1. Introduction
Identifying patients who satisfy predefined criteria with a particular condition from electronic health records (EHRs) can be used in numerous research studies, such as clinical trial recruitments, survival analysis, or outcome predictions, etc. [
1,
2]. Importantly, EHRs-based cohorts allows studies on rare conditions or genomic studies to have the potential of scaling to large populations with a sufficiently powered analysis. Larger cohorts are a key contribution to novel discoveries in genetic medicine [
3]. But the central question is: how can we define or identify the criteria that accurately describe the condition of interest within the EHRs?
Currently, the patient records held in primary care contain the most comprehensive medical history for a population, including information on symptoms, diagnoses, referrals, treatment, and changes in an individual’s health over time, representing an incredible resource for cohort identification research. However, there are many challenges in using primary care health records to identify cohorts of patients with a particular condition. First, the quality of the records can vary [
1]; in particular, the diagnostic codes in primary care may be inaccurate. For example, using medical records in Medicare database (USA), the positive predictive values (PPVs) were 55.7% for at least two claims coded for RA, 65.5% for at least three claims for RA, and 66.7% for at least two rheumatology claims for RA [
4]. From the Clinical Practice Research Datalink, a routinely collected primary care database (UK), the accuracy of identifying someone as definite RA by a single RA Read code was 78.7% [
5]. A recent study in the Netherlands suggests that more than half of the children with asthma diagnosed and treated by their general practitioners (GPs) may not actually have the condition [
6], leading to unnecessary treatment, disease burden, and impact on quality of life and putting them at risk from the side effects of their medication. These findings suggest that some conditions may appear under-reported in primary care and others over-reported. In contrast, patient records held in secondary care contain more robust diagnostic data than primary data, but they are sparse, often contain only severe active disease, and are not easily available. Therefore, an ideal and natural scheme is to link the primary care records with the secondary care ones at patient level, in which the secondary care records act as a gold standard to enhance the accuracy of the codes selected from primary care for a population.
However, distinguishing patients with a particular condition from their records can be extremely time consuming and resource costly. This is because the criteria that characterise a condition, particularly a chronic condition, are buried within complex hierarchical terminology structures across multiple data points in the records of a patient, such as the Read codes [
7] used in General Practice in the United Kingdom or the SNOMED CT (Systematized Nomenclature of Medicine Clinical Terms) [
8], which merge the SNOMED Reference Terminology and Read codes and is promoted in many countries. These hierarchical terminology structures with extensive overlap of classes lead to a huge number of codes (terms) describing the condition of a patient across multiple data points. In addition, prevalence of a certain condition within a population is comparatively very small. This results in a both highly dimensional (large number of medical codes/characteristics) and imbalanced (few positive patients) EHR data space. All these issues present a big methodological challenge in identifying cohorts from EHRs, making the classical statistical modelling techniques no longer feasible due to the curse of dimensionality [
9,
10]. Thus, to distinguish patients with a condition in primary care, it becomes crucial to identify the most predictive code patterns buried in their EHRs rather than consider the whole set of overlapping codes. To achieve this, researchers have turned to experts’ review of records to manually select the most relevant codes in safeguarding the accuracy of patient identification [
11,
12]. However, this methodology is expensive, inefficient, and highly subjective and depends on the local healthcare system and the level of clinicians’ knowledge.
Different from the expert-knowledge-driven methods, AI- and machine-learning-based, data-driven methods offer a promising application in improving diagnostic performance [
13,
14]. In this paper, we propose to use a transparent machine-learning framework to mine EHRs for automatic phenotype identification by parsimonious set of useful clinical signals. The proposed methodology is grounded in the theory and methods of text mining [
15]. Each patient is treated as a document consisting of coded terms and other variables (words); prediction of health outcomes for each patient is thus treated as a problem of text categorisation. One efficient way of representing words in vector space is to embed words in a latent factor vector space [
16,
17]. Although latent representation of words may significantly improve performance of word classification, it is difficult or impossible to know the logic involved about relationships between the raw words and the outcome. As the General Data Protection Regulation (GDPR) [
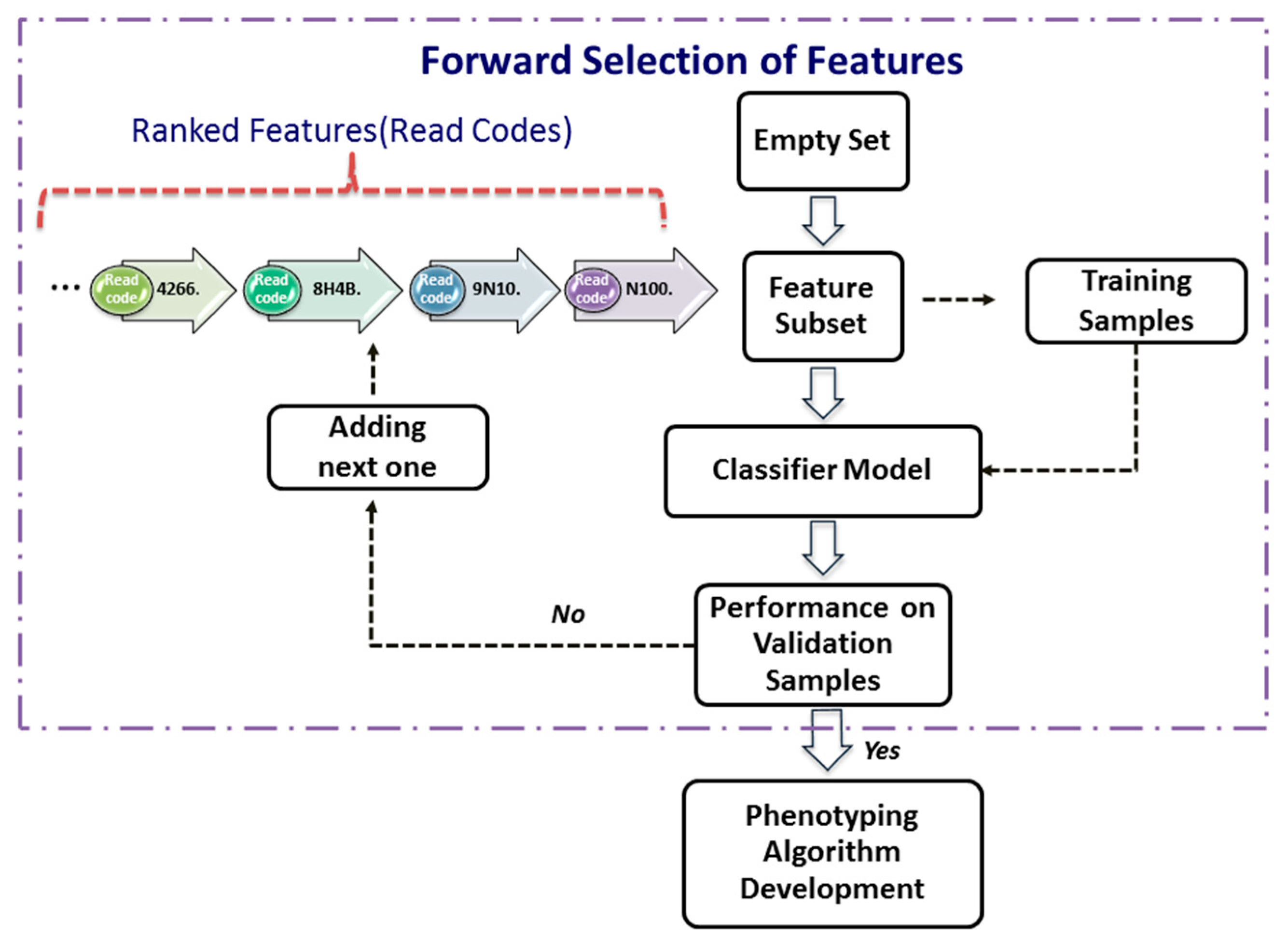
18] takes effect, data subjects have the right to have meaningful information about the logic involved and the envisaged consequences of such automated decision making for the data subject. It is of pressing importance to develop transparent data-driven models that have explanatory power. This is essential in order for machine-learning-based tools to be sufficiently trusted by medical community in modifying protocols for diagnosis and treatment. Therefore, in this paper, we directly work on raw words rather than their latent representations by integrating data-mining techniques to offer a transparent disease-phenotyping framework that generates decision rules with clear inferencing logic involved about relationships between clinical concepts and health outcome. To reduce the large number of raw words to the minimum combination of features able to phenotype a condition and with each feature representing a single EHR within a patient’s medical history, feature selection (FS) is used to score and rank the clinical codes (terms) so that only the most relevant codes are kept.
Clinical data are commonly imbalanced with a small number of positive patients within a large patient cohort. However, most health statistical analysis studies did not consider the problem of imbalance of clinical data; these studies inherently and inexplicitly put more emphasis on learning data observations with more occurrences. Instead, our proposed methodology suggested to use cost-sensitive machine-learning techniques to tackle this challenge. In this way, this framework enables the patterns examined in the primary care records to be able to identify the presence or absence of the condition as accurately as possible, at best with a secondary care diagnosis.
In this study, two arthritis conditions are used as exemplar of this framework: rheumatoid arthritis (RA) and ankylosing spondylitis (AS). Moreover, we compared this data-driven framework with the standard clinical expert-knowledge-driven algorithms for both RA and AS.
In summary, the contribution of this study includes: (1) the proposed methodology focused on generating transparent knowledge from data; (2) from a large number of factors, a parsimonious set of influential clinical signals with the fewest number of variables were identified while system predictive performance was maintained; (3) the proposed framework worked efficiently with a large and very high dimensional dataset, which allows the predictive models to avoid the challenges of dimensionality; and (4) the proposed framework suggested to use cost-sensitive machine-learning techniques to address the common but ignored challenge of the imbalance of clinical diagnostic data.
4. Discussion
By selecting the most relevant features from a huge number of primary care codes, this framework can bring multiple benefits to health informatics research. Firstly, dimensionality reduction makes many machine-learning classifiers feasible for large data problems due to avoiding the curse of dimensionality. Secondly, using only the most relevant codes (terms) can significantly speed up the training and testing process of the classifiers, which makes the development of a phenotyping algorithm effective. Thirdly, using those relevant codes can remove the noises and reduce biases that distort the true relationship between the risk factors and the outcome. Fourthly, using only the most relevant features can avoid the over-fitting dilemma, a common challenge in the development of data-driven approaches, which would help improve the generalisation performance of the classifiers applied to un-seen patients’ data. Importantly, with only the most relevant features, the developed classifiers themselves and their prediction results would achieve better transparency and interpretation in clinical decision supports [
15,
33,
34,
35,
36].
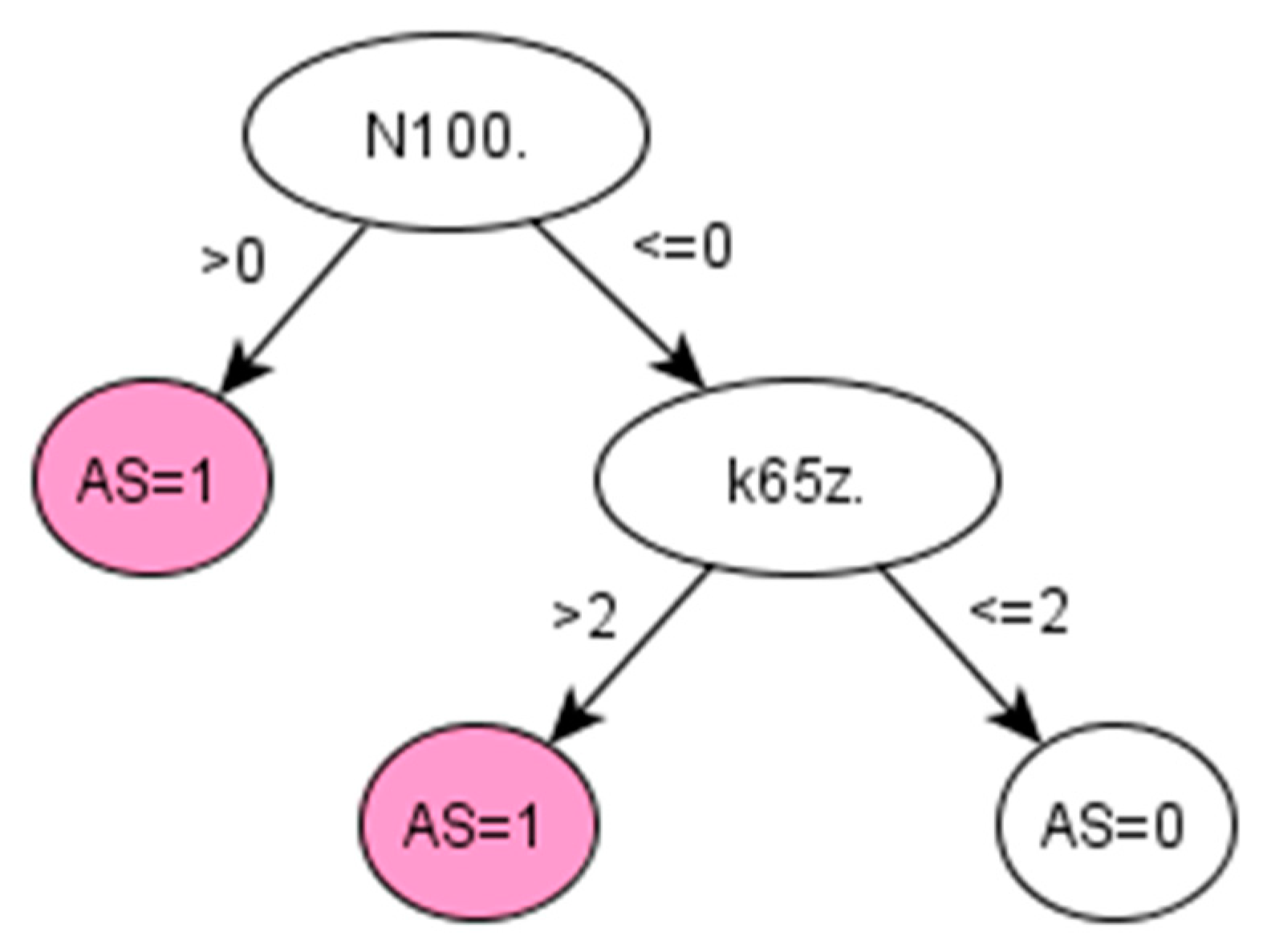
On the other hand, chronic diseases, like RA and AS, often take years to develop and be properly diagnosed. For instance, AS can take more than 10 years to be diagnosed, and when a formal diagnosis is made, the disease is in a very advanced state [
37]. The proposed method is used in retrospective to extract knowledge from the medical history of already diagnosed AS patients and therefore discover early symptoms of AS. In this way, this framework provides an efficient option for identifying cohort of patients with a particular condition, especially where knowledge-driven approaches, such as QOF-based rulesets, do not yet exist. In addition, by learning directly from the data, the data-driven algorithm generated by the proposed framework might potentially reduce bias and/or errors derived from applying other algorithms implemented using a specific population with different characteristics.
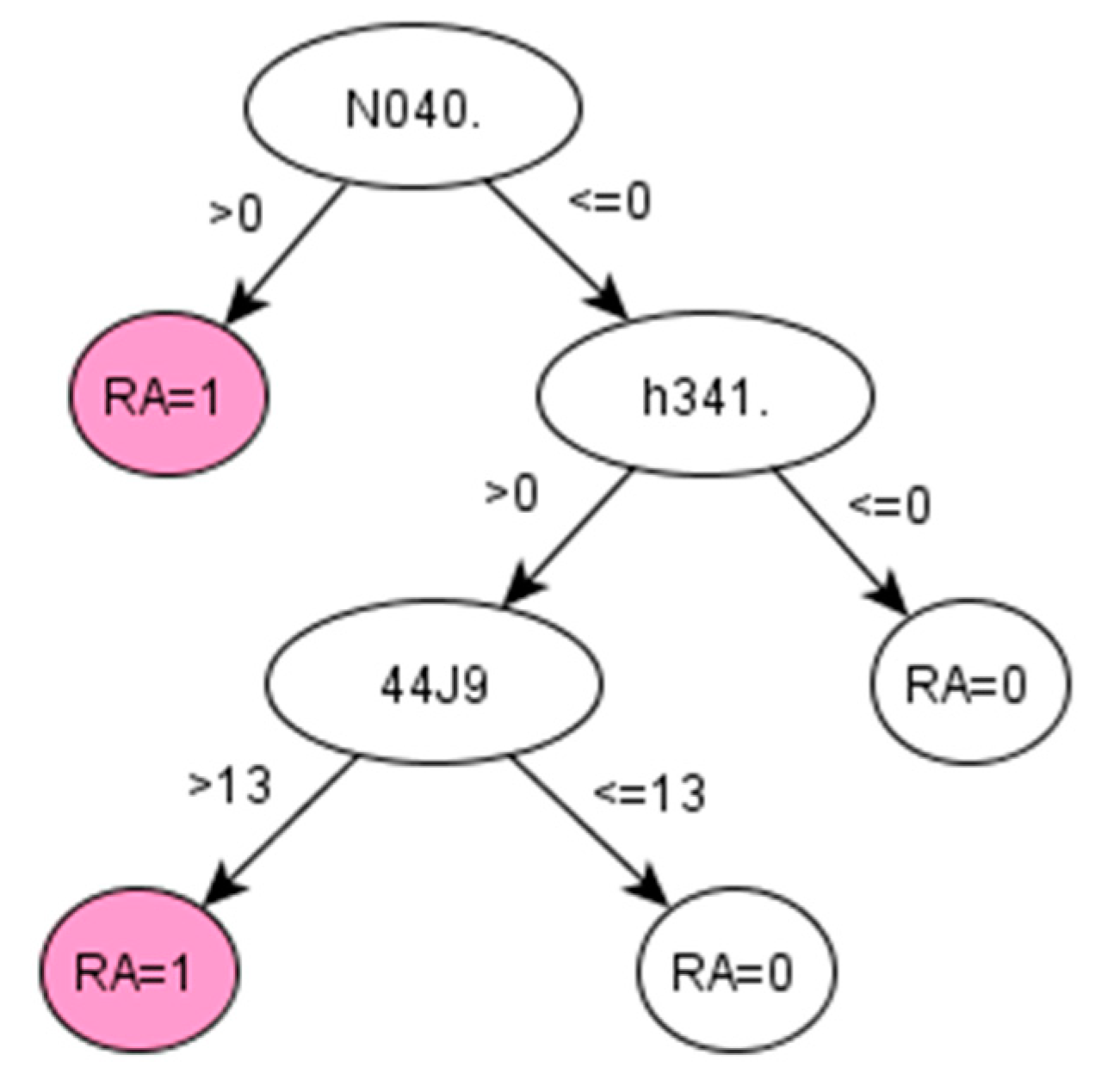
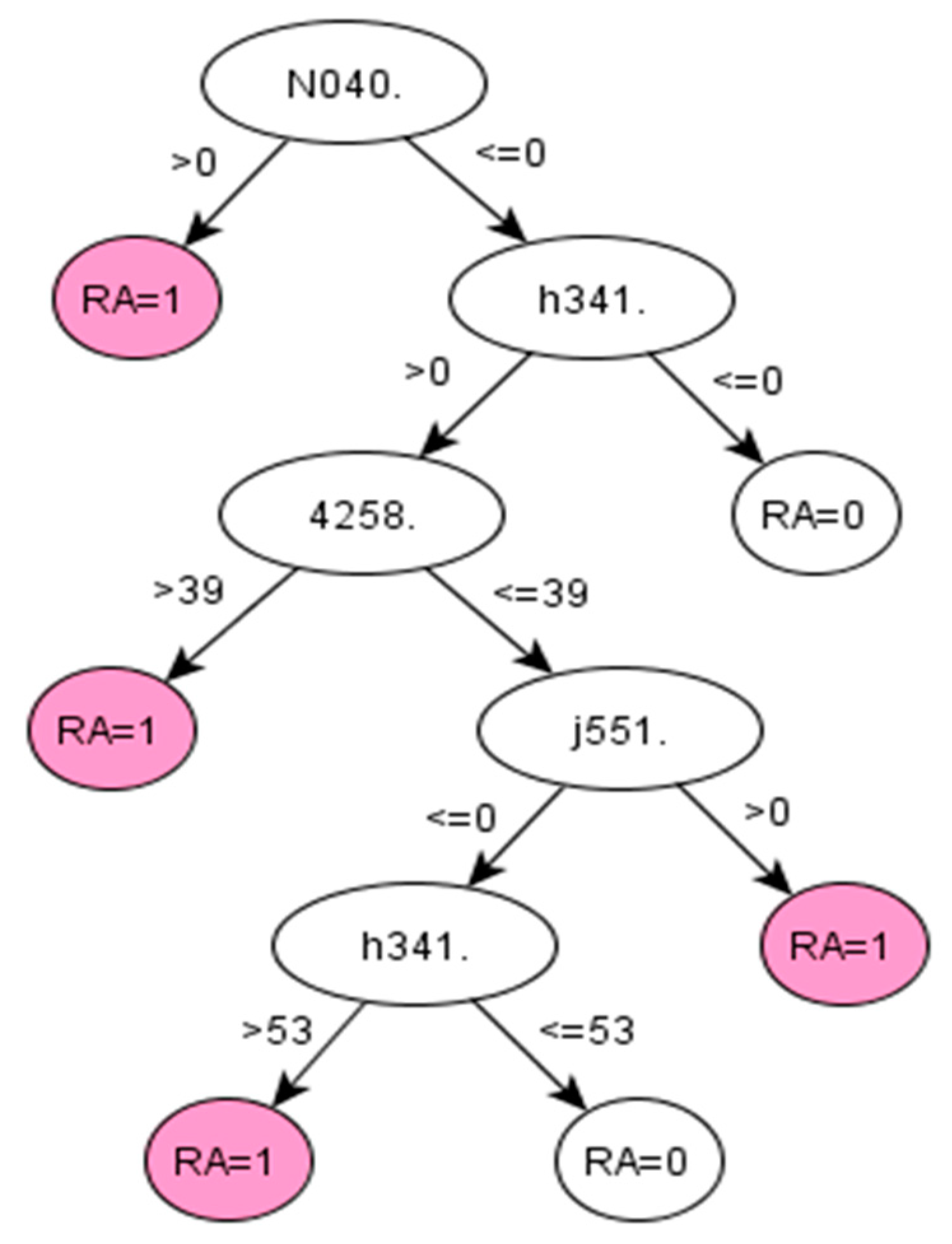
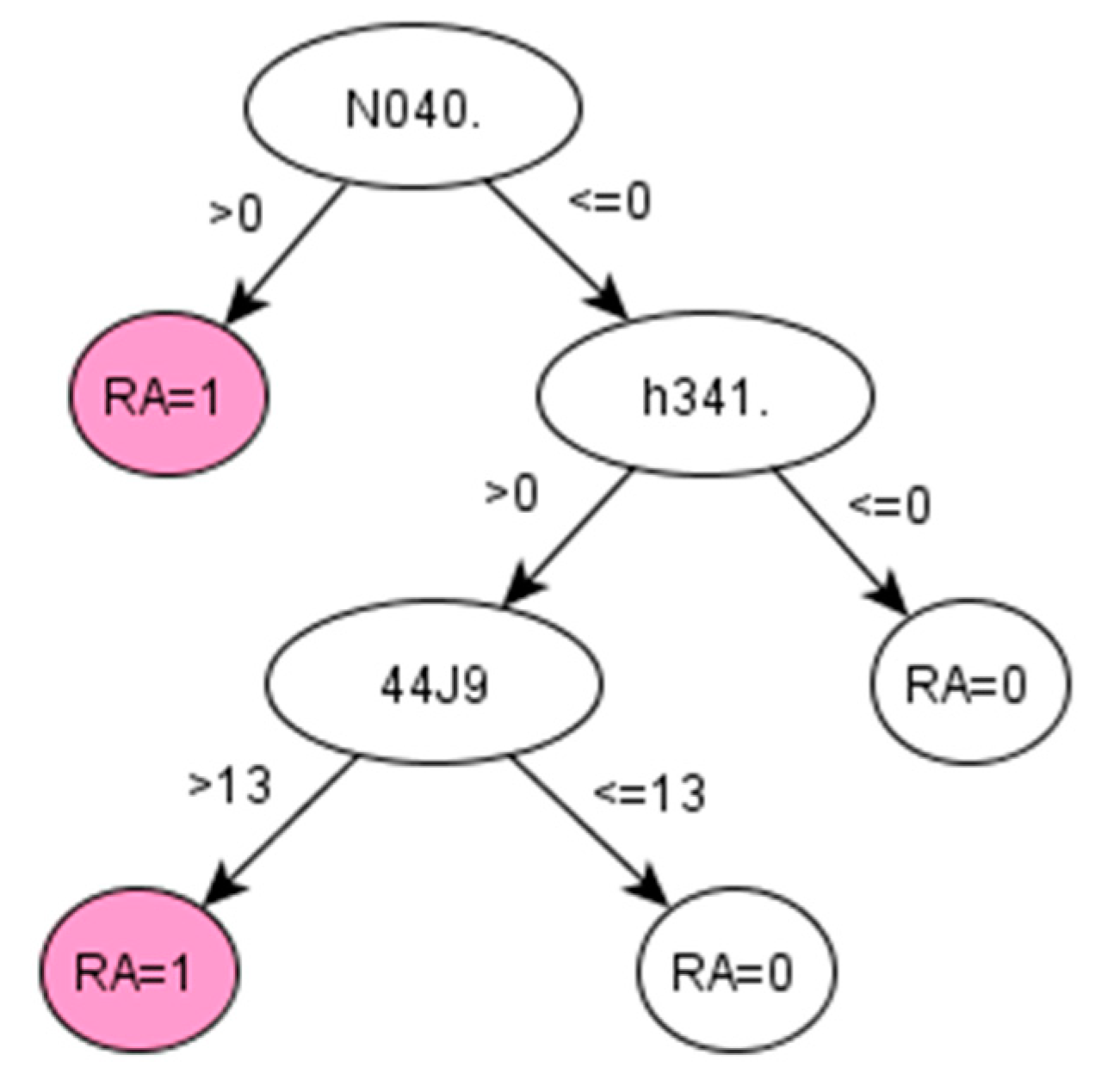
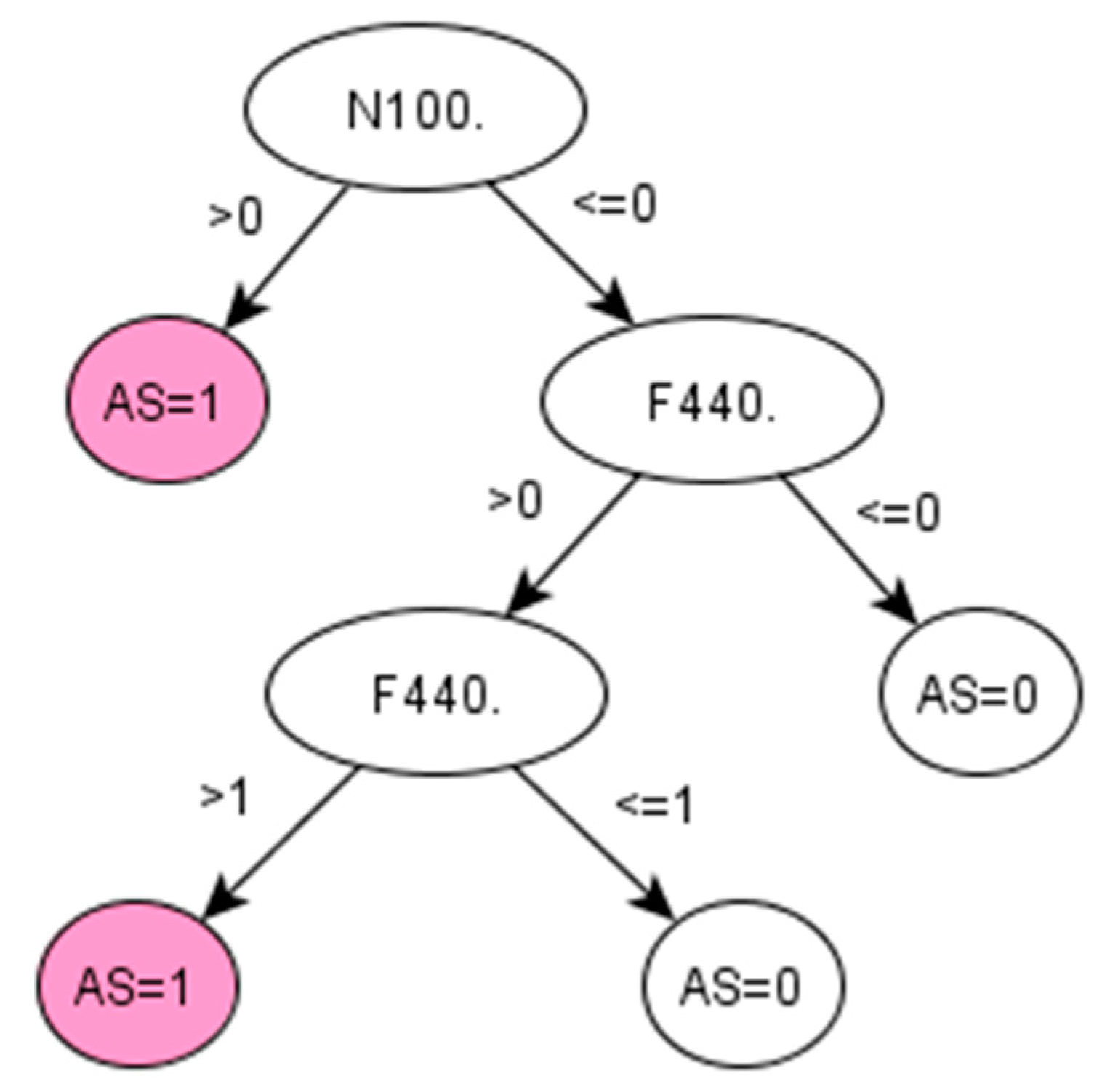
For RA, the algorithms development discovered a set of codes related to drugs that are commonly given to RA patients [
38]. The particular Read codes identified by the data-driven method may be specific characteristics of the studied population across ABMU and Cardiff regions. Reasonably, such a unique set of codes may vary for other populations (datasets). Promisingly, the proposed framework was demonstrated to identify these unique sets of codes related to certain populations from given datasets.
Our results indicate that the χ
2 statistic and the Gini index generated the same ranking of features in both datasets. This is mainly because the χ
2 statistic coefficient for a 2 by 2 contingency table can be also interpreted as the Gini coefficient [
39]. Each feature-selection method works differently depending on the nature of the data. To identify patients with a particular profile, a single feature-section method may generate incomplete results. Our recommendation is to use multiple feature-selection methods to generate a bigger picture of prediction performances and select an optimal algorithm with consideration of clinical practices.
We stress that the proposed framework aiming at identifying influential features actually fulfils the task of dimensionality reduction in nature. In a high-dimensional data space, dimensionality reduction can be performed by two different schemes. One scheme is to directly select influential features from original features, in which the selected features work together to demonstrate good prediction performance, such as our proposed framework. The other scheme is to transform all original features into a low-dimensional space, in which each dimension of this space works as a latent variable while holding certain statistical property of original features, such as the principal component analysis. Each scheme has its own advantages and disadvantages, but in disease phenotyping and patient identification, the first scheme is preferred. This is mainly because the first scheme can maintain the physical meanings of original variables in a low-dimensional space so that the finally selected features in this space can gain clear interpretations in prediction. The second scheme uses latent variables in a low-dimensional space that are often difficult for which to gain clear physical meanings.
As a baseline method for identifying optimal hyper-parameters, grid-search can provide coarse but uniform exploration of the parameter space. The performances of decision tree models varied greatly for different values of the parameters. The C5.0 trees are very sensitive to the two parameters of minimum cases for split and asymmetric cost. For the RA and AS, we found that the optimal parameters are particular to each dataset. That is to say, one cannot expect a single optimal parameter set can be generalised to different datasets. In the future, heuristic optimisation procedures will be used to reduce the computing loads in identifying optimal hyper-parameters.
Medical data sets are often predominately composed of “normal” cases (a large number of population without a condition of interest) together with a small percentage of “abnormal” cases (a small number of cases with this condition of interest, such as RA or AS). Such an imbalance presents methodological challenges in developing and verifying machine-learning algorithms. This study used the C5.0 tree model to tackle imbalanced classification problems by benefiting from the asymmetric costs implemented in majority class and minority class.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}