Assessment of Peripheral Nervous System Alterations in Patients with the Fabry Related GLA-Variant p.A143T

 , , ,
, , ,
Abstract
:1. Introduction
2. Patients and Methods
2.1. Clinical and Demographic Patient Data
2.2. Imaging Protocol
- A three-dimensional (3D) T2-weighted (T2w) sampling perfection with application-optimized contrasts using different flip-angle evolution (SPACE) short-tau-inversion-recovery (STIR) sequence of the lumbosacral plexus: repetition time/echo time 3.000/208 ms, inversion time 210 ms, effective echo time 68 ms, matrix size 320 × 320 × 104, field of view 305 × 305 mm2, slice thickness 1.0 mm, no gap, voxel size 1.0 × 1.0 × 1.0 mm3, and acquisition time 8:35 min, imaging the lumbosacral spine from the second lumbar vertebra to the coccyx.
- A T1-weighted, dynamic contrast enhanced (DCE) volumetric-interpolated breath-hold examination (VIBE) sequence (repetition time/echo time 3.3/1.11 ms, flip angle 158, 24 slices, resolution 1.3 × 1.3 × 3.0 mm3) covering the pelvis from the upper plate of the fifth lumbar vertebra to the second sacral vertebra. Contrast agent (Dotarem, Guerbert, France) was administered intravenously at a concentration of 0.1 mmol/kg with a flow rate of 3.5 mL/s by automated injection. A total of 24 frames were recorded with a rate of 7.46 s per frame.
2.3. Imaging Analysis
2.4. Skin Biopsies
2.5. Normative Reference Values and Statistical Analysis
2.6. Statistical Analysis
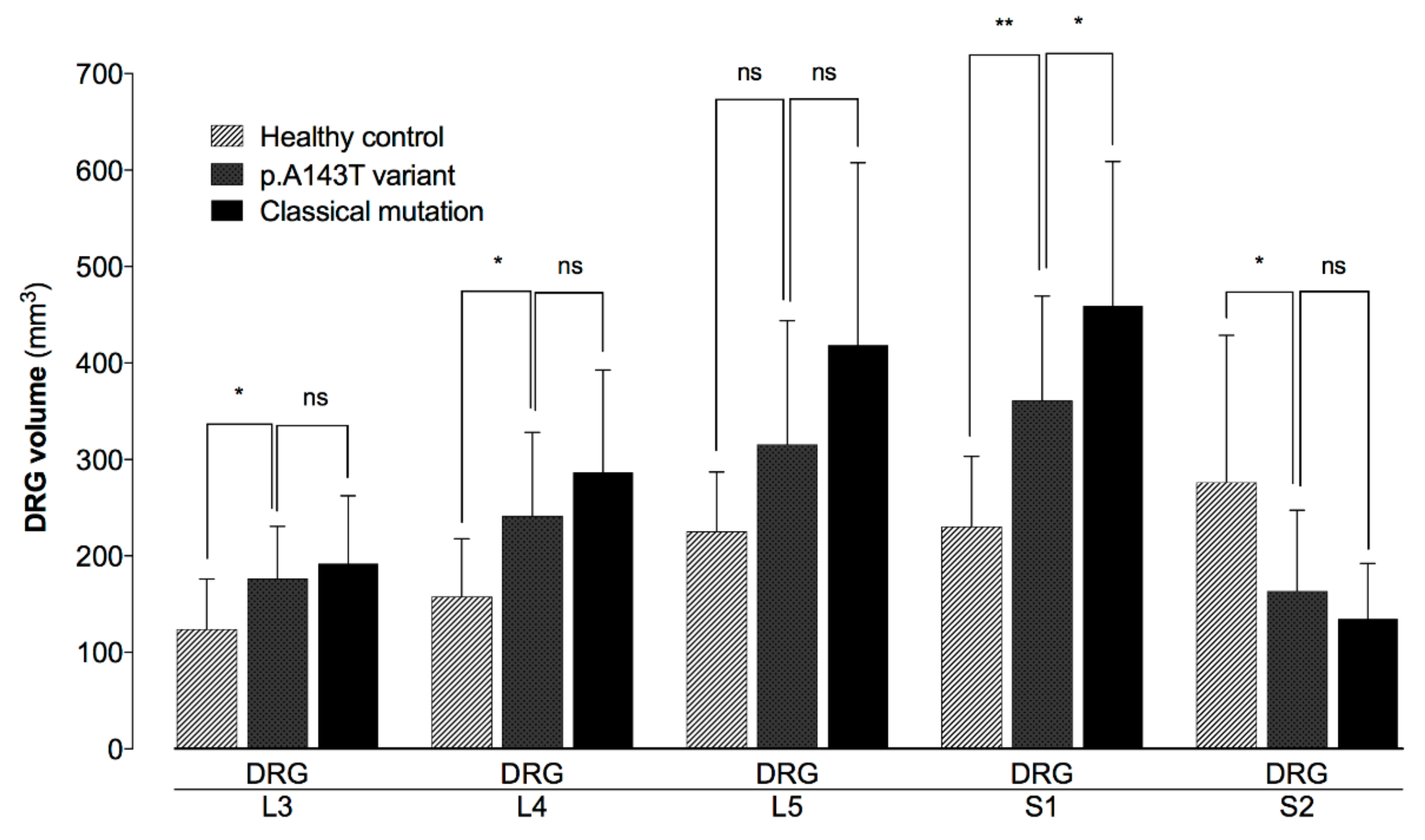
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Üçeyler, N.; He, L.; Schönfeld, D.; Kahn, A.-K.; Reiners, K.; Hilz, M.J.; Breunig, F.; Sommer, C.; Sommer, C. Small fibers in Fabry disease: Baseline and follow-up data under enzyme replacement therapy. J. Peripher. Nerv. Syst. 2011, 16, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Liguori, R.; Di Stasi, V.; Bugiardini, E.; Mignani, R.; Burlina, A.; Borsini, W.; Baruzzi, A.; Montagna, P.; Donadio, V. Small fiber neuropathy in female patients with fabry disease. Muscle Nerve 2010, 41, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Ries, M. Fabry Disease: A Disorder of Childhood Onset. Pediatr. Neurol. 2016, 64, 10–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biegstraaten, M.; Hollak, C.E.; Bakkers, M.; Faber, C.G.; Aerts, J.M.; van Schaik, I.N. Small fiber neuropathy in Fabry disease. Mol. Genet. Metab. 2012, 106, 135–141. [Google Scholar] [CrossRef]
- Onishi, A.; Dyck, P.J. Loss of small peripheral sensory neurons in Fabry disease. Histologic and morphometric evaluation of cutaneous nerves, spinal ganglia, and posterior columns. Arch. Neurol. 1974, 31, 120–127. [Google Scholar] [CrossRef]
- Godel, T.; Bäumer, P.; Pham, M.; Köhn, A.; Muschol, N.; Kronlage, M.; Kollmer, J.; Heiland, S.; Bendszus, M.; Mautner, V.-F. Human dorsal root ganglion in vivo morphometry and perfusion in Fabry painful neuropathy. Neurology 2017, 89, 1274–1282. [Google Scholar] [CrossRef]
- Godel, T.; Köhn, A.; Muschol, N.; Kronlage, M.; Schwarz, D.; Kollmer, J.; Heiland, S.; Bendszus, M.; Mautner, V.-F.; Bäumer, P. Dorsal root ganglia in vivo morphometry and perfusion in female patients with Fabry disease. J. Neurol. 2018, 265, 2723–2729. [Google Scholar] [CrossRef]
- Zarate, Y.A.; Hopkin, R.J. Fabry’s disease. Lancet 2008, 372, 1427–1435. [Google Scholar] [CrossRef]
- Van Der Tol, L.; Smid, B.E.; Poorthuis, B.J.H.M.; Biegstraaten, M.; Deprez, R.H.L.; Linthorst, G.E.; Hollak, C.E.M. A systematic review on screening for Fabry disease: Prevalence of individuals with genetic variants of unknown significance. J. Med. Genet. 2014, 51, 1–9. [Google Scholar] [CrossRef]
- Smid, B.E.; Hollak, C.E.M.; Poorthuis, B.J.; Weerman, M.A.; Florquin, S.; Kok, W.E.M.; Deprez, R.H.L.; Timmermans, J.; Linthorst, G.E. Diagnostic dilemmas in Fabry disease: A case series study on GLA mutations of unknown clinical significance. Clin. Genet. 2015, 88, 161–166. [Google Scholar] [CrossRef]
- Du Moulin, M.; Koehn, A.; Golsari, A.; Dulz, S.; Atiskova, Y.; Patten, M.; Münch, J.; Avanesov, M.; Ullrich, K.; Muschol, N. The mutation p.D313Y is associated with organ manifestation in Fabry disease. Clin. Genet. 2017, 92, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Du Moulin, M.; Muschol, N. p.D313Y is more than just a polymorphism in Fabry disease. Clin. Genet. 2018, 93, 1258. [Google Scholar] [CrossRef] [PubMed]
- Niemann, M.; Rolfs, A.; Giese, A.; Mascher, H.; Breunig, F.; Ertl, G.; Wanner, C.; Weidemann, F. Lyso-Gb3 Indicates that the Alpha-Galactosidase A Mutation D313Y is not Clinically Relevant for Fabry Disease. JIMD Rep. 2013, 7, 99–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oder, D.; Üçeyler, N.; Liu, D.; Hu, K.; Petritsch, B.; Sommer, C.; Ertl, G.; Wanner, C.; Nordbeck, P. Organ manifestations and long-term outcome of Fabry disease in patients with the GLA haplotype D313Y. BMJ Open 2016, 6, e010422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oder, D.; Wanner, C.; Nordbeck, P. The D313Y genotype-Pathogenic mutation or polymorphism? Clin. Genet. 2018, 93, 1257. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Shabbeer, J.; Benson, S.D.; Maire, I.; Burnett, R.M.; Desnick, R.J. Fabry disease: Characterization of alpha-galactosidase A double mutations and the D313Y plasma enzyme pseudodeficiency allele. Hum. Mutat. 2003, 22, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Godel, T.; Bäumer, P.; Stumpfe, K.; Muschol, N.; Kronlage, M.; Brunnée, M.; Kollmer, J.; Heiland, S.; Bendszus, M.; Mautner, V.-F. Dorsal root ganglia volume is increased in patients with the Fabry-related GLA variant p.D313Y. J. Neurol. 2019, 266, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Whybra, C.; Kampmann, C.; Krummenauer, F.; Ries, M.; Mengel, E.; Miebach, E.; Baehner, F.; Kim, K.; Bajbouj, M.; Schwarting, A.; et al. The Mainz Severity Score Index: A new instrument for quantifying the Anderson-Fabry disease phenotype, and the response of patients to enzyme replacement therapy. Clin. Genet. 2004, 65, 299–307. [Google Scholar] [CrossRef]
- Godel, T.; Pham, M.; Heiland, S.; Bendszus, M.; Baumer, P. Human dorsal-root-ganglion perfusion measured in-vivo by MRI. Neuroimage 2016, 141, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Lauria, G.; Bakkers, M.; Schmitz, C.; Lombardi, R.; Penza, P.; Devigili, G.; Smith, A.G.; Hsieh, S.-T.; Mellgren, S.I.; Umapathi, T.; et al. Intraepidermal nerve fiber density at the distal leg: A worldwide normative reference study. J. Peripher. Nerv. Syst. 2010, 15, 202–207. [Google Scholar] [CrossRef]
- Farschtschi, S.; Kluwe, L.; Schön, G.; Friedrich, R.E.; Matschke, J.; Glatzel, M.; Weis, J.; Hagel, C.; Mautner, V. Distinctive low epidermal nerve fiber density in schwannomatosis patients provides a major parameter for diagnosis and differential diagnosis. Brain Pathol. 2020, 30, 386–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauth, L.; Kerstens, J.; Yperzeele, L.; Eyskens, F.; Parizel, P.M.; Willekens, B. Galactosidase Alpha p.A143T Variant Fabry Disease May Result in a Phenotype With Multifocal Microvascular Cerebral Involvement at a Young Age. Front. Neurol. 2018, 9, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenders, M.; Weidemann, F.; Kurschat, C.; Canaan-Kühl, S.; Duning, T.; Stypmann, J.; Schmitz, B.; Reiermann, S.; Krämer, J.; Blaschke, D.; et al. Alpha-Galactosidase A p.A143T, a non-Fabry disease-causing variant. Orphanet. J. Rare Dis. 2016, 11, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terryn, W.; Vanholder, R.; Hemelsoet, D.; Leroy, B.P.; Van Biesen, W.; De Schoenmakere, G.; Wuyts, B.; Claes, K.B.M.; De Backer, J.; De Paëpe, G.; et al. Questioning the Pathogenic Role of the GLA p.Ala143Thr “Mutation” in Fabry Disease: Implications for Screening Studies and ERT. JIMD Rep. 2013, 8, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Brabander, I.; Yperzeele, L.; Groote, C.C.-D.; Brouns, R.; Baker, R.; Belachew, S.; Delbecq, J.; De Keulenaer, G.W.; Dethy, S.; Eyskens, F.; et al. Phenotypical characterization of alpha-galactosidase A gene mutations identified in a large Fabry disease screening program in stroke in the young. Clin. Neurol. Neurosurg. 2013, 115, 1088–1093. [Google Scholar] [CrossRef]
- Noel, L.H.; Laurent, B.; Grunfeld, J.P. Renal biopsies in Fabry disease: A multicenter French study. Nephrol. Ther. 2012, 8, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Rolfs, A.; Fazekas, F.; Grittner, U.; Dichgans, M.; Martus, P.; Holzhausen, M.; Böttcher, T.; Heuschmann, P.U.; Tatlisumak, T.; Tanislav, C.; et al. Acute cerebrovascular disease in the young: The Stroke in Young Fabry Patients study. Stroke 2013, 44, 340–349. [Google Scholar] [CrossRef] [Green Version]
- Brouns, R.; Thijs, V.; Eyskens, F.; De Deyn, P.P. Belgian Fabry study: Prevalence of Fabry disease in a cohort of 1000 young patients with cerebrovascular disease. Stroke 2010, 41, 863–868. [Google Scholar] [CrossRef] [Green Version]
- Desnick, R.J.; Brady, R.O. Fabry disease in childhood. J. Pediatr. 2004, 144, S20–S26. [Google Scholar] [CrossRef]
- Dubuc, V.; Moore, D.F.; Gioia, L.C.; Saposnik, G.; Selchen, D.; Lanthier, S. Prevalence of Fabry disease in young patients with cryptogenic ischemic stroke. J. Stroke Cereb. Dis. 2013, 22, 1288–1292. [Google Scholar] [CrossRef]
- Lenders, M.; Duning, T.; Schelleckes, M.; Schmitz, B.; Ständer, S.; Rolfs, A.; Brand, S.-M.; Brand, E. Multifocal white matter lesions associated with the D313Y mutation of the alpha-galactosidase A gene. PLoS ONE 2013, 8, e55565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tøndel, C.; Bostad, L.; Larsen, K.K.; Hirth, A.; Vikse, B.E.; Houge, G.; Svarstad, E. Agalsidase benefits renal histology in young patients with Fabry disease. J. Am. Soc. Nephrol. 2013, 24, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Patient | Age (at Diagnosis) | Neurological Symptoms/Clinical Features | MSSI | Lyso-Gb3 Level (ng/mL) a | IENFD | <5% Norm/Decade | <5% Norm/Age | IENFD Change b | DRG Volume (L3-S2) c | DRG Permeability (L5-S1) c |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 71 (61) | Hypohidrosis, angiokeratosis, tinnitus, cardiomyopathy (<15 mm), valve insufficiency, hypertension, proteinuria | 18 | 1.3 | 1.33 | 2.0 | 2.3 | −43% | +29% | −63% |
| 2 | 59 (56) | Myocardial infarction, occasional acroparesthesia, migraine | 3 | 1.1 | 2.8 | 4.1 | 3.6 | −23% | +54% | −35% |
| 3 | 69 (62) | Valve insufficiency, hypertonia, cerebral white matter lesions, tinnitus | 3 | 1.1 | n.a | n.a. | n.a. | n.a. | +55% | −77% |
| 4 | 37 (34) | Edema, hyperhidrosis, vertigo | 4 | 0.8 | 1.6 | 6.1 | 6.6 | −6% | +17% | −12% |
| 5 | 56 (52) | Myocarditis, valve insufficiency, renal insufficiency (stadium II), pruritus, acroparesthesia, depression, hypertonia, NYHA I | 5 | 1.1 | 7.5 | 3.2 | 4.0 | +88% | +43% | −49% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godel, T.; von Cossel, K.; Friedrich, R.E.; Glatzel, M.; Canaan-Kühl, S.; Duning, T.; Kronlage, M.; Heiland, S.; Bendszus, M.; Muschol, N.; et al. Assessment of Peripheral Nervous System Alterations in Patients with the Fabry Related GLA-Variant p.A143T. Diagnostics 2020, 10, 1027. https://doi.org/10.3390/diagnostics10121027
Godel T, von Cossel K, Friedrich RE, Glatzel M, Canaan-Kühl S, Duning T, Kronlage M, Heiland S, Bendszus M, Muschol N, et al. Assessment of Peripheral Nervous System Alterations in Patients with the Fabry Related GLA-Variant p.A143T. Diagnostics. 2020; 10(12):1027. https://doi.org/10.3390/diagnostics10121027
Chicago/Turabian StyleGodel, Tim, Katharina von Cossel, Reinhard E. Friedrich, Markus Glatzel, Sima Canaan-Kühl, Thomas Duning, Moritz Kronlage, Sabine Heiland, Martin Bendszus, Nicole Muschol, and et al. 2020. "Assessment of Peripheral Nervous System Alterations in Patients with the Fabry Related GLA-Variant p.A143T" Diagnostics 10, no. 12: 1027. https://doi.org/10.3390/diagnostics10121027
APA StyleGodel, T., von Cossel, K., Friedrich, R. E., Glatzel, M., Canaan-Kühl, S., Duning, T., Kronlage, M., Heiland, S., Bendszus, M., Muschol, N., & Mautner, V.-F. (2020). Assessment of Peripheral Nervous System Alterations in Patients with the Fabry Related GLA-Variant p.A143T. Diagnostics, 10(12), 1027. https://doi.org/10.3390/diagnostics10121027