
A Review of Selected IBD Biomarkers: From Animal Models to Bedside


Abstract
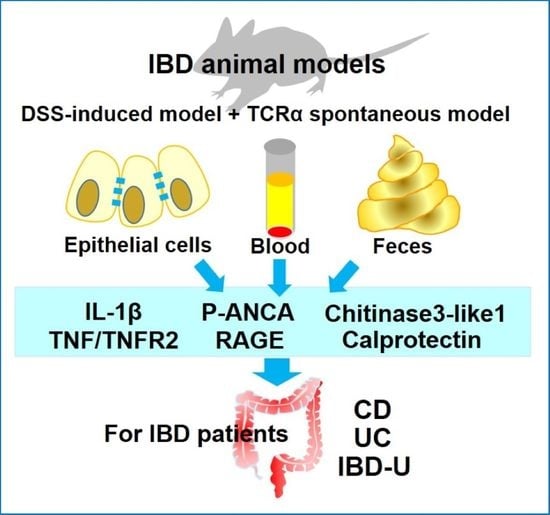
:
1. Introduction
2. A Few Selected IBD Biomarkers, Which Are Identified by Murine Experimental Models of DSS-Induced Colitis and/or TCRα KO Mice
2.1. pANCA
2.2. Chitinase 3-Like 1 (CHI3L1)
2.3. S100A12/RAGE
2.4. S100A8/S100A9: Calprotectin

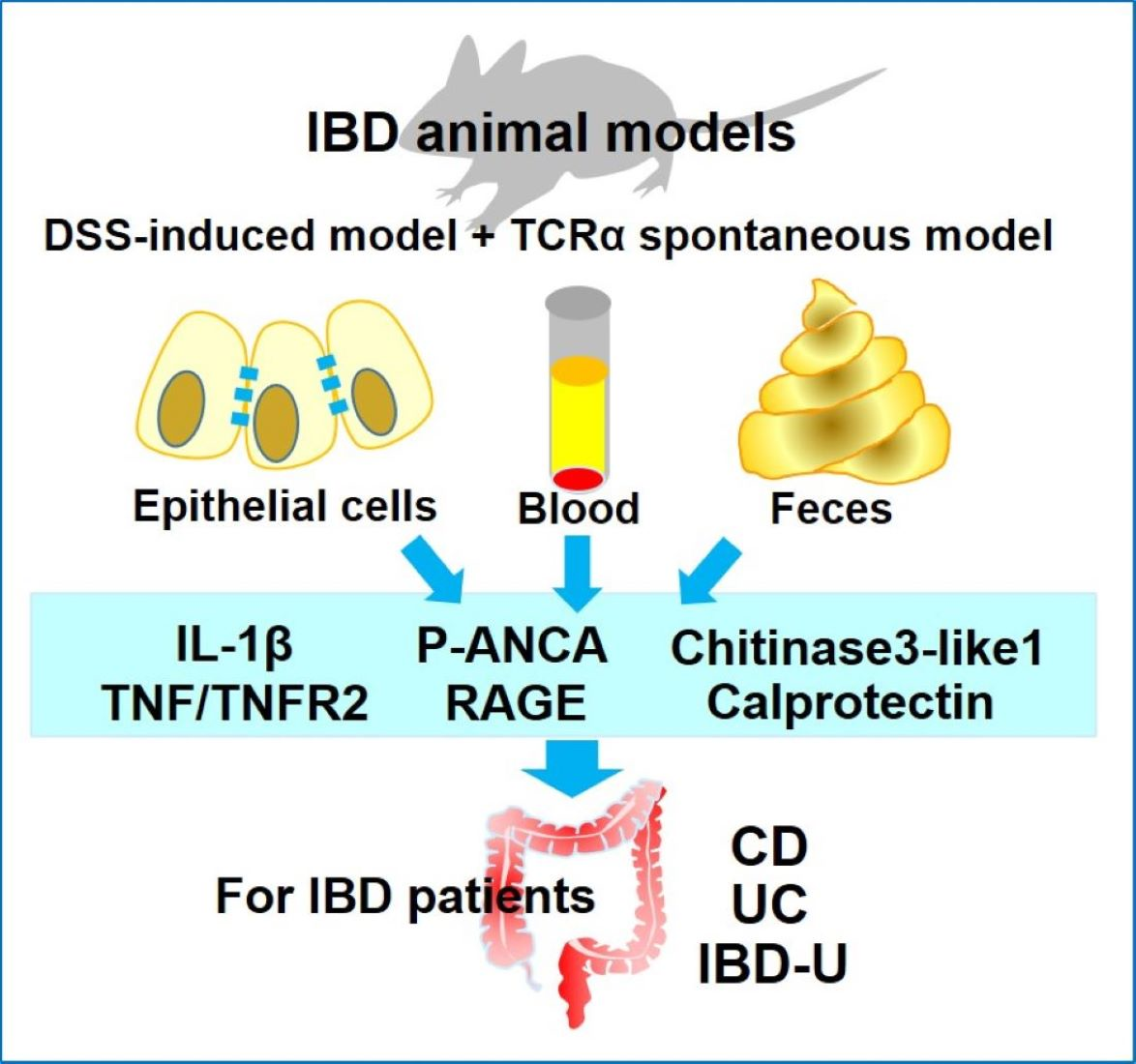{kind=link}
| Calprotectin | S100A12 | |
|---|---|---|
| Expression | granulocyte, monocyte and macrophage [62] epithelial cell under inflammatory conditions [48,61] | neutrophil, monocyte, and macrophage [35,52] endothelial cell, keratinocyte, and epithelial cell under inflammatory conditions [35,48] |
| Function | modulate intracellular calcium signaling [65] antibacterial infections [71] regulation of the intestinal microbiota and immune system in neonate [86] | promotion of cytokine production [35,65] regulation of leukocyte adhesion and migration [35,65] |
| Receptors | TLR4 [67] Heparensulfate proteoglycans [62] N-glycans [62] RAGE [65] EMMPRIN [66] (only S100A9 binds) | TLR4 [37,38] RAGE [42] CD36 [39,40] |
| Related diseases | Inflammatory diseases [67] IBD Psoriasis Rheumatoid arthritis Transplantation Infections | IBD IBS Arthritis Vasculitis Periodontitis Kawasaki disease Infections [39,40,56,65] |
2.5. Proinflammatory Factors Including IL-1β, TNFα and TNFR2
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Schirmer, M.; Garner, A.; Vlamakis, H.; Xavier, R.J. Microbial genes and pathways in inflammatory bowel disease. Microbial genes and pathways in inflammatory bowel disease. Nat. Rev. Microbiol. 2019, 17, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Fang, M.; Jostins, L.; Mirkov, M.U.; Boucher, G.; Anderson, C.A.; Anderson, V.; Cleynen, I.; Cortes, A.; Crins, F.; et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 2017, 547, 173–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubstov, Y.P.; Rasmussen, J.P.; Chi, E.Y.; Fontenot, J.; Castelli, L.; Ye, X.; Treuting, P.; Siewe, L.; Roers, A.; Henderson, W.R.; et al. Regulatory T cell-derived interleukin-10 limits binflammation at environmental interfaces. Immunity 2008, 28, 546–558. [Google Scholar]
- Norouzionia, M.; Chaleshi, V.; Alizadeh, A.H.M.; Zali, M.R. Biomarkers in inflammatory bowel diseases: Insight into diagnosis, prognosis and treatment. Gastroenterol. Hepatol. Bed Bench 2017, 10, 155–167. [Google Scholar]
- Mombaerts, P.; Mizoguchi, E.; Grusby, M.J.; Glimcher, L.H.; Bhan, A.K.; Tonegawa, S. Spontaneius development of inflammatory bowel disease in T cell receptor mutant mice. Cell 1993, 75, 274–282. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Mizoguchi, E.; Chiba, C.; Spiekermann, G.M.; Tonegawa, S.; Nagler-Anderson, C.; Bhan, A.K. Cytokine imbalance and autoantibody production in T cell receptor-alpha mutant mice with inflammatory bowel disease. J. Exp. Med. 1996, 183, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, E.; Mizoguchi, A.; Chiba, C.; Niles, J.L.; Bhan, A.K. Anti-neutrophil cytoplasmic antibodies in T-cell receptor α-deficient mice with chronic colitis. Gastroenterology 1997, 113, 1828–1835. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Mizoguchi, E. Suppressive role of B cells in chronic colitis of T cell receptor α mutant mice. J. Exp. Med. 1997, 186, 1749–1756. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Mizoguchi, E.; Smith, R.N.; Preffer, F.I.; Bhan, A.K. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity 2002, 16, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Low, D.; Nguyen, D.D.; Mizoguchi, E. Animal models of ulcerative colitis and their application in drug research. Drug. Des. Dev. Ther. 2013, 7, 1341–1357. [Google Scholar]
- Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702. [Google Scholar] [CrossRef]
- Kawada, M.; Arihiro, A.; Mizoguchi, E. Insights from advances in research of chemically induced experimental models of human inflammatory bowel disease. World. J. Gastroenterol. 2007, 13, 5581–5593. [Google Scholar] [CrossRef]
- Dieleman, L.A.; Ridwan, B.U.; Tennyson, G.S.; Beagley, K.W.; Bucy, R.P.; Elson, C.O. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 1994, 107, 1643–1652. [Google Scholar] [CrossRef]
- Rump, J.A.; Schölmerich, J.; Gross, V.; Roth, M.; Helfesrieder, R.; Rautmann, A.; Lüdemann, J.; Gross, W.L.; Peter, H.H. A new type of perinuclear anti-neutrophil cytoplasmic antibody (p-ANCA) in active ulcerative colitis but not in Crohn’s disease. Immunobiology 1990, 181, 406–413. [Google Scholar] [CrossRef]
- Eggena, M.P.; Targan, S.R.; Vidrich, A.; Clemens, D.F.; Iwanczyk, L.; Braun, J. Histone HI: The ulcerative colitis specific pANCA target antigen. FASEB J. 1996, 10, 463. [Google Scholar]
- Reese, G.E.; Constantinides, V.A.; Simillis, C.; Darzi, A.W.; Orchard, T.R.; Fazio, V.W.; Tekkis, P.P. Diagnostic precision of anti-Saccharomyces cerevisiae antibodies and perinuclear antineutrophil cytoplasmic antibodies in inflammatory bowel disease. Am. J. Gastroenterol. 2006, 10, 2410–2422. [Google Scholar] [CrossRef]
- Roozendaal, C.; Kallenberg, C.G.M. Are anti-neutrophil cytoplasmic antibodies (ANCA) clinically useful in inflammatory bowel disease (IBD)? Clin. Exp. Immunol. 1999, 116, 206–213. [Google Scholar] [CrossRef]
- Gionchetti, P.; Vecchi, M.; Rizzello, F.; Ferretti, M.; Calabresi, C.; Venturi, A.; Bianchi, M.B.; Brignola, C.; Sinico, R.A.; De Franchis, R.; et al. Lack of effect of antineutrophil cytoplasmic antibodies associated with ulcerative colitis on superoxide anion production from neutrophils. Gut 1997, 40, 102–104. [Google Scholar] [CrossRef]
- Quinton, J.F.; Sendid, B.; Reumaux, D.; Duthilleul, P.; Cortot, A.; Grandbastien, B.; Charrier, G.; Targan, S.R.; Colombel, J.F.; Poulain, D. Anti-Saccharomyces cerevisiae mannaa antibodies combined with antineutrophil cytoplasmic autoantibodies in inflammatory bowel disease: Prevalence and diagnostic role. Gut 1998, 42, 788–791. [Google Scholar] [CrossRef] [Green Version]
- Mitsuyama, K.; Niwa, M.; Takedatsu, H.; Yamasaki, H.; Kuwaki, K.; Yoshioka, S.; Yamauchi, R.; Fukunaga, S.; Torimura, T. Antibody markers in the diagnosis ofiInflammatory bowel disease. World J. Gastroenterol. 2016, 22, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Smids, C.; Horjus Talabur Horje, C.S.; Groenen, M.J.M.; van Koolwijk, E.H.M.; Wahab, P.J.; van Lochem, E.G. The value of serum antibodies in differentiating inflammatory bowel disease, Predicting disease activity and disease course in the newly diagnosed patient. Scand. J. Gastroenterol. 2017, 52, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Birimberg-Schwartz, L.; Wilson, D.C.; Kolho, K.L.; Karolewska-Bochenek, K.; Afzal, N.A.; Spray, C.; Romano, C.; Lionetti, P.; Hauer, A.C.; Martinez-Vinson, C.; et al. PANCA and ASCA in children with IBD-unclassified, Crohn’s colitis, and ulcerative colitis—A longitudinal report from the IBD porto group of ESPGHAN. Inflamm. Bowel Dis. 2016, 22, 1908–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrance, I.C.; Murray, K.; Hall, A.; Sung, J.J.Y.; Leong, R. A prospective comparative study of ASCA and pANCA in chinese and caucasian IBD patients. Am. J. Gastroenterol. 2004, 99, 2186–2194. [Google Scholar] [CrossRef] [PubMed]
- Takedatsu, H.; Mitsuyama, K.; Fukunaga, S.; Yoshioka, S.; Yamauchi, R.; Mori, A.; Yamasaki, H.; Kuwaki, K.; Sakisaka, H.; Sakisaka, S.; et al. Diagnostic and clinical role of serum proteinase 3 antineutrophil cytoplasmic antibodies in inflammatory bowel disease. J. Gastroenterol. Hepatol. 2018, 33, 1603–1607. [Google Scholar] [CrossRef]
- Subramaniam, R.; Mizoguchi, A.; Mizoguchi, E. Mechanistic roles of epithelial and immune cell signaling during the development of colitis-associated cancer. Cancer Res. Front. 2016, 2, 1–21. [Google Scholar] [CrossRef]
- Mizoguchi, E. Chitinase 3-Like-1 exacerbates intestinal inflammation by enhancing bacterial adhesion and invasion in colonic epithelial cells. Gastroenterology 2006, 130, 398–411. [Google Scholar] [CrossRef]
- Low, D.; Tran, H.T.; Lee, I.A.; Dreux, N.; Kamba, A.; Reinecker, H.C.; Darfeuille-Michaud, A.; Barnich, N.; Mizoguchi, E. Chitin-binding domains of Escherichia coli ChiA mediate interactions with intestinal epithelial cells in Mice with Colitis. Gastroenterology 2013, 145, 602–612. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.; Pekow, J.; Llado, V.; Kanneganti, M.; Lau, C.W.; Mizoguchi, A.; Mino-Kenudson, M.; Bissonnette, M.; Mizoguchi, E. Chitinase 3-like-1 expression in colonic epithelial cells as a potentially novel marker for colitis-associated neoplasia. Am. J. Pathol. 2011, 179, 1494–1503. [Google Scholar] [CrossRef]
- Low, D.; Subramaniam, R.; Lin, L.; Aomatsu, T.; Mizoguchi, A.; Ng, A.; DeGruttola, A.K.; Lee, C.G.; Elias, J.A.; Andoh, A.; et al. Chitinase 3-like 1 induces survival and proliferation of intestinal epithelial cells during chronic inflammation and colitis-associated cancer by regulating S100A9. Oncotarget 2015, 6, 36535–36550. [Google Scholar] [CrossRef] [Green Version]
- Aomatsu, T.; Imaeda, H.; Matsumoto, K.; Kimura, E.; Yoden, A.; Tamai, H.; Fujiyama, Y.; Mizoguchi, E.; Andoh, A. Faecal Chitinase 3-like-1: A novel biomarker of disease activity in paediatric inflammatory bowel disease. Aliment. Pharmacol. Ther. 2011, 34, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Koutroubakis, I.E.; Petinaki, E.; Dimoulios, P.; Vardas, E.; Roussomoustakaki, M.; Maniatis, A.N.; Kouroumalis, E.A. Increased serum levels of YKL-40 in patients with inflammatory bowel disease. Int. J. Colorectal Dis. 2003, 18, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Erzin, Y.; Uzun, H.; Karatas, A.; Celik, A.F. Serum YKL-40 as a marker of disease activity and stricture formation inpPatients with Crohn’s disease. J. Gastroenterol. Hepatol. 2008, 23, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Vind, I.; Johansen, J.S.; Price, P.A.; Munkholm, P. Serum YKL-40, a potential new marker of disease activity in patients with inflammatory bowel disease. Scand. J. Gastroenterol. 2003, 38, 599–605. [Google Scholar] [PubMed]
- Xia, C.; Braunstein, Z.; Toomey, A.C.; Zhong, J.; Rao, X. S100 proteins as an important regulator of macrophage inflammation. Front. Immunol. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dell’Angelica, E.C.; Schleicher, C.H.; Santome, J.A. Primary structure and binding properties of calgranulin C, a novel S100-like calcium-binding protein from pig granulocytes. J. Biol. Chem. 1994, 269, 28929–28936. [Google Scholar] [CrossRef]
- Foell, D.; Wittkowski, H.; Kessel, C.; Lüken, A.; Weinhage, T.; Varga, G.; Vogl, T.; Wirth, T.; Viemann, D.; Björk, P.; et al. Proinflammatory S100A12 can activate human monocytes via toll-like receptor 4. Am. J. Respir. Crit. Care Med. 2013, 187, 1324–1334. [Google Scholar] [CrossRef]
- Kessel, C.; Fuehner, S.; Zell, J.; Zimmermann, B.; Drewianka, S.; Brockmeyer, S.; Holzinger, D.; Hinze, C.; Wittkowski, H.; Foell, D. Calcium and zinc tune autoinflammatory Toll-like receptor 4 signaling by S100A12. J. Allergy Clin. Immunol. 2018, 142, 1370–1373. [Google Scholar] [CrossRef] [Green Version]
- Tondera, C.; Laube, M.; Pietzsch, J. Insights into binding of S100 proteins to scavenger receptors: Class B scavenger receptor CD36 binds S100A12 with high affinity. Amino Acids 2017, 49, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Farokhzadian, J.; Mangolian Shahrbabaki, P.; Bagheri, V. S100A12-CD36 axis: A novel player in the pathogenesis of atherosclerosis? Cytokine 2019, 122, 2017–2019. [Google Scholar] [CrossRef]
- Moroz, O.V.; Antson, A.A.; Dodson, E.J.; Burrell, H.J.; Grist, S.J.; Lloyd, R.M.; Maitland, N.J.; Dodson, G.G.; Wilson, K.S.; Lukanidin, E.; et al. The structure of S100A12 in a hexameric form and its proposed role in receptor signalling. Acta Crystallogr. Sect. D Biol. Cristallogr. 2002, 58, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Burz, D.S.; He, W.; Bronstein, I.B.; Lednev, I.; Shekhtman, A. Hexameric calgranulin C (S100A12) binds to the receptor for advanced glycated end products (RAGE) using symmetric hydrophobic target-binding patches. J. Biol. Chem. 2007, 282, 4218–4231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokkola, R.; Andersson, Å.; Mullins, G.; Östberg, T.; Treutiger, C.J.; Arnold, B.; Nawroth, P.; Andersson, U.; Harris, R.A.; Harris, H.E. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand. J. Immunol. 2005, 61, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wautier, J.L.; Wautier, M.P.; Schmidt, A.M.; Anderson, G.M.; Hori, O.; Zoukourian, C.; Capron, L.; Chappey, O.; Yan, S.D.; Brett, J.; et al. Advanced glycation end products (AGEs) on the surface of diabetic erythrocytes bind to the vessel wall via a specific receptor inducing oxidant stress in the vasculature: A link between surface-associated AGEs and diabetic complications. Proc. Natl. Acad. Sci. USA 1994, 91, 7742–7746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, M.M.; Du Yan, S.; Stern, D.; Saraiva, M.J. Interaction of the receptor for advanced glycation end products (RAGE) with transthyretin triggers nuclear transcription factor kB (NF-kB) activation. Lab. Investig. 2000, 80, 1101–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirois, C.M.; Jin, T.; Miller, A.L.; Bertheloot, D.; Nakamura, H.; Horvath, G.L.; Mian, A.; Jiang, J.; Schrum, J.; Bossaller, L.; et al. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J. Exp. Med. 2013, 210, 2447–2463. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Kang, J.H.; Hwang, S.M.; Chung, I.Y. S100A8, S100A9 and S100A12 activate airway epithelial cells to produce MUC5AC via extracellular signal-regulated kinase and nuclear factor-κB pathways. Immunology 2015, 144, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Yan, W.X.; Cai, H.; Tedla, N.; Armishaw, C.; Di Girolamo, N.; Wang, H.W.; Hampartzoumian, T.; Simpson, J.L.; Gibson, P.G.; et al. S100A12 provokes mast cell activation: A potential amplification pathway in asthma and innate immunity. J. Allergy Clin. Immunol. 2007, 119, 106–114. [Google Scholar] [CrossRef]
- Vogl, T.; Pröpper, C.; Hartmann, M.; Strey, A.; Strupat, K.; Van Den Bos, C.; Sorg, C.; Roth, J. S100A12 is expressed exclusively by granulocytes and acts independently from MRP8 and MRP14. J. Biol. Chem. 1999, 274, 25291–25296. [Google Scholar] [CrossRef] [Green Version]
- Foell, D.; Wittkowski, H.; Ren, Z.; Turton, J.; Pang, G.; Daebritz, J.; Ehrchen, J.; Heidermann, J.; Borody, T.; Roth, J.; et al. Phagocyte-specific S100 proteins are released from affected mucosa and promote immune responses during inflammatory bowel disease. J. Pathol. J. Pathol. Soc. Great Br. Irel. 2008, 216, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Foell, D.; Kucharzik, T.; Kraft, M.; Vogl, T.; Sorg, C.; Domschke, W.; Roth, J. Neutrophil derived human S100A12 (EN-RAGE) is strongly expressed during chronic active inflammatory bowel disease. Gut 2003, 52, 847–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boon, G.J.A.M.; Day, A.S.; Mulder, C.J.; Gearry, R.B. Are faecal markers good indicators of mucosal healing in inflammatory bowel disease? World J. Gastroenterol. 2015, 21, 11469–11480. [Google Scholar] [CrossRef] [PubMed]
- Däbritz, J.; Langhorst, J.; Lügering, A.; Heidemann, J.; Mohr, M.; Wittkowski, H.; Krummenerl, T.; Foell, D. Improving relapse prediction in inflammatory bowel disease by neutrophil-derived S100A12. Inflamm. Bowel Dis. 2013, 19, 1130–1138. [Google Scholar] [CrossRef] [PubMed]
- Kopylov, U.; Rosenfeld, G.; Bressler, B.; Seidman, E. Clinical utility of fecal biomarkers for the diagnosis and management of inflammatory bowel disease. Inflamm. Bowel Dis. 2014, 20, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, T.; Langhorst, J.; Wittkowski, H.; Becker, K.; Friedrich, A.W.; Rueffer, A.; Dobos, G.J.; Roth, J.; Foell, D. Faecal S100A12 as a non-invasive marker distinguishing inflammatory bowel disease from irritable bowel syndrome. Gut 2007, 56, 1706–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judd, T.A.; Day, A.S.; Lemberg, D.A.; Turner, D.; Leach, S.T. Update of fecal markers of inflammation in inflammatory bowel disease. J. Gastroenterol. Hepatol. 2011, 26, 1493–1499. [Google Scholar] [CrossRef]
- Fagerhol, M.K.; Dale, I.; Anderson, T. Release and Quantitation of a Leucocyte Derived Protein (L1). Scand. J. Haematol. 1980, 24, 393–398. [Google Scholar] [CrossRef]
- Steinbakk, M.; Naess-Andresen, C.F.; Lingaas, E.; Dale, I.; Brandtzaeg, P.; Fagerhol, M.K. Antimicrobial actions of calcium binding leucocyte L1 protein, calprotectin. Lancet 1990, 336, 763–765. [Google Scholar] [CrossRef]
- Ikhtaire, S.; Shajib, M.S.; Reinisch, W.; Khan, W.I. Fecal calprotectin: Its scope and utility in the management of inflammatory bowel disease. J. Gastroenterol. 2016, 51, 434–446. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Xavier, R.J.; Reinecker, H.C.; Uchino, H.; Bhan, A.K.; Podolsky, D.K.; Mizoguchi, A. Colonic epithelial functional phenotype varies with type and phase of experimental colitis. Gastroenterology 2003, 125, 148–161. [Google Scholar] [CrossRef]
- Boyapati, R.K.; Rossi, A.G.; Satsangi, J.; Ho, G.T. Gut mucosal DAMPs in IBD: From mechanisms to therapeutic implications. Mucosal Immunol. 2016, 9, 567–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagerhol, M.K.; Dale, I.; Andersson, T. A radioimmunoassay for a granulocyte protein as a marker in studies on the turnover of such cells. In Biochemistry, Pathology and Genetics of Pulmonary Emphysema; Bignon, J., Scarpa, G.L., Eds.; Pergamon: Oxford, UK, 1981; pp. 273–282. ISBN 978-0-08-027379-2. [Google Scholar]
- Walsham, N.E.; Sherwood, R.A. Fecal calprotectin in inflammatory bowel disease. Clin. Exp. Gastroenterol. 2016, 9, 21–29. [Google Scholar] [PubMed] [Green Version]
- Austermann, J.; Spiekermann, C.; Roth, J. S100 proteins in rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, T. Basigin (CD147), a multifunctional transmembrane glycoprotein with various binding partners. J. Biochem. 2016, 159, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef]
- Hobbs, J.A.R.; May, R.; Tanousis, K.; McNeill, E.; Mathies, M.; Gebhardt, C.; Henderson, R.; Robinson, M.J.; Hogg, N. Myeloid Cell Function in MRP-14 (S100A9) Null Mice. Mol. Cell. Biol. 2003, 23, 2564–2576. [Google Scholar] [CrossRef] [Green Version]
- Manitz, M.-P.; Horst, B.; Seeliger, S.; Strey, A.; Skryabin, B.V.; Gunzer, M.; Frings, W.; Schönlau, F.; Roth, J.; Sorg, C.; et al. Loss of S100A9 (MRP14) Results in Reduced Interleukin-8-Induced CD11b Surface Expression, a Polarized Microfilament System, and Diminished Responsiveness to Chemoattractants In Vitro. Mol. Cell. Biol. 2003, 23, 1034–1043. [Google Scholar] [CrossRef] [Green Version]
- Lugering, N.; Stoll, R.; Schmid, K.W.; Kucharzik, T.; Stein, H.; Burmeister, G.; Sorg, C.; Domschke, W. The myeloic related protein MRP8/14 (27E10 antigen)— usefulness as a potential marker for disease activity in ulcerative colitis and putative biological function. Eur. J. Clin. Investig. 1995, 25, 659–664. [Google Scholar] [CrossRef]
- Pruenster, M.; Vogl, T.; Roth, J.; Sperandio, M. S100A8/A9: From basic science to clinical application. Pharmacol. Ther. 2016, 167, 120–131. [Google Scholar] [CrossRef]
- RØseth, A.G.; Fagerhol, M.K.; Aadland, E.; Schjønsby, H. Assessment of the Neutrophil Dominating Protein Calprotectin in Feces: A Methodologic Study. Scand. J. Gastroenterol. 1992, 27, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Lasson, A.; Stotzer, P.O.; Öhmanb, L.; Isakssonc, S.; Sapnara, M.; Strid, H. The intra-individual variability of faecal calprotectin: A prospective study in patients with active ulcerative colitis. J. Crohn’s Colitis 2015, 9, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calafat, M.; Cabré, E.; Mañosa, M.; Lobatón, T.; Marín, L.; Domènech, E. High within-day variability of fecal calprotectin levels in patients with active ulcerative colitis: What is the best timing for stool sampling? Inflamm. Bowel Dis. 2015, 21, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- Schoepfer, A.M.; Beglinger, C.; Straumann, A.; Trummler, M.; Renzulli, P.; Seibold, F. Ulcerative colitis: Correlation of the Rachmilewitz Endoscopic Activity Index with fecal calprotectin, clinical activity, C-reactive protein, and blood leukocytes. Inflamm. Bowel Dis. 2009, 15, 1851–1858. [Google Scholar] [CrossRef]
- Dulai, P.S.; Peyrin-Biroulet, L.; Danese, S.; Sands, B.E.; Dignass, A.; Turner, D.; Mantzaris, G.; Schölmerich, J.; Mary, J.Y.; Reinisch, W.; et al. Approaches to Integrating Biomarkers Into Clinical Trials and Care Pathways as Targets for the Treatment of Inflammatory Bowel Diseases. Gastroenterology 2019, 157, 1032–1043. [Google Scholar] [CrossRef]
- Manceau, H.; Chicha-Cattoir, V.; Puy, H.; Peoc’h, K. Fecal calprotectin in inflammatory bowel diseases: Update and perspectives. Clin. Chem. Lab. Med. 2017, 55, 474–483. [Google Scholar] [CrossRef]
- Gecse, K.B.; Brandse, J.F.; van Wilpe, S.; Löwenberg, M.; Ponsioen, C.; van den Brink, G.; D’Haens, G. Impact of disease location on fecal calprotectin levels in Crohn’s disease. Scand. J. Gastroenterol. 2015, 50, 841–847. [Google Scholar] [CrossRef]
- Ye, L.; Chen, W.; Chen, B.Q.; Lan, X.; Wang, S.D.; Wu, X.C.; Huang, W.; Wang, F.Y. Levels of Faecal Calprotectin and Magnetic Resonance Enterocolonography Correlate with Severity of Small Bowel Crohn’s Disease: A Retrospective Cohort Study. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Heida, A.; Knol, M.; Kobold, A.M.; Bootsman, J.; Dijkstra, G.; van Rheenen, P.F. Agreement between home-based measurement of stool Calprotectin and ELISA results for monitoring inflammatory bowel disease activity. Clin. Gastroenterol. Hepatol. 2017, 15, 1742–1749. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.C.; Huang, V.W.; Bourdages, R.; Fedorak, R.N.; Reinhard, C.; Leung, Y.; Bressler, B.; Rosenfeld, G. IBDOC Canadian user performance evaluation. Inflamm. Bowel Dis. 2019, 25, 1107–1114. [Google Scholar] [CrossRef]
- Mumolo, M.G.; Bertani, L.; Ceccarelli, L.; Laino, G.; Di Fluri, G.; Albano, E.; Tapete, G.; Costa, F. From bench to bedside: Fecal calprotectin in inflammatory bowel diseases clinical setting. World J. Gastroenterol. 2018, 24, 3681–3694. [Google Scholar] [CrossRef] [PubMed]
- Willers, M.; Ulas, T.; Völlger, L.; Vogl, T.; Heinemann, A.S.; Pirr, S.; Pagel, J.; Fehlhaber, B.; Halle, O.; Schöning, J.; et al. S100A8 and S100A9 Are Important for Postnatal Development of Gut Microbiota and Immune System in Mice and Infants. Gastroenterology 2020, 159, 2130–2145. [Google Scholar] [CrossRef] [PubMed]
- Roca, M.; Rodriguez Varela, A.; Carvajal, E.; Donat, E.; Cano, F.; Armisen, A.; Vaya, M.J.; Ekoff, H.; Hervas, D.; Rydell, N.; et al. Fecal calprotectin in healthy children aged 4–16 years. Sci. Rep. 2020, 10, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Roca, M.; Varela, A.R.; Donat, E.; Cano, F.; Hervas, D.; Armisen, A.; Vaya, M.J.; Sjölander, A.; Ribes-Koninckx, C. Fecal Calprotectin and Eosinophil-derived Neurotoxin in Healthy Children between 0 and 12 Years. J. Pediatr. Gastroenterol. Nutr. 2017, 65, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Lira-Junior, R.; Holmström, S.B.; Clark, R.; Zwicker, S.; Majster, M.; Johannsen, G.; Axtelius, B.; Åkerman, S.; Svensson, M.; Klinge, B.; et al. S100A12 Expression Is Modulated During Monocyte Differentiation and Reflects Periodontitis Severity. Front. Immunol. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Mizoguchi, A.; Bhan, A.K. Role of cytokines in the early stage of chronic coltis in TCRα-mutant mice. Lab. Investig. 1997, 76, 385–397. [Google Scholar]
- Menghini, P.; Corridoni, D.; Butto, L.F.; Osme, A.; Shivaswamy, S.; Lam, M.; Bamias, G.; Pizarro, T.T.; Rodriguez-Palacios, A.; Dinarello, C.A.; et al. Neutralization of IL-1α ameliorates Crohn’s disease-like ileitis by functional alterations of the gut microbiome. Proc. Natl. Acad. Sci. USA 2019, 116, 26717–26726. [Google Scholar] [CrossRef]
- Aschenbrenner, D.; Quaranta, M.; Banerjee, S.; Ilott, N.; Jansen, J.; Steere, B.; Chen, Y.H.; Ho, S.; Cox, K.; Arancibia-Carcamo, C.V.; et al. Deconvolution of monocyte responses in inflammatory bowel disease reveals an IL-1 cytokine network that regulates IL-23 in genetic and acquired IL-10 resistance. Gut 2020, in press. [Google Scholar] [CrossRef]
- Lopetuso, L.R.; Petito, V.; Cufino, V.; Arena, V.; Stigliano, E.; Gerardi, V.; Gaetani, G.; Poscia, A.; Amato, A.; Cammarota, G.; et al. Locally injected Infliximab ameriorates murine DSS colitis: Diffrences in serum and intestinal levels of drug between healthy and colitic mice. Dig. Liver Dis. 2013, 45, 1017–1021. [Google Scholar] [CrossRef]
- Present, D.H.; Rutgeerts, P.; Targan, S.; Hanauer, S.B.; Mayer, L.; van Hogezand, R.A.; Podolsky, D.K.; Sands, B.E.; Braakman, T.; Dewoody, K.L. Infliximab for the treatment of fistulas in patients with Crohn’s disease. N. Engl. J. Med. 1999, 340, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Aardoom, M.A.; Veereman, G.; de Ridder, L. A review on the use of anti-TNF in children and aldolescents with inflammatory bowel disease. Int. J. Mol. Sci. 2019, 20, 2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dense, S.; Vuitton, L.; Peyrin-Biroulet, L. Biologic agents for IBD: Practical insights. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Wallanch, D.; Varfolomeev, E.E.; Malinin, N.L.; Goltsev, Y.V.; Kovalenko, A.V.; Boldin, M.P. Tumor necrosis factor receptor and Fas signaling mechanisms. Ann. Rev. Immunol. 1999, 17, 331–367. [Google Scholar] [CrossRef]
- Urbano, U.C.M.; Aquirre-Gamboa, R.; Ashikov, A.; van Heeswijk, B.; Krippner-Heidenreich, A.; Tijssen, H.; Li, T.; Azevedo, V.F.; Smits, L.J.T.; Hoentjen, F. TNF-α-induced protein 3 (TNFAIP3)/A20 acts as a master switch in aTNF-α blockade-driven IL-17A expression. J. Allergy Clin. Immunol. 2018, 142, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizoguchi, E.; Mizoguchi, A.; Takedatsu, H.; Cario, E.; de Jong, Y.P.; Ooi, C.J.; Xavier, R.J.; Terhorst, C.; Podolsky, D.K.; Bhan, A.K. Role of tumor necrosis factor receptor 2 (TNFR2) in colonic epithelial hyperplasia and chronic intestinal inflammation in mice. Gastroenterology 2002, 122, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, G.C.; Polk, D.B. Tumor necrosis factor α regulates proliferation in a mouse intestinal cell line. Gastroenterology 1997, 112, 1231–1240. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Hachiya, Y.; Kawada, M.; Nagatani, K.; Ogawa, A.; Sugimoto, K.; Mizoguchi, A.; Podolsky, D.K. TNF receptor type I-dependent activation of innate responses to reduce intestinal damage-associated mortality. Gastroenterology 2008, 134, 470–480. [Google Scholar] [CrossRef]
- Spoettl, T.; Hausmann, M.; Klebl, F.; Dirmeir, A.; Klump, B.; Hoffmann, J.; Herfarth, H.; Timmer, A.; Rogler, G. Serum soluble TNF receptor I and II levels correlate with disease activity in IBD patients. Inflamm. Bowel Dis. 2007, 13, 727–732. [Google Scholar] [CrossRef]
- Low, D.; DeGruttola, A.K.; Poltrak, A.; Mizoguchi, A.; Mino-Kenudson, M.; Mizoguchi, E. High endogenous expression of chitinase 3-like 1 and excessive epithelial proliferation with colonic tumor formation in MOLF/EiJ mice. PLoS ONE 2015, 10, 0139149. [Google Scholar] [CrossRef] [Green Version]
| References | Age | Disease Type and Number of Enrolled Patients | Chitinase 3-like 1 Levels | |
|---|---|---|---|---|
| Feces | ||||
| Aomatsu et al. (2011) [31] | P | UC (n = 94) CD (n = 87) HC (n = 56) | Active UC | 366.6 ng/g (median) |
| Active CD | 632.7 ng/g | |||
| Inactive UC | 15.8 ng/g | |||
| Inactive CD | 18.4 ng/g | |||
| HC | 2.2 ng/g | |||
| Serum | ||||
| Koutroubakis et al. (2002) [32] | A | UC (n = 94) | UC | 102.6 ± 82.7 ng/mL |
| CD (n = 85) | CD | 112.2 ± 83.7 ng/mL | ||
| HC (n = 70) | HC | 64.1 ± 21.4 ng/mL | ||
| Non-IBD (n = 23) | Non-IBD | 77.8 ± 23.1 ng/mL | ||
| Erzin et al. (2007) [33] | A | CD (n = 41) HC (n = 46) | CD | 105.69 ± 88.08 ng/mL |
| CD with stricture | 167.60 ± 119.3 ng/mL | |||
| CD w/o stricture | 80.12 ± 56.38 ng/mL | |||
| HC | 44.92 ± 24.89 ng/mL | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizoguchi, E.; Subramaniam, R.; Okada, T.; Mizoguchi, A. A Review of Selected IBD Biomarkers: From Animal Models to Bedside. Diagnostics 2021, 11, 207. https://doi.org/10.3390/diagnostics11020207
Mizoguchi E, Subramaniam R, Okada T, Mizoguchi A. A Review of Selected IBD Biomarkers: From Animal Models to Bedside. Diagnostics. 2021; 11(2):207. https://doi.org/10.3390/diagnostics11020207
Chicago/Turabian StyleMizoguchi, Emiko, Renuka Subramaniam, Toshiyuki Okada, and Atsushi Mizoguchi. 2021. "A Review of Selected IBD Biomarkers: From Animal Models to Bedside" Diagnostics 11, no. 2: 207. https://doi.org/10.3390/diagnostics11020207
APA StyleMizoguchi, E., Subramaniam, R., Okada, T., & Mizoguchi, A. (2021). A Review of Selected IBD Biomarkers: From Animal Models to Bedside. Diagnostics, 11(2), 207. https://doi.org/10.3390/diagnostics11020207