Vaginal Microbiome-Based Bacterial Signatures for Predicting the Severity of Cervical Intraepithelial Neoplasia

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Sample Collection
2.2. HPV-Assay and HPV Genotyping
2.3. DNA Extraction and Ion Torrent Sequencing
2.4. Bioinformatics Analysis
2.5. Data and Statistical Analysis
3. Results
3.1. Participants’ Characteristics
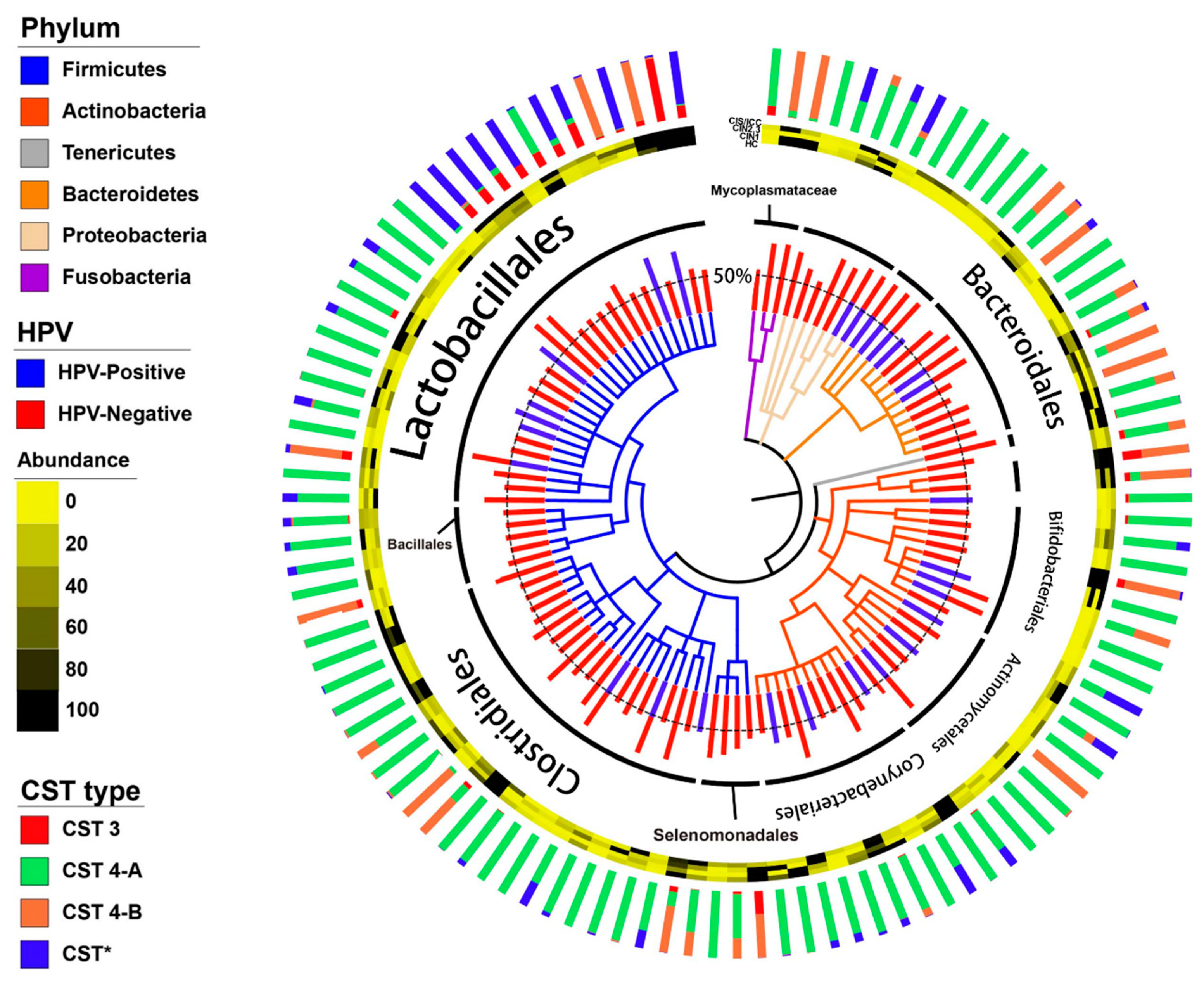
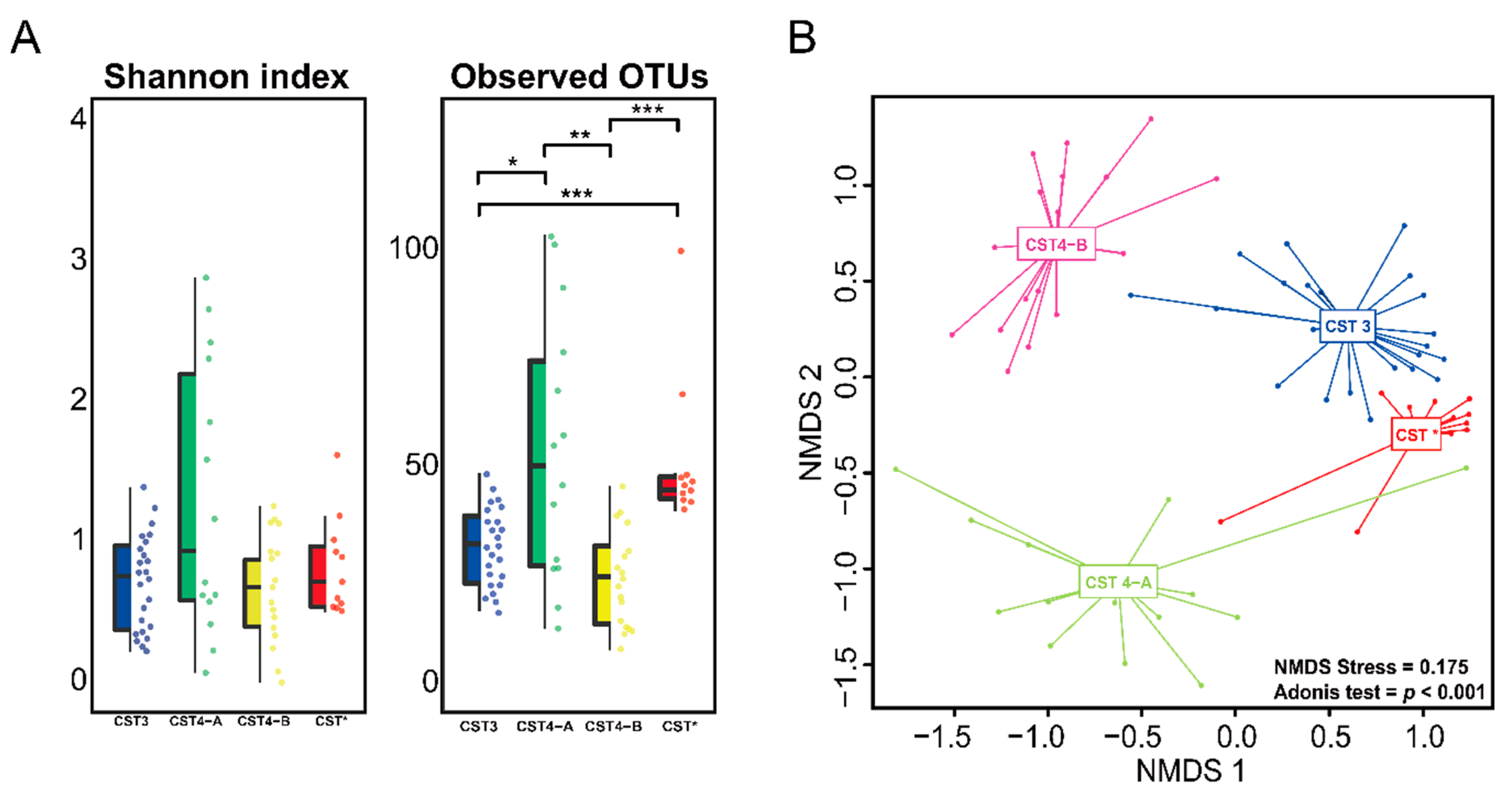
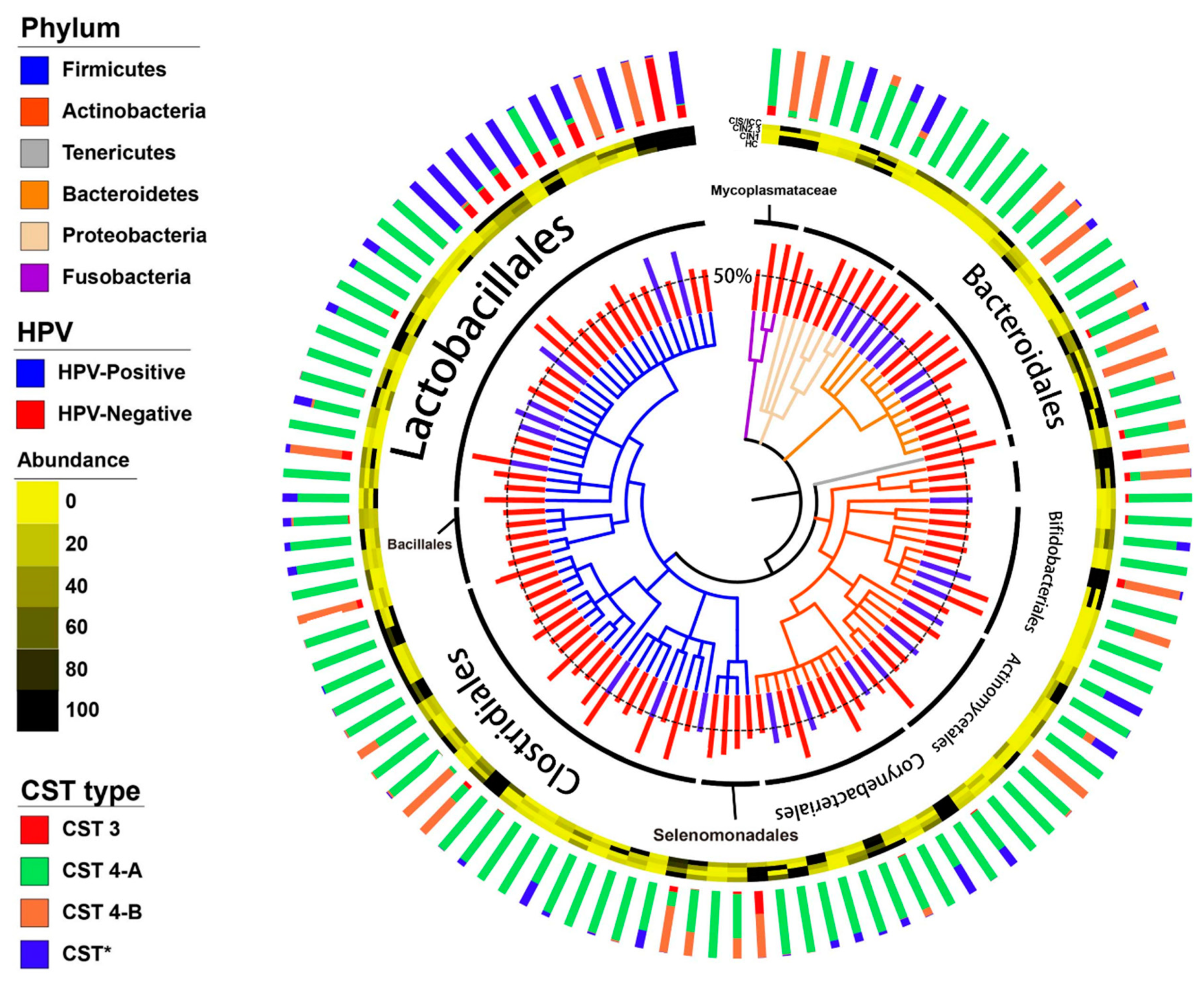
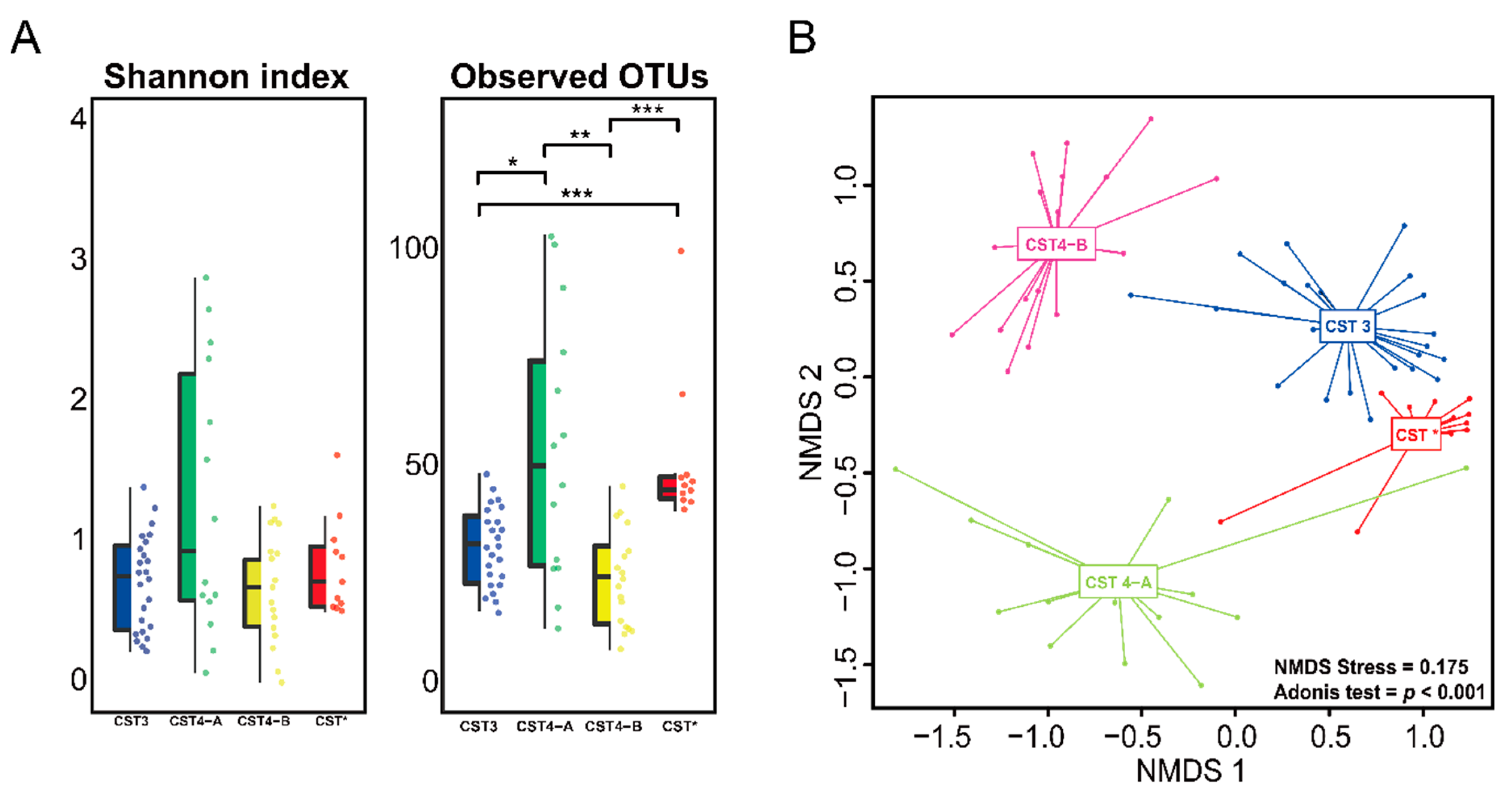
3.2. Vaginal Microbiome
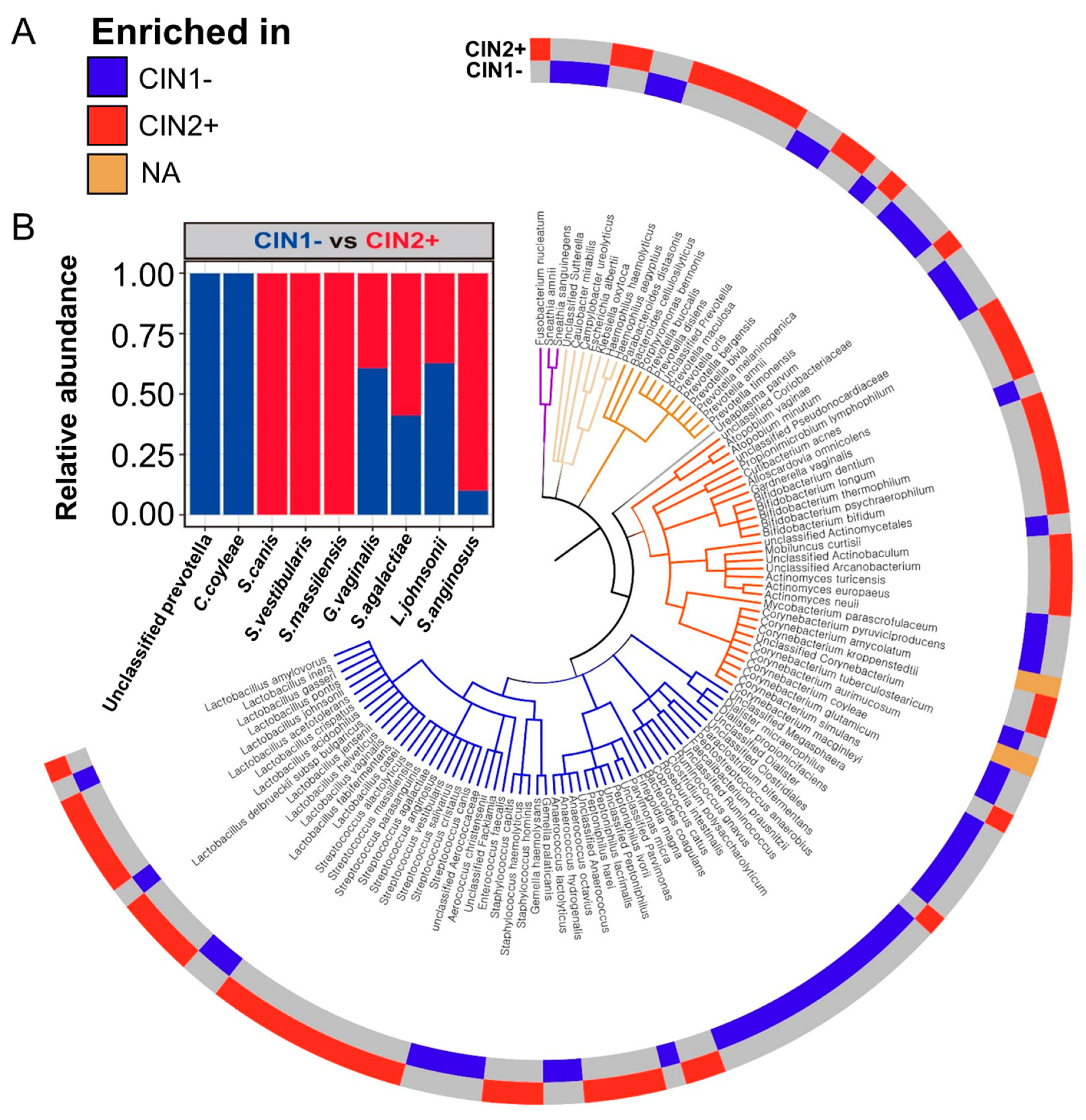
3.3. Differences between the Vaginal Microbiomes of CIN 1− and CIN 2+
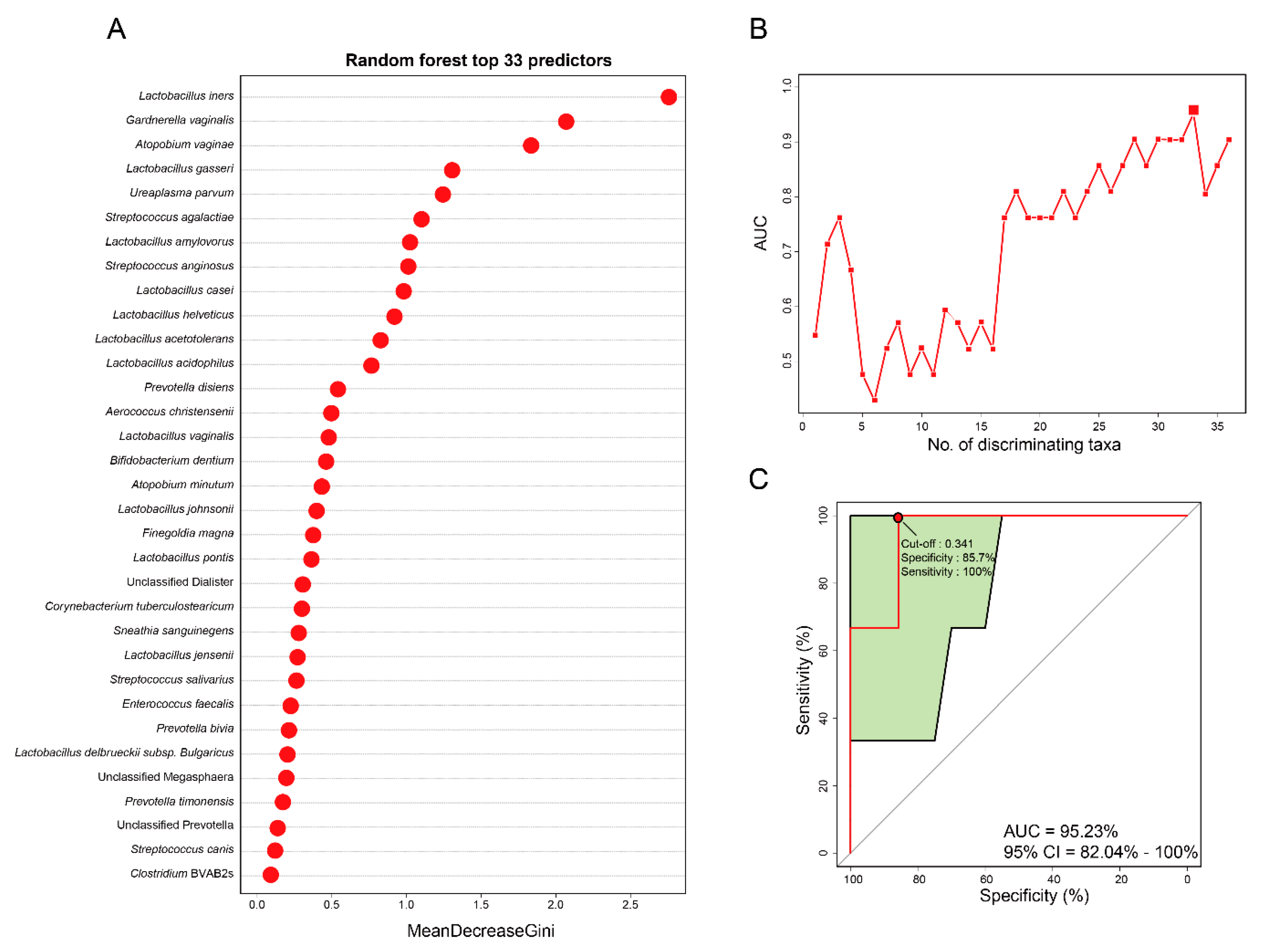
3.4. Vaginal Microbiome-Derived Signature Can Predict the Severity of CIN
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CIN | Cervical intraepithelial neoplasia |
| RF | Random Forest |
| ROC | Receiver operating characteristic |
| AUC | Area under curve |
| CST | Community state type |
| HPV | Human papillomavirus |
References
- Arbyn, M.; Castellsagué, X.; de Sanjosé, S.; Bruni, L.; Saraiya, M.; Bray, F.; Ferlay, J. Worldwide burden of cervical cancer in 2008. Ann. Oncol. 2011, 22, 2675–2686. [Google Scholar] [CrossRef] [PubMed]
- Okuma, K.; Yamashita, H.; Yokoyama, T.; Nakagawa, K.; Kawana, K. Undetected human papillomavirus DNA and uterine cervical carcinoma. Strahlentherapie und Onkologie 2016, 192, 55–62. [Google Scholar] [CrossRef]
- Ronco, G.; Dillner, J.; Elfström, K.M.; Tunesi, S.; Snijders, P.J.; Arbyn, M.; Kitchener, H.; Segnan, N.; Gilham, C.; Giorgi-Rossi, P. Efficacy of HPV-based screening for prevention of invasive cervical cancer: Follow-up of four European randomised controlled trials. Lancet 2014, 383, 524–532. [Google Scholar] [CrossRef]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25, 2–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westrich, J.A.; Warren, C.J.; Pyeon, D. Evasion of host immune defenses by human papillomavirus. Virus Res. 2017, 231, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Hellberg, D. Sex steroids and cervical cancer. Anticancer Res. 2012, 32, 3045–3054. [Google Scholar] [PubMed]
- Zhu, H.; Shen, Z.; Luo, H.; Zhang, W.; Zhu, X. Chlamydia trachomatis infection-associated risk of cervical cancer: A meta-analysis. Medicine 2016, 95, e3077. [Google Scholar] [CrossRef] [PubMed]
- Kyrgiou, M.; Mitra, A.; Moscicki, A.-B. Does the vaginal microbiota play a role in the development of cervical cancer? Transl. Res. 2017, 179, 168–182. [Google Scholar] [CrossRef] [Green Version]
- King, C.C.; Jamieson, D.J.; Wiener, J.; Cu-Uvin, S.; Klein, R.S.; Rompalo, A.M.; Shah, K.V.; Sobel, J.D. Bacterial vaginosis and the natural history of human papillomavirus. Infect. Dis. Obstetr. Gynecol. 2011, 2011, 319460. [Google Scholar]
- Piyathilake, C.J.; Ollberding, N.J.; Kumar, R.; Macaluso, M.; Alvarez, R.D.; Morrow, C.D. Cervical microbiota associated with higher grade cervical intraepithelial neoplasia in women infected with high-risk human papillomaviruses. Cancer Prev. Res. 2016, 9, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.; Kim, B.-S.; Seo, S.-S.; Kong, J.-S.; Lee, J.-K.; Park, S.-Y.; Hong, K.-M.; Kim, H.-K.; Kim, M. The association of uterine cervical microbiota with an increased risk for cervical intraepithelial neoplasia in Korea. Clin. Microbiol. Infect. 2015, 21, 674.e671–674.e679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Liu, Y.; Gao, W.; Pan, Y.; Gao, Y.; Shen, J.; Xiong, H. The direct and indirect association of cervical microbiota with the risk of cervical intraepithelial neoplasia. Cancer Med. 2018, 7, 2172–2179. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.; Foster, J.A. Machine learning techniques accurately classify microbial communities by bacterial vaginosis characteristics. PLoS ONE 2014, 9, e87830. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armour, C.R.; Nayfach, S.; Pollard, K.S.; Sharpton, T.J. A metagenomic meta-analysis reveals functional signatures of health and disease in the human gut microbiome. MSystems 2019, 4, e00332-18. [Google Scholar] [CrossRef] [Green Version]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. 2017, 25, 1054–1062.e1055. [Google Scholar] [CrossRef]
- De Seta, F.; Campisciano, G.; Zanotta, N.; Ricci, G.; Comar, M. The vaginal community state types microbiome-immune network as key factor for bacterial vaginosis and aerobic vaginitis. Front. Microbiol. 2019, 10, 2451. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Comeau, A.M.; Douglas, G.M.; Langille, M.G. Microbiome helper: A custom and streamlined workflow for microbiome research. mSystems 2017, 2, e00127-16. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Fettweis, J.M.; Serrano, M.G.; Sheth, N.U.; Mayer, C.M.; Glascock, A.L.; Brooks, J.P.; Jefferson, K.K.; Buck, G.A.; Consortium, V.M. Species-level classification of the vaginal microbiome. BMC Genom. 2012, 13, S17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennard, K.; Dabee, S.; Barnabas, S.L.; Havyarimana, E.; Blakney, A.; Jaumdally, S.Z.; Botha, G.; Mkhize, N.N.; Bekker, L.-G.; Lewis, D.A. Microbial composition predicts genital tract inflammation and persistent bacterial vaginosis in South African adolescent females. Infect. Immunity 2018, 86, e00410-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Foundation for Statistical Computing. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]
- Youngblut, N.D.; Reischer, G.H.; Walters, W.; Schuster, N.; Walzer, C.; Stalder, G.; Ley, R.E.; Farnleitner, A.H. Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108, 4680–4687. [Google Scholar] [CrossRef] [Green Version]
- Gajer, P.; Brotman, R.; Bai, G.; Sakamoto, J.; Schütte, U.; Zhong, X.; Koenig, S.; Fu, L.; Ma, Z.; Zhou, X. Temporal dynamics of the human vaginal microbiota. Sci. Transl. Med. 2012, 4, 132ra52. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Geng, R.; Liu, L.; Jin, X.; Yan, W.; Zhao, F.; Wang, S.; Guo, X.; Ghimire, G.; Wei, Y. Gut microbiota-based algorithms in the prediction of metachronous adenoma in colorectal cancer patients following surgery. Front. Microbiol. 2020, 11, 1106. [Google Scholar] [CrossRef]
- Lennard, K.; Dabee, S.; Barnabas, S.L.; Havyarimana, E.; Blakney, A.; Jaumdally, S.Z.; Botha, G.; Mkhize, N.N.; Bekker, L.-G.; Lewis, D.A. Vaginal microbiota varies by geographical location in South African women. arXiv 2019, arXiv:1905.11946. [Google Scholar]
- Schloss, P.D.; Jenior, M.L.; Koumpouras, C.C.; Westcott, S.L.; Highlander, S.K. Sequencing 16S rRNA gene fragments using the PacBio SMRT DNA sequencing system. PeerJ 2016, 4, e1869. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lu, J.; Lu, Y.; Cai, Q.; Liu, H.; Xu, C. Cervical microbiome is altered in cervical intraepithelial neoplasia after loop electrosurgical excision procedure in china. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- MacIntyre, D.A.; Chandiramani, M.; Lee, Y.S.; Kindinger, L.; Smith, A.; Angelopoulos, N.; Lehne, B.; Arulkumaran, S.; Brown, R.; Teoh, T.G. The vaginal microbiome during pregnancy and the postpartum period in a European population. Sci. Rep. 2015, 5, 8988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, K.; Oki, A.; Furuta, R.; Maeda, H.; Yasugi, T.; Takatsuka, N.; Mitsuhashi, A.; Fujii, T.; Hirai, Y.; Iwasaka, T. Predicting the progression of cervical precursor lesions by human papillomavirus genotyping: A prospective cohort study. Int. J. Cancer 2011, 128, 2898–2910. [Google Scholar] [CrossRef] [PubMed]
- Gil, N.F.; Martinez, R.C.; Gomes, B.C.; Nomizo, A.; De Martinis, E.C. Vaginal lactobacilli as potential probiotics against Candida spp. Braz. J. Microbiol. 2010, 41, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Marrazzo, J.M.; Thomas, K.K.; Fiedler, T.L.; Ringwood, K.; Fredricks, D.N. Relationship of specific vaginal bacteria and bacterial vaginosis treatment failure in women who have sex with women. Ann. Intern. Med. 2008, 149, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Verhelst, R.; Verstraelen, H.; Claeys, G.; Verschraegen, G.; Delanghe, J.; Van Simaey, L.; De Ganck, C.; Temmerman, M.; Vaneechoutte, M. Cloning of 16S rRNA genes amplified from normal and disturbed vaginal microflora suggests a strong association between Atopobium vaginae, Gardnerella vaginalis and bacterial vaginosis. BMC Microbiol. 2004, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Boris, S.; Suárez, J.E.; Vázquez, F.; Barbés, C. Adherence of human vaginal lactobacilli to vaginal epithelial cells and interaction with uropathogens. Infect. Immunity 1998, 66, 1985–1989. [Google Scholar] [CrossRef] [Green Version]
- Vásquez, A.; Jakobsson, T.; Ahrné, S.; Forsum, U.; Molin, G. Vaginal Lactobacillus flora of healthy Swedish women. J. Clin. Microbiol. 2002, 40, 2746–2749. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Da, M.; Zhang, W.; Qi, Q.; Zhang, C.; Han, S. Role of Lactobacillus in cervical cancer. Cancer Manag. Res. 2018, 10, 1219. [Google Scholar] [CrossRef] [Green Version]
- Kaambo, E.; Africa, C.; Chambuso, R.; Passmore, J.-A.S. Vaginal microbiomes associated with aerobic vaginitis and bacterial vaginosis. Front. Public Health 2018, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Zhang, L.; Zhang, Q.; Lv, T.; Chen, R.; Wang, L.; Huang, Z.; Hu, L.; Liao, Q. The Pathogenesis Of Streptococcus anginosus In Aerobic Vaginitis. Infect. Drug Resist. 2019, 12, 3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masood, U.; Sharma, A.; Lowe, D.; Khan, R.; Manocha, D. Colorectal cancer associated with streptococcus anginosus bacteremia and liver abscesses. Case Rep. Gastroenterol. 2016, 10, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Hui, M. Streptococcus anginosus bacteremia: Sutton’s law. J. Clin. Microbiol. 2005, 43, 6217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, H.; Ishizuka, T.; Muto, M.; Nezu, M.; Nakanishi, Y.; Inagaki, Y.; Watanabe, H.; Watanabe, H.; Terada, M. Presence of Streptococcus anginosus DNA in esophageal cancer, dysplasia of esophagus, and gastric cancer. Cancer Res. 1998, 58, 2991–2995. [Google Scholar] [PubMed]
- Sasaki, M.; Yamaura, C.; Ohara-Nemoto, Y.; Tajika, S.; Kodama, Y.; Ohya, T.; Harada, R.; Kimura, S. Streptococcus anginosus infection in oral cancer and its infection route. Oral Dis. 2005, 11, 151–156. [Google Scholar] [CrossRef]
- Kelly, H.; Benavente, Y.; Pavon, M.A.; De Sanjose, S.; Mayaud, P.; Lorincz, A.T. Performance of DNA methylation assays for detection of high-grade cervical intraepithelial neoplasia (CIN2+): A systematic review and meta-analysis. Br. J. Cancer 2019, 121, 954–965. [Google Scholar] [CrossRef]
- Uleberg, K.-E.; Øvestad, I.T.; Munk, A.C.; Brede, C.; Diermen, B.v.; Gudlaugsson, E.; Janssen, E.A.; Hjelle, A.; Baak, J. Prediction of spontaneous regression of cervical intraepithelial neoplasia lesions grades 2 and 3 by proteomic analysis. Int. J. Proteomics 2014, 2014, 129064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, D.; Foster, J.A. Machine learning classifiers provide insight into the relationship between microbial communities and bacterial vaginosis. BioData Min. 2015, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Perrotta, A.R.; Borrelli, G.M.; Martins, C.O.; Kallas, E.G.; Sanabani, S.S.; Griffith, L.G.; Alm, E.J.; Abrao, M.S. The Vaginal Microbiome as a Tool to Predict rASRM Stage of Disease in Endometriosis: A Pilot Study. Reproductive Sciences 2020, 27, 1064–1073. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Total (n = 66) | CIN 1− (n = 24) | CIN 2+ (n = 42) | p Value |
|---|---|---|---|---|
| Age (years) | 45.1 ± 11.7 | 49.2 ± 7.3 | 42.7 ± 13.2 | 0.0313 |
| Menopause (n, %) | 20 (30.3) | 9 (37.5) | 11 (26.2) | 0.3399 |
| Marriage (n, %) | 58 (87.9) | 22 (91.7) | 36 (85.7) | 0.4794 |
| Parity (n) | 1.7 ± 1.0 | 1.8 ± 0.9 | 1.6 ± 1.1 | 0.4169 |
| Smoker (n, %) | 4 (6.1) | 1 (4.8) | 3 (12.0) | 0.6139 |
| Contraceptive use (n, %) | 16 (24.6) | 7 (29.2) | 9 (22.0) | 0.5178 |
| Human papillomavirus (HPV) positive (n, %) | 48 (72.7) | 7 (29.2) | 41 (97.6) | <0.0001 |
| HPV16/18 positive (n, %) | 24 (36.4) | 2 (8.3) | 22 (52.4) | 0.0004 |
| CST Type | Total (n = 66) | CIN 1− (n = 24) | CIN 2+ (n = 42) | p Value |
|---|---|---|---|---|
| CST3 (n, %) | 24 (36.4) | 9 (37.5) | 15 (35.7) | 0.8855 |
| CST 4-A (n, %) | 14 (21.2) | 4 (16.7) | 10 (23.8) | 0.4980 |
| CST 4-B (n, %) | 17 (25.8) | 8 (33.3) | 9 (21.4) | 0.2911 |
| CST* (n, %) | 11 (16.7) | 3 (12.5) | 8 (19.0) | 0.4956 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.H.; Kang, G.-U.; Jeon, S.Y.; Tagele, S.B.; Pham, H.Q.; Kim, M.-S.; Ahmad, S.; Jung, D.-R.; Park, Y.-J.; Han, H.S.; et al. Vaginal Microbiome-Based Bacterial Signatures for Predicting the Severity of Cervical Intraepithelial Neoplasia. Diagnostics 2020, 10, 1013. https://doi.org/10.3390/diagnostics10121013
Lee YH, Kang G-U, Jeon SY, Tagele SB, Pham HQ, Kim M-S, Ahmad S, Jung D-R, Park Y-J, Han HS, et al. Vaginal Microbiome-Based Bacterial Signatures for Predicting the Severity of Cervical Intraepithelial Neoplasia. Diagnostics. 2020; 10(12):1013. https://doi.org/10.3390/diagnostics10121013
Chicago/Turabian StyleLee, Yoon Hee, Gi-Ung Kang, Se Young Jeon, Setu Bazie Tagele, Huy Quang Pham, Min-Sueng Kim, Sajjad Ahmad, Da-Ryung Jung, Yeong-Jun Park, Hyung Soo Han, and et al. 2020. "Vaginal Microbiome-Based Bacterial Signatures for Predicting the Severity of Cervical Intraepithelial Neoplasia" Diagnostics 10, no. 12: 1013. https://doi.org/10.3390/diagnostics10121013