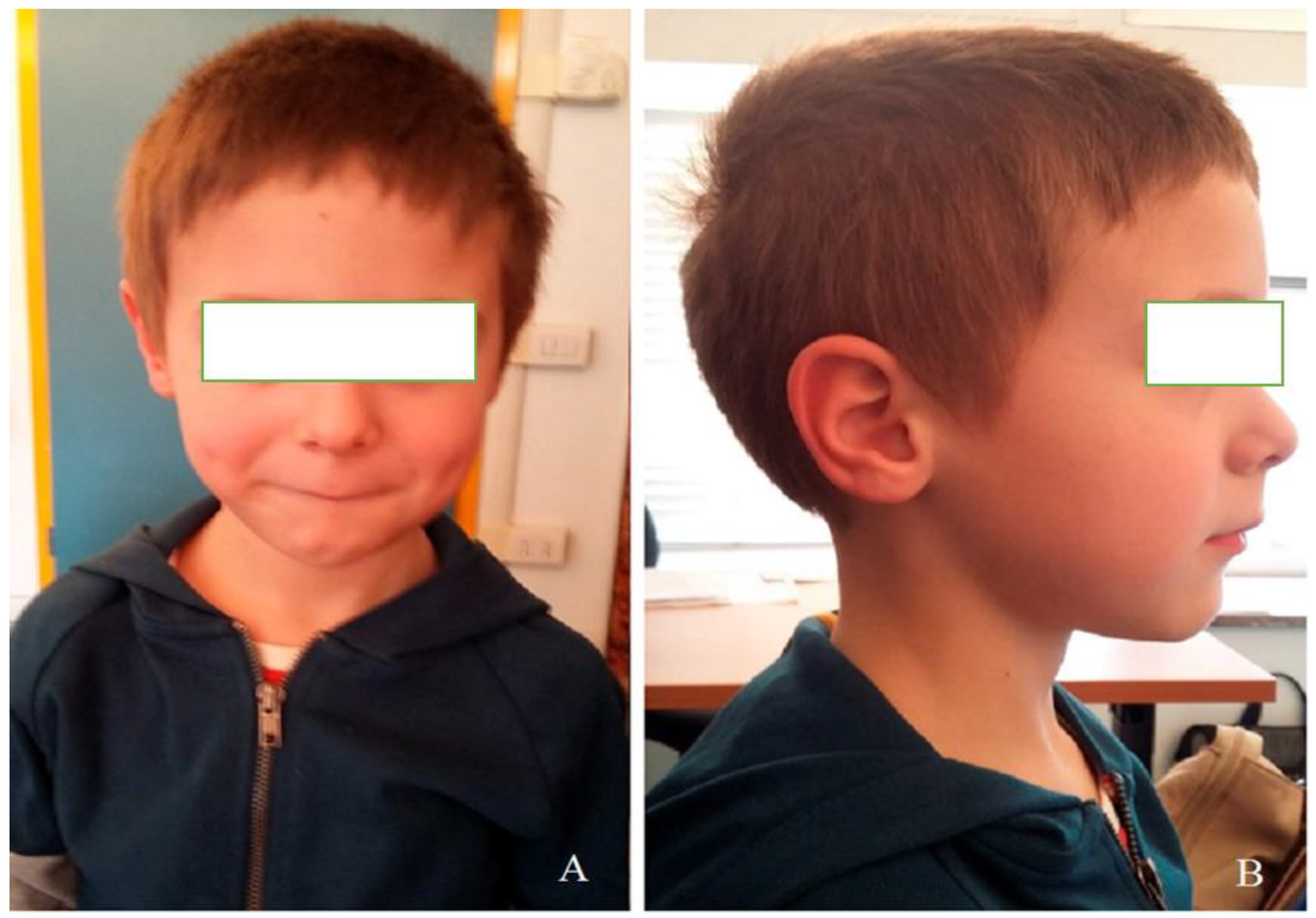{kind=link}
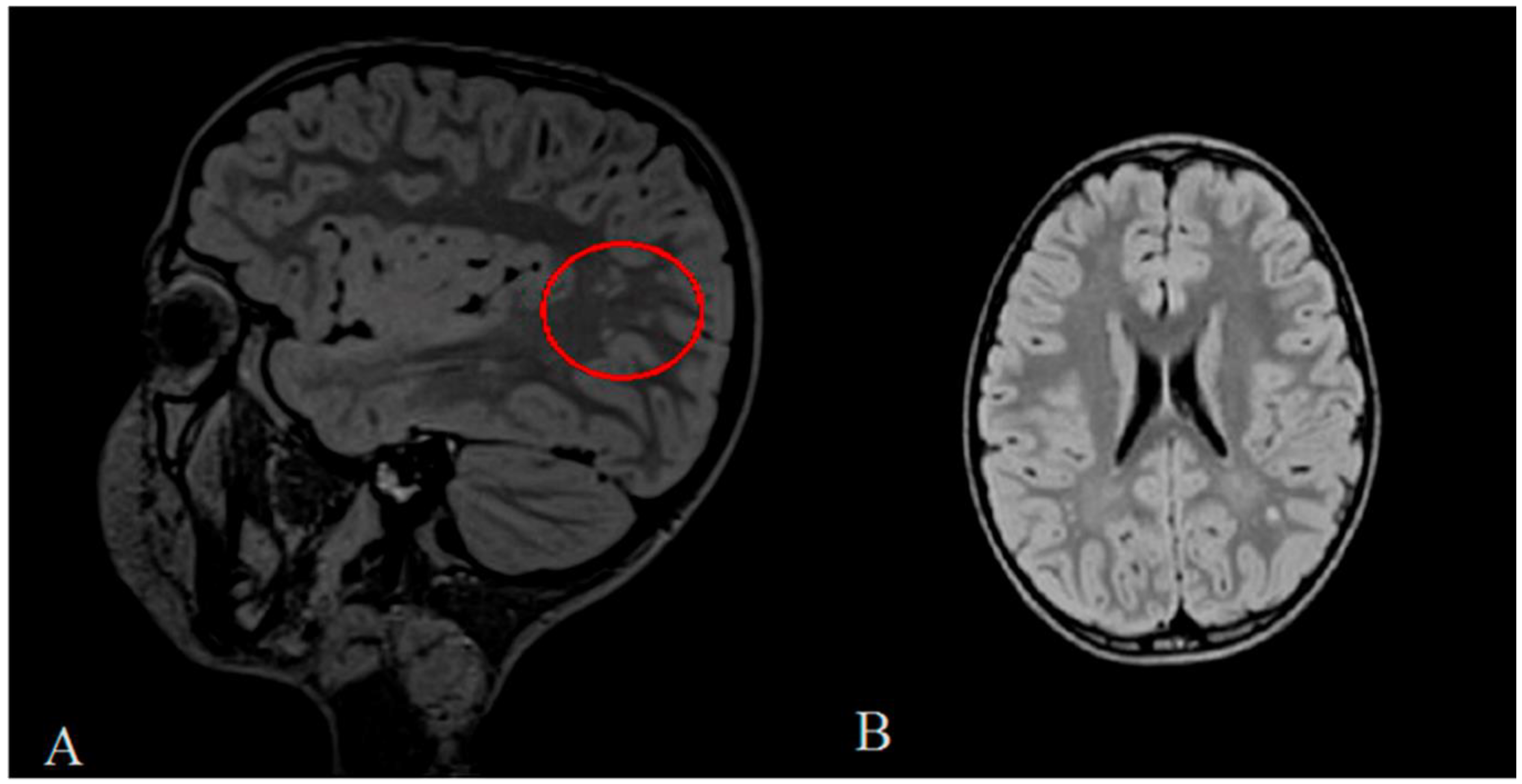{kind=link}
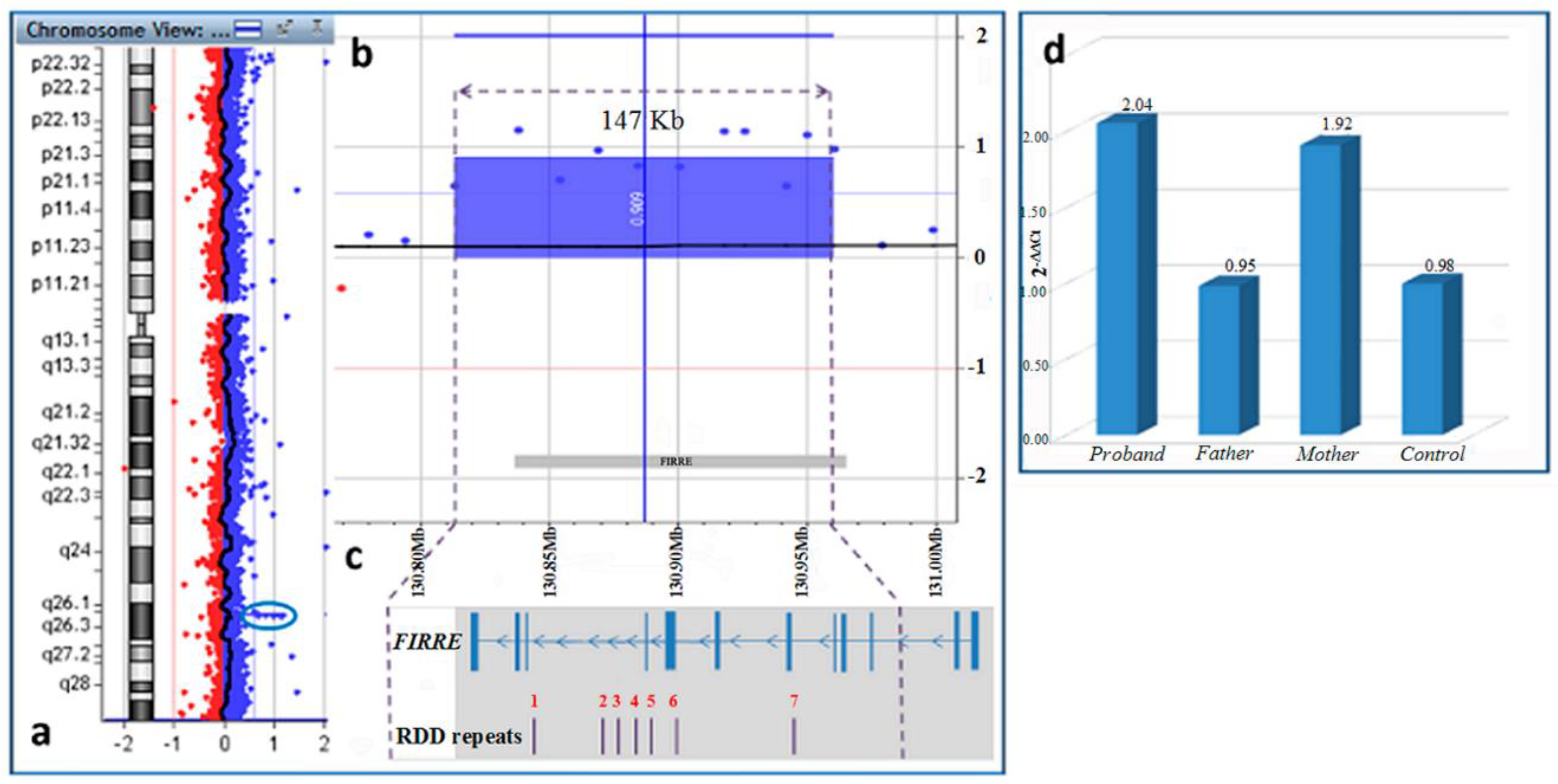{kind=link}
Abstract
Share and Cite
Miolo, G.; Bernardini, L.; Capalbo, A.; Favia, A.; Goldoni, M.; Pivetta, B.; Tessitori, G.; Corona, G. Identification of a De Novo Xq26.2 Microduplication Encompassing FIRRE Gene in a Child with Intellectual Disability. Diagnostics 2020, 10, 1009. https://doi.org/10.3390/diagnostics10121009
Miolo G, Bernardini L, Capalbo A, Favia A, Goldoni M, Pivetta B, Tessitori G, Corona G. Identification of a De Novo Xq26.2 Microduplication Encompassing FIRRE Gene in a Child with Intellectual Disability. Diagnostics. 2020; 10(12):1009. https://doi.org/10.3390/diagnostics10121009
Chicago/Turabian StyleMiolo, Gianmaria, Laura Bernardini, Anna Capalbo, Anna Favia, Marina Goldoni, Barbara Pivetta, Giovanni Tessitori, and Giuseppe Corona. 2020. "Identification of a De Novo Xq26.2 Microduplication Encompassing FIRRE Gene in a Child with Intellectual Disability" Diagnostics 10, no. 12: 1009. https://doi.org/10.3390/diagnostics10121009
APA StyleMiolo, G., Bernardini, L., Capalbo, A., Favia, A., Goldoni, M., Pivetta, B., Tessitori, G., & Corona, G. (2020). Identification of a De Novo Xq26.2 Microduplication Encompassing FIRRE Gene in a Child with Intellectual Disability. Diagnostics, 10(12), 1009. https://doi.org/10.3390/diagnostics10121009