
In Vitro Fertilization Using Preimplantation Genetic Testing in a Romanian Couple Carrier of Mutations in the TTN Gene: A Case Report and Literature Review

 ,
,  , and
, and
Abstract
:1. Introduction
2. Case Presentation Section
2.1. First Unsuccessful Pregnancy
2.2. Initial Diagnoses of the Newborn
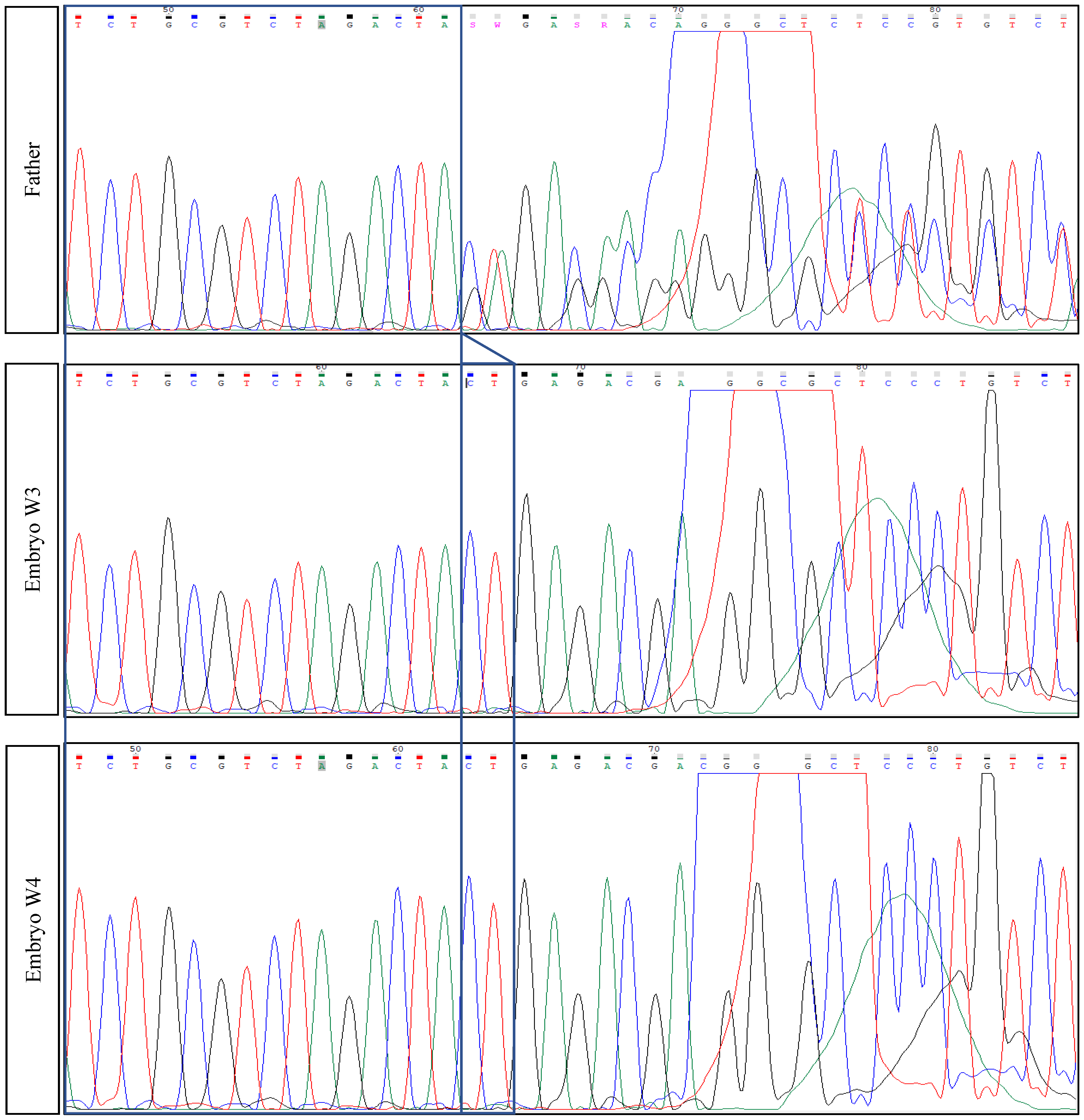
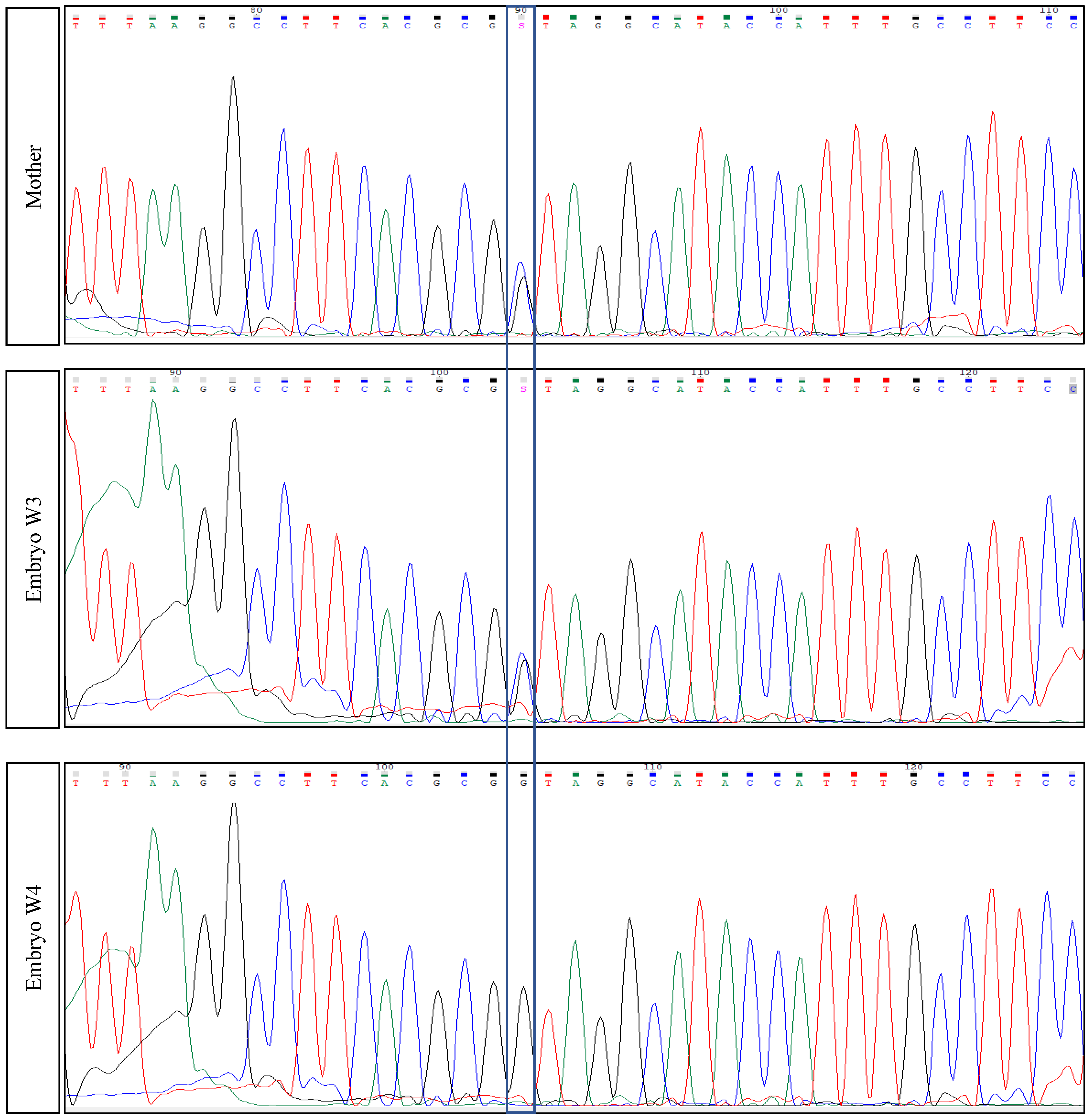
2.3. Parents’ Testing
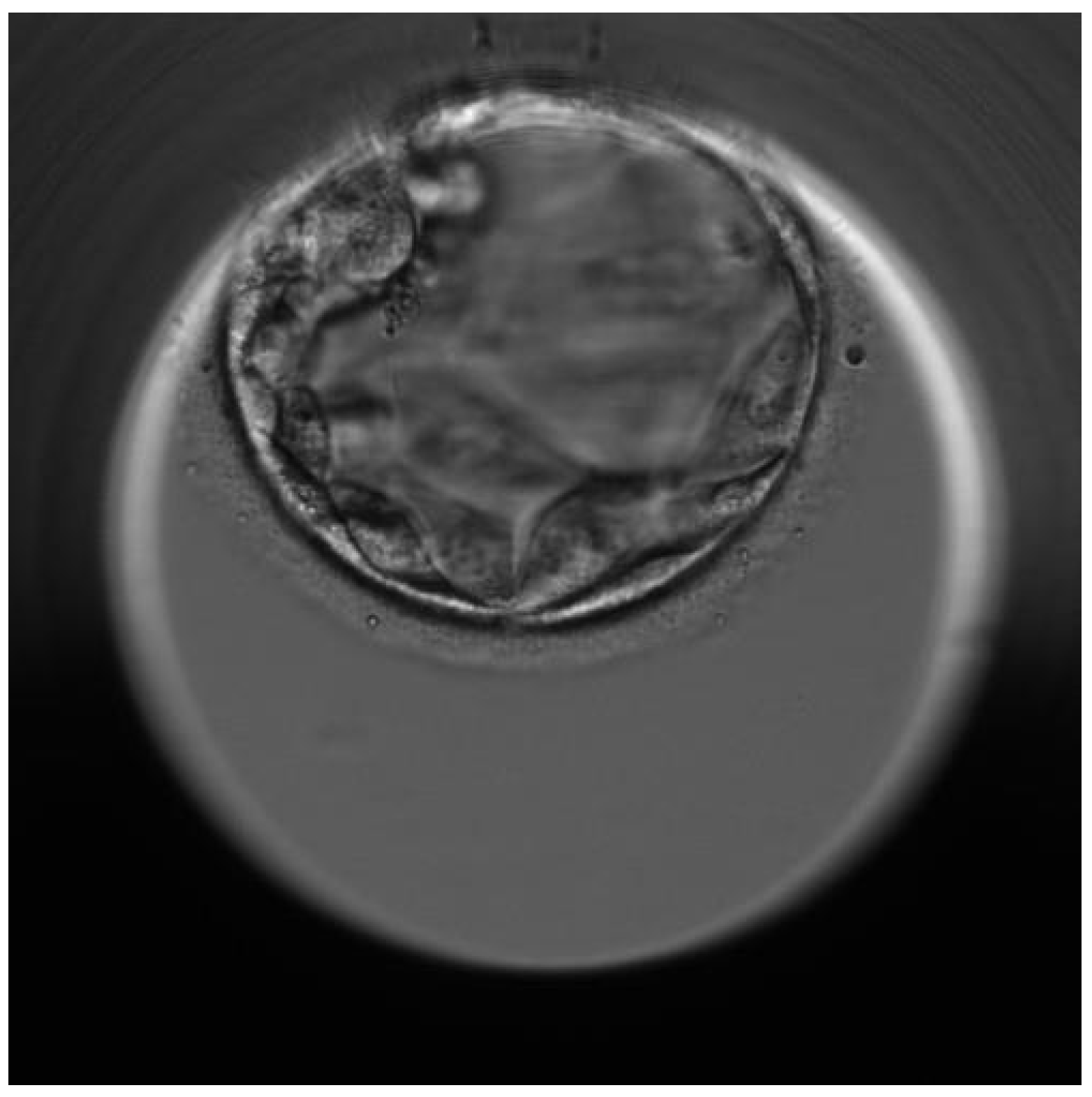
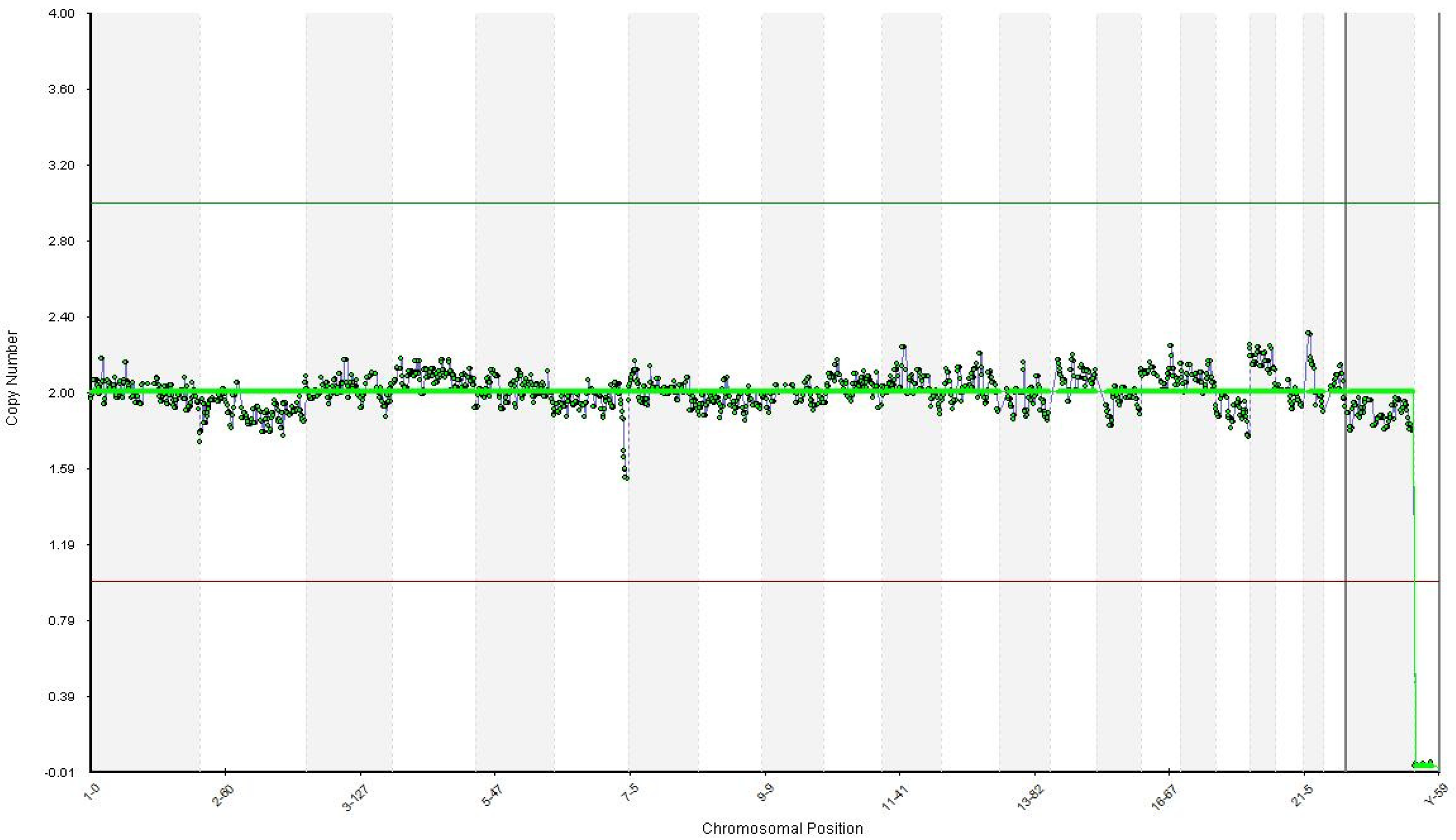
2.4. Successful Pregnancy
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Haravuori, H.; Vihola, A.; Straub, V.; Auranen, M.; Richard, I.; Marchand, S.; Voit, T.; Labeit, S.; Somer, H.; Peltonen, L.; et al. Secondary calpain3 deficiency in 2q-linked muscular dystrophy. Neurology 2001, 56, 869–877. [Google Scholar] [CrossRef]
- Hackman, P.; Vihola, A.; Haravuori, H.; Marchand, S.; Sarparanta, J.; De Seze, J.; Labeit, S.; Witt, C.; Peltonen, L.; Richard, I.; et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am. J. Hum. Genet. 2002, 71, 492–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, M.-L.; Centner, T.; Fornoff, F.; Geach, A.J.; Gotthardt, M.; McNabb, M.; Witt, C.C.; Dietmar, L.; Gregorio, C.C.; Granzier, H.; et al. The Complete Gene Sequence of Titin, Expression of an Unusual ≈700-kDa Titin Isoform, and Its Interaction With Obscurin Identify a Novel Z-Line to I-Band Linking System. Circ. Res. 2001, 89, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Savarese, M.; Sarparanta, J.; Vihola, A.; Udd, B.; Hackman, P. Increasing Role of Titin Mutations in Neuromuscular Disorders. J. Neuromuscul. Dis. 2016, 3, 293–308. [Google Scholar] [CrossRef] [Green Version]
- Hackman, P.; Udd, B.; Bönnemann, C.G.; Ferreiro, A.; Udd, B.; Hackman, P.; Ferreiro, A.; Bonnemann, C.; Beggs, A.; Gautel, M.; et al. 219th ENMC International Workshop Titinopathies International database of titin mutations and phenotypes, Heemskerk, The Netherlands, 29 April–1 May 2016. Neuromuscul. Disord. 2017, 27, 396–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, D.S.; Lam, L.; Taylor, M.R.G.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of Titin Causing Dilated Cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerull, B.; Gramlich, M.; Atherton, J.; McNabb, M.; Trombitás, K.; Sasse-Klaassen, S.; Seidman, J.G.; Seidman, C.; Granzier, H.; Labeit, S.; et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 2002, 30, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, G.; Elliott, H.R.; Griffin, H.; Barresi, R.; Miller, J.; Marsh, J.; Evilä, A.; Vihola, A.; Hackman, P.; Straub, V.; et al. Titin mutation segregates with hereditary myopathy with early respiratory failure. Brain 2012, 135, 1695–1713. [Google Scholar] [CrossRef]
- Pfeffer, G.; Barresi, R.; Wilson, I.J.; Hardy, S.A.; Griffin, H.; Hudson, J.; Elliott, H.R.; Ramesh, A.V.; Radunovic, A.; Winer, J.B.; et al. Titin founder mutation is a common cause of myofibrillar myopathy with early respiratory failure. J. Neurol. Neurosurg. Psychiatry 2014, 85, 331–338. [Google Scholar] [CrossRef]
- Palmio, J.; Evilä, A.; Chapon, F.; Tasca, G.; Xiang, F.; Brådvik, B.; Eymard, B.; Echaniz-Laguna, A.; Laporte, J.; Kärppä, M.; et al. Hereditary myopathy with early respiratory failure: Occurrence in various populations. J. Neurol. Neurosurg. Psychiatry 2014, 85, 345–353. [Google Scholar] [CrossRef]
- De Cid, R.; Ben Yaou, R.; Roudaut, C.; Charton, K.; Baulande, S.; Leturcq, F.; Romero, N.B.; Malfatti, E.; Beuvin, M.; Vihola, A.; et al. A new titinopathy: Childhood-juvenile onset Emery-Dreifuss-like phenotype without cardiomyopathy. Neurology 2015, 85, 2126–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmignac, V.; Salih, M.A.M.; Quijano-Roy, S.; Marchand, S.; Al Rayess, M.M.; Mukhtar, M.M.; Urtizberea, J.A.; Labeit, S.; Guicheney, P.; Leturcq, F.; et al. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann. Neurol. 2007, 61, 340–351. [Google Scholar] [CrossRef]
- Salih, M.A.M.; Al Rayess, M.; Cutshall, S.; Urtizberea, J.A.; Al-Turaiki, M.H.S.; Ozo, C.O.; Straub, V.; Akbar, M.; Abid, M.; Andeejani, A.; et al. A Novel Form of Familial Congenital Muscular Dystrophy in Two Adolescents. Neuropediatrics 1998, 29, 289–293. [Google Scholar] [CrossRef]
- Subahi, S.A. Distinguishing Cardiac Features of a Novel Form of Congenital Muscular Dystrophy (Salih CMD). Pediatr. Cardiol. 2001, 22, 297–301. [Google Scholar] [CrossRef]
- Fukuzawa, A.; Lange, S.; Holt, M.; Vihola, A.; Carmignac, V.; Ferreiro, A.; Udd, B.; Gautel, M. Interactions with titin and myomesin target obscurin and obscurin-like 1 to the M-band—Implications for hereditary myopathies. J. Cell Sci. 2008, 121, 1841–1851. [Google Scholar] [CrossRef] [Green Version]
- Pernigo, S.; Fukuzawa, A.; Bertz, M.; Holt, M.; Rief, M.; Steiner, R.A.; Gautel, M. Structural insight into M-band assembly and mechanics from the titin-obscurin-like-1 complex. Proc. Natl. Acad. Sci. USA 2010, 107, 2908–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauveau, C.; Bonnemann, C.G.; Julien, C.; Kho, A.L.; Marks, H.; Talim, B.; Maury, P.; Arne-Bes, M.C.; Uro-Coste, E.; Alexandrovich, A.; et al. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum. Mol. Genet. 2014, 23, 980–991. [Google Scholar] [CrossRef]
- Salih, M.; Hamad, M.; Savarese, M.; Alorainy, I.; Alkhalidi, H.; Alotaibi, A.; Alsagob, M.; Colak, D.; Udd, B.; Kaya, N.; et al. Delineating the phenotypes of early onset myopathy due to novel titin gene mutations. J. Neurol. Sci. 2019, 405, 38–39. [Google Scholar] [CrossRef] [Green Version]
- Oates, E.C.; Jones, K.J.; Donkervoort, S.; Charlton, A.; Brammah, S.; Smith, J.E., III; Ware, J.S.; Yau, K.S.; Swanson, L.C.; Whiffin, N.; et al. Congenital Titinopathy: Comprehensive characterization and pathogenic insights. Ann. Neurol. 2018, 83, 1105–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rees, M.; Nikoopour, R.; Fukuzawa, A.; Kho, A.L.; Fernandez-Garcia, M.A.; Wraige, E.; Bodi, I.; Deshpande, C.; Özdemir, Ö.; Daimagüler, H.-S.; et al. Making sense of missense variants in TTN-related congenital myopathies. Acta Neuropathol. 2021, 141, 431–453. [Google Scholar] [CrossRef]
- Savarese, M.; Maggi, L.; Vihola, A.; Jonson, P.H.; Tasca, G.; Ruggiero, L.; Bello, L.; Magri, F.; Giugliano, T.; Torella, A.; et al. Interpreting Genetic Variants in Titin in Patients With Muscle Disorders. JAMA Neurol. 2018, 75, 557–565. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-W.; Liu, Y.-L.; Chen, C.-H. Targeting myotonic dystrophy by preimplantation genetic diagnosis-karyomapping. Taiwan J. Obstet. Gynecol. 2019, 58, 891–894. [Google Scholar] [CrossRef]
- Kalman, L.; Tarleton, J.; Hitch, M.; Hegde, M.; Hjelm, N.; Berry-Kravis, E.; Zhou, L.; Hilbert, J.E.; Luebbe, E.A.; Moxley, R.T., III; et al. Development of a Genomic DNA Reference Material Panel for Myotonic Dystrophy Type 1 (DM1) Genetic Testing. J. Mol. Diagn. 2013, 15, 518–525. [Google Scholar] [CrossRef] [Green Version]
- Senba, H.; Sueoka, K.; Sato, S.; Higuchi, N.; Mizuguchi, Y.; Sato, K.; Tanaka, M. The impact of parental unaffected allele combination on the diagnostic outcome in the preimplantation genetic testing for myotonic dystrophy type 1 in Japanese ancestry. Reprod. Med. Biol. 2020, 19, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Lian, M.; Lee, C.G.; Chong, S.S. Robust Preimplantation Genetic Testing Strategy for Myotonic Dystrophy Type 1 by Bidirectional Triplet-Primed Polymerase Chain Reaction Combined with Multi-microsatellite Haplotyping Following Whole-Genome Amplification. Front. Genet. 2019, 10, 589. [Google Scholar] [CrossRef]
- Mongkolchaipak, S.; Piyamongkol, W.; Piyamongkolx, W. 62. Successful Strategy of Comprehensive Pre-Implantation Genetic Testing for Duchenne Muscular Dystrophy and Chromosome Balance Using Karyomapping. Reprod. Biomed. Online 2019, 39, e64–e65. [Google Scholar] [CrossRef]
- Bianco, B.; Christofolini, D.M.; Conceição, G.S.; Barbosa, C.P. Preimplantation genetic diagnosis associated to Duchenne muscular dystrophy. Einstein 2017, 15, 489–491. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Shen, X.; Wu, H.; Chen, D.; Zhou, C. Preimplantation Genetic Testing for Monogenic Disease of Spinal Muscular Atrophy by Multiple Displacement Amplification: 11 unaffected livebirths. Int. J. Med. Sci. 2019, 16, 1313–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, C.E.; Lewis, A.; Willats, E.; Mullen, J. Preimplantation genetic testing using Karyomapping for a paternally inherited reciprocal translocation: A case study. J. Assist. Reprod. Genet. 2019, 36, 951–963. [Google Scholar] [CrossRef]
- Shi, D.; Xu, J.; Niu, W.; Liu, Y.; Shi, H.; Yao, G.; Shi, S.; Li, G.; Song, W.; Jin, H.; et al. Live births following preimplantation genetic testing for dynamic mutation diseases by karyomapping: A report of three cases. J. Assist. Reprod. Genet. 2020, 37, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Pratt, M.; Garritty, C.; Thuku, M.; Esmaeilisaraji, L.; Hamel, C.; Hartley, T.; Millar, K.; Skidmore, B.; Dougan, S.; Armour, C.M. Application of exome sequencing for prenatal diagnosis: A rapid scoping review. Genet. Med. 2020, 22, 1925–1934. [Google Scholar] [CrossRef]
- Dempsey, E.; Haworth, A.; Ive, L.; Dubis, R.; Savage, H.; Serra, E.; Kenny, J.; Elmslie, F.; Greco, E.; Thilaganathan, B.; et al. A report on the impact of rapid prenatal exome sequencing on the clinical management of 52 ongoing pregnancies: A retrospective review. BJOG Int. J. Obstet. Gynaecol. 2021, 128, 1012–1019. [Google Scholar] [CrossRef]
- Lord, J.; McMullan, D.J.; Eberhardt, R.Y.; Rinck, G.; Hamilton, S.J.; Quinlan-Jones, E.; Prigmore, E.; Keelagher, R.; Best, S.K.; Carey, G.K.; et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): A cohort study. Lancet 2019, 393, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Chen, J.; Wang, C.; Chen, F.; Xie, Y.; Li, Y.; Li, N.; Wang, J.; Zhang, V.W.; Chen, D. Clinical application of medical exome sequencing for prenatal diagnosis of fetal structural anomalies. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 251, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Drury, S.; Williams, H.; Trump, N.; Boustred, C.; GOSGene; Lench, N.; Scott, R.H.; Chitty, L.S. Exome sequencing for prenatal diagnosis of fetuses with sonographic abnormalities. Prenat. Diagn. 2015, 35, 1010–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaghan, K.G.; Leach, N.T.; Pekarek, D.; Prasad, P.; Rose, N.C. on behalf of the ACMG Professional Practice and Guidelines Committee. The use of fetal exome sequencing in prenatal diagnosis: A points to consider document of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2020, 22, 675–680. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family Member | Phenotype | Genotype |
|---|---|---|
| First newborn | At 33 weeks of pregnancy minor clubfoot altered fetal biophysical profile | Three areas associated with loss of heterozygosity on chromosomes 1(q25.1–q25.3) of 6115 kb, 5(p15.2–p15.1) of 2589 kb and 8(q11.21–q11.23) of 4830 kb, a duplication of 1104 kb on chromosome 10 in the position q11.22, and a duplication of 1193 kb on chromosome 16 in the position p11.2p11.1. |
| At 36 weeks of pregnancy abnormal fetal heart rate abnormal fetal movement poor muscular tonus | ||
| At birth generalized congenital muscular atony right diaphragmatic hernia cerebral atrophy neonatal anemia bilateral varus equinus neonatal hypocalcemia prematurity and low birth weight ostium secundum atrial heart defect tricuspid valve dysplasia right diaphragmatic hernia moderate cerebral atrophy | ||
| Mother | Healthy carrier | Chr2(GRCh37):g.179479653G>C—TTN variant c.48681C>G p.(Tyr16227*)—Exon 260 |
| Father | Healthy carrier | Chr2(GRCh37):g.179396832_179396833del—TTN variant c.104509_104510del p.(Leu34837Glufs*12)—Exon 358 |
| First euploid embryo (W3) | Healthy embryo | Carrier of the mother’s mutation |
| Second euploid embryo (W4) | Healthy embryo | Healthy embryo (Wild-type) for both mutations |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doroftei, B.; Maftei, R.; Ilie, O.-D.; Armeanu, T.; Puiu, M.; Ivanov, I.; Nemtanu, L. In Vitro Fertilization Using Preimplantation Genetic Testing in a Romanian Couple Carrier of Mutations in the TTN Gene: A Case Report and Literature Review. Diagnostics 2021, 11, 2328. https://doi.org/10.3390/diagnostics11122328
Doroftei B, Maftei R, Ilie O-D, Armeanu T, Puiu M, Ivanov I, Nemtanu L. In Vitro Fertilization Using Preimplantation Genetic Testing in a Romanian Couple Carrier of Mutations in the TTN Gene: A Case Report and Literature Review. Diagnostics. 2021; 11(12):2328. https://doi.org/10.3390/diagnostics11122328
Chicago/Turabian StyleDoroftei, Bogdan, Radu Maftei, Ovidiu-Dumitru Ilie, Theodora Armeanu, Maria Puiu, Iuliu Ivanov, and Loredana Nemtanu. 2021. "In Vitro Fertilization Using Preimplantation Genetic Testing in a Romanian Couple Carrier of Mutations in the TTN Gene: A Case Report and Literature Review" Diagnostics 11, no. 12: 2328. https://doi.org/10.3390/diagnostics11122328