
The Rapidly Expanding Group of RB1-Deleted Soft Tissue Tumors: An Updated Review


Abstract
:1. Introduction
2. Review
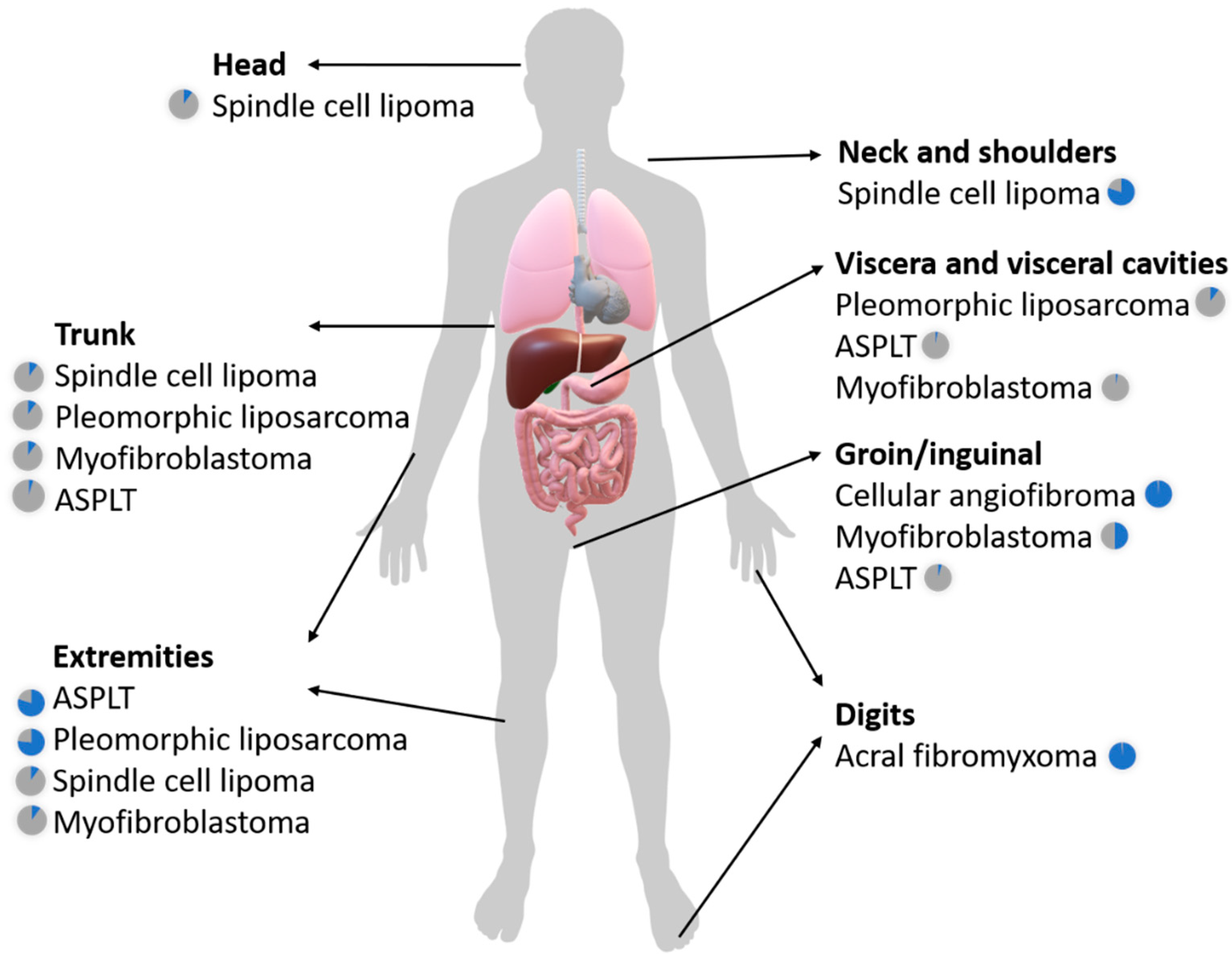
2.1. The Broad Clinicopathologic Spectrum of RB1-Deleted Mesenchymal Neoplasms
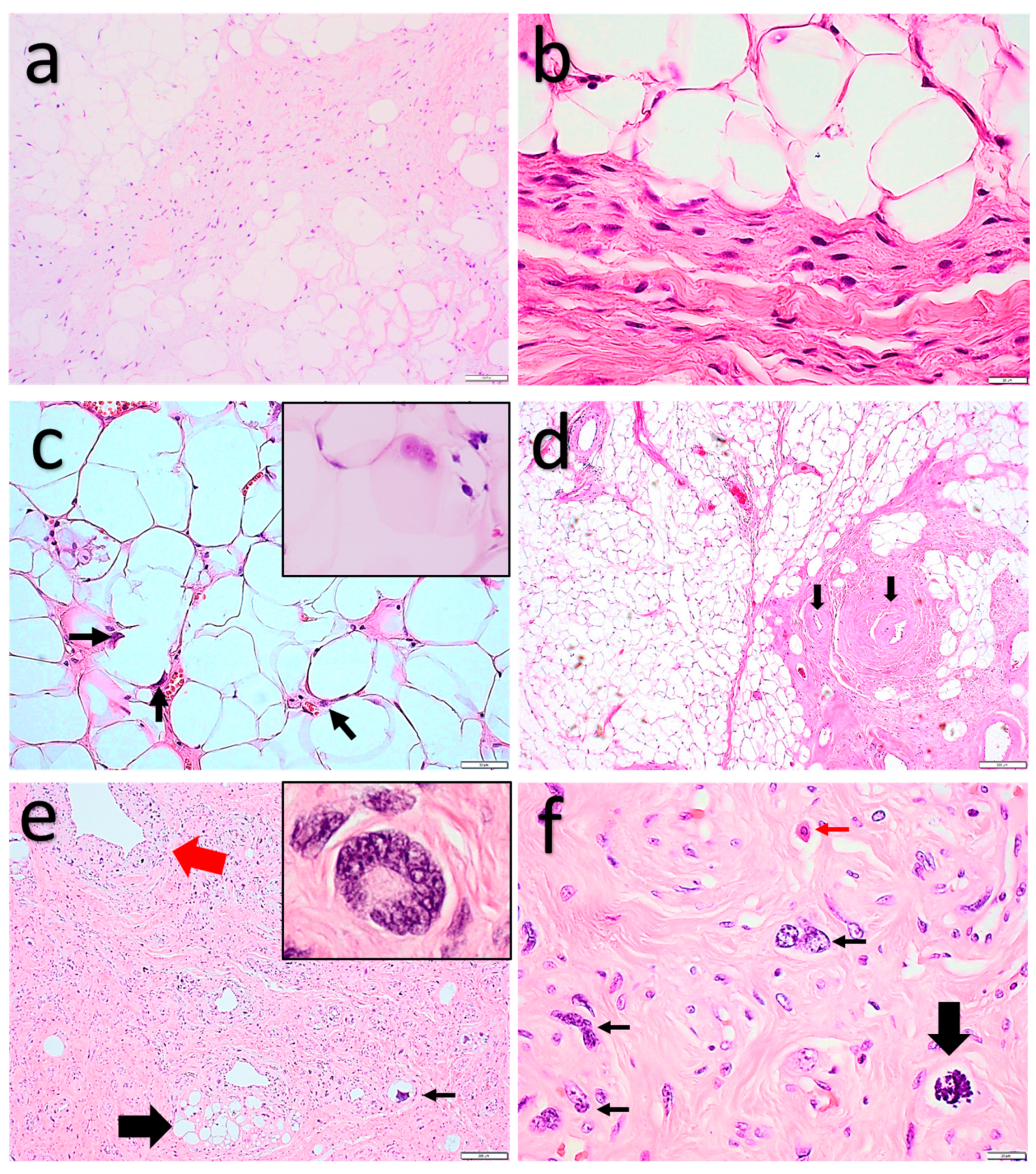
2.1.1. Spindle Cell Lipoma and Pleomorphic Lipoma
2.1.2. Atypical Spindle Cell/Pleomorphic Lipomatous Tumor
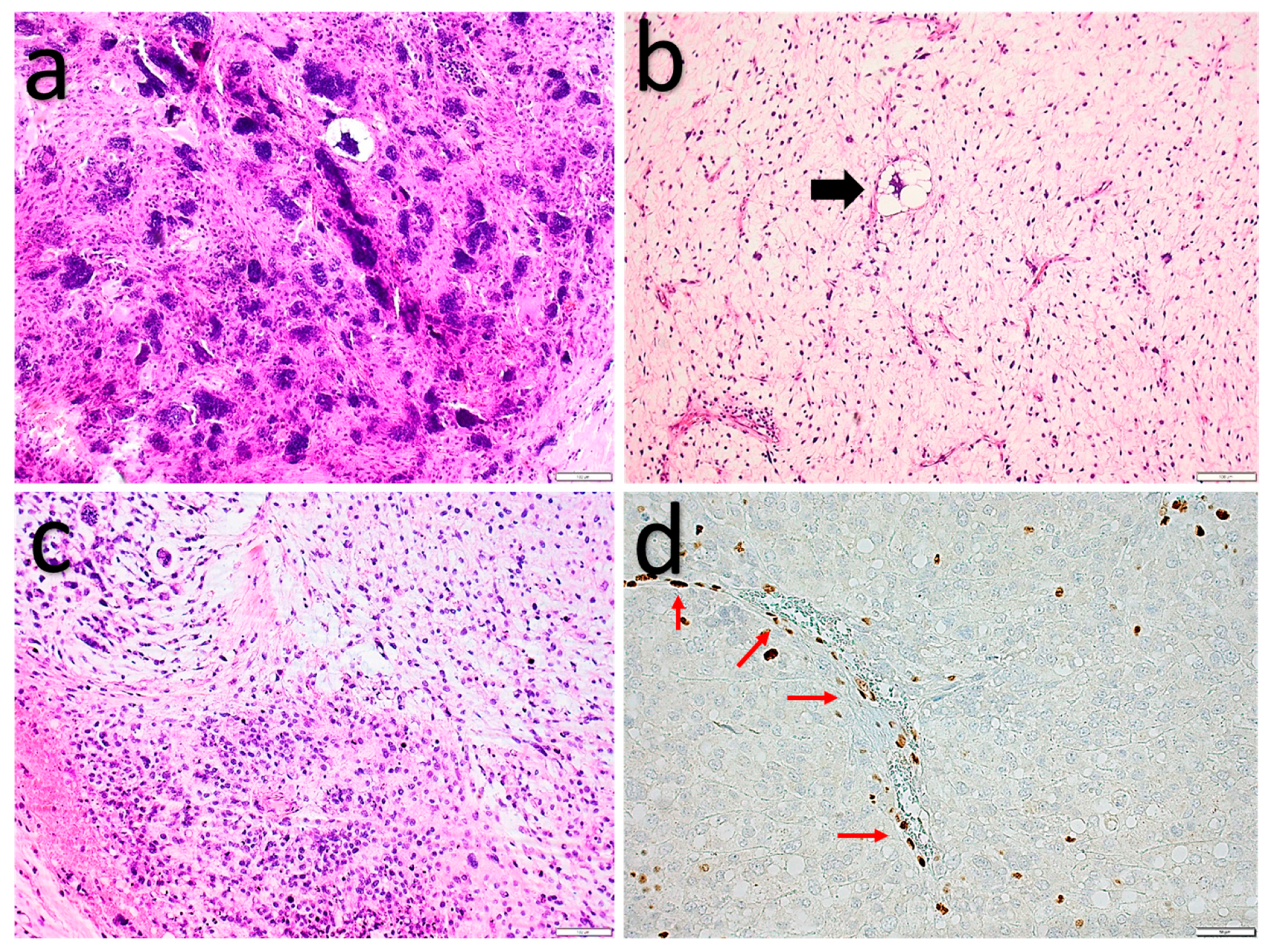
2.1.3. Pleomorphic Liposarcoma
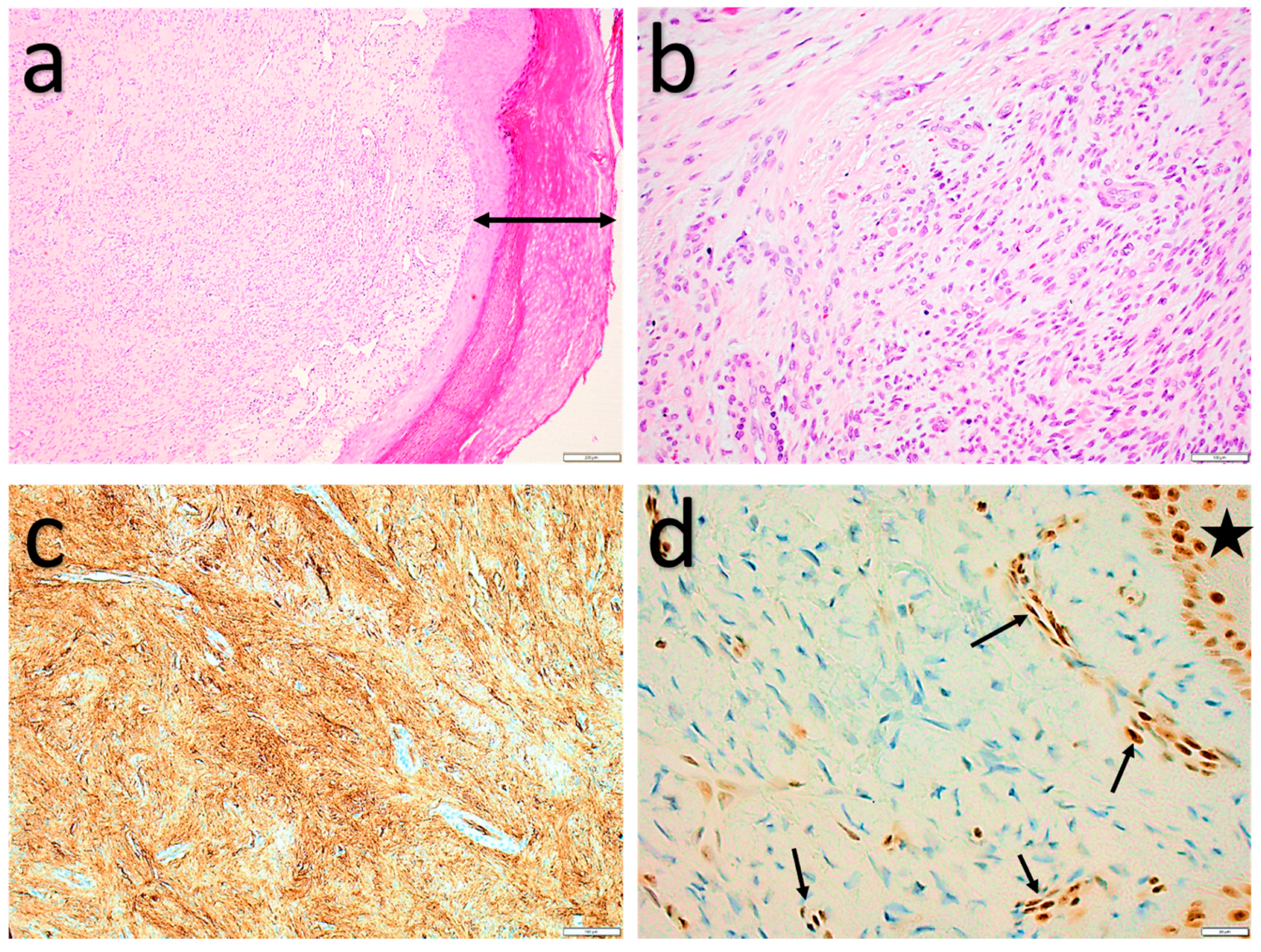
2.1.4. Myofibroblastoma (of Soft Tissue)
2.1.5. Cellular angiofibroma
2.1.6. Acral Fibromyxoma
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Delattre, O.; Zucman, J.; Melot, T.; Garau, X.S.; Zucker, J.-M.; Lenoir, G.M.; Ambros, P.F.; Sheer, D.; Turc-Carel, C.; Triche, T.J.; et al. The ewing family of tumors—A subgroup of small-round-cell tumors defined by specific chimeric transcripts. N. Engl. J. Med. 1994, 331, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Karakousis, C.P.; Cin, P.D.; Turc-Carel, C.; Limon, J.; Sandberg, A.A. Chromosomal changes in soft-tissue sarcomas: A new diagnostic parameter. Arch. Surg. 1987, 122, 1257–1260. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef] [PubMed]
- WHO. Classification of Tumours of Soft Tissue and Bone, 5th ed.; IARC Press: Lyon, France, 2020. [Google Scholar]
- Knudson, A.G. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, J.L.; Polski, A.; Cavenee, W.K.; Dryja, T.P.; Murphree, A.L.; Gallie, B.L. The RB1 Story: Characterization and cloning of the first tumor suppressor Gene. Genes 2019, 10, 879. [Google Scholar] [CrossRef] [Green Version]
- Dyson, N.J. RB1: A prototype tumor suppressor and an enigma. Genes Dev. 2016, 30, 1492–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedesco, N.S.; Henshaw, R.M. Unplanned Resection of Sarcoma. J. Am. Acad. Orthop. Surg. 2016, 24, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, R.; D’Anneo, A.; Tesoriere, G.; Vento, R. RB1 in cancer: Different mechanisms of RB1 inactivation and alterations of pRb pathway in tumorigenesis. J. Cell. Physiol. 2013, 228, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Takahira, T.; Oda, Y.; Tamiya, S.; Yamamoto, H.; Kobayashi, C.; Izumi, T.; Ito, K.; Iwamoto, Y.; Kobayashi, C.; Izumi, T.; et al. Alterations of the RB1 gene in dedifferentiated liposarcoma. Mod. Pathol. 2005, 18, 1461–1470. [Google Scholar] [CrossRef] [Green Version]
- Schneider-Stock, R.; Boltze, C.; Jaeger, V.; Stumm, M.; Seiler, C.; Rys, J.; Schütze, K.; Roessner, A. Significance of loss of heterozygsisty of the RB1 gene during tumour progression in well-differentiated liposarcomas. J. Pathol. 2002, 197, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Z.; Okada, T.; Kim, Y.M.; Agaram, N.P.; Sanchez-Vega, F.; Shen, Y.; Tsubokawa, N.; Rios, J.; Martin, A.S.; Dickson, M.A.; et al. Rb and p53-deficient myxofibrosarcoma and undif-ferentiated pleomorphic sarcoma require Skp2 for survival. Cancer Res. 2020, 80, 2461–2471. [Google Scholar] [CrossRef] [Green Version]
- Billings, S.; Ud Din, N. Spindle cell lipoma and pleomorphic lipoma. In WHO Classification of Soft Tissue and Bone Tumours; IARC Press: Lyon, France, 2020. [Google Scholar]
- Fletcher, C.; Martin-Bates, E. Spindle cell lipoma: A clinicopathological study with some original observations. Histopathology 1987, 11, 803–817. [Google Scholar] [CrossRef]
- Van Treeck, B.J.; Fritchie, K.J. Updates in spindle cell/pleomorphic lipomas. Semin. Diagn. Pathol. 2019, 36, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Ud Din, N.; Zhang, P.; Sukov, W.R.; Sattler, C.A.; Jenkins, S.M.; Doyle, L.A.; Folpe, A.L.; Fritchie, K.J. Spindle Cell Lipomas Arising at Atypical Locations. Am. J. Clin. Pathol. 2016, 146, 487–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, C.A.; Mentzel, T.; Kutzner, H.; Fletcher, C.D. Intradermal spindle cell/pleomorphic lipoma: A distinct subset. Am. J. Dermatopathol. 2000, 22, 496–502. [Google Scholar] [CrossRef]
- Harvell, J.D. Multiple spindle cell lipomas and dermatofibrosarcoma protuberans within a single patient: Evidence for a common neoplastic process of interstitial dendritic cells? J. Am. Acad. Dermatol. 2003, 48, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Cheah, A.; Billings, S.; Goldblum, J.; Hornick, J.; Uddin, N.; Rubin, B. Spindle cell/pleomorphic lipomas of the face: An under-recognized diagnosis. Histopathology 2015, 66, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Fanburg-Smith, J.C.; Devaney, K.O.; Miettinen, M.; Weiss, S.W. Multiple spindle cell lipomas: A report of 7 familial and 11 nonfamilial cases. Am. J. Surg. Pathol. 1998, 22, 40–48. [Google Scholar] [CrossRef]
- Enzinger, F.M.; Harvey, D.A. Spindle cell lipoma. Cancer 1975, 36, 1852–1859. [Google Scholar] [CrossRef]
- Shmookler, B.M.; Enzinger, F.M. Pleomorphic lipoma: A benign tumor simulating liposarcoma. A clinicopathologic analysis of 48 cases. Cancer 1981, 47, 126–133. [Google Scholar] [CrossRef]
- Michal, M.; Kazakov, D.V.; Hadravsky, L.; Michalova, K.; Grossmann, P.; Steiner, P.; Vanecek, T.; Renda, V.; Suster, S.; Michal, M. Lipoblasts in spindle cell and pleomorphic lipomas: A close scrutiny. Hum. Pathol. 2017, 65, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Mariño-Enríquez, A.; Fletcher, C.D.M.; Hornick, J.L. Loss of retinoblastoma protein expression in spindle cell/pleomorphic lipomas and cytogenetically related tumors: An immunohistochemical study with diagnostic implications. Am. J. Surg. Pathol. 2012, 36, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Mentzel, T.; Rütten, A.; Hantschke, M.; Hornick, J.L.; Brenn, T. S-100 protein expressing spindle cells in spindle cell lipoma: A diagnostic pitfall. Virchows Arch. 2016, 469, 435–438. [Google Scholar] [CrossRef]
- Lecoutere, E.; Creytens, D. Atypical spindle cell/pleomorphic lipomatous tumor. Histol. Histopathol. 2020, 35, 18210. [Google Scholar]
- Creytens, D.; Mariño-Enriquez, A. Atypical spindle cell/pleomorphic lipomatous tumour. In WHO Classification of Soft Tissue and Bone Tumours; IARC Press: Lyon, France, 2020. [Google Scholar]
- Dei Tos, A.P.; Mentzel, T.; Newman, P.L.; Fletcher, C.D.M. Spindle cell liposarcoma, a hitherto unrecognized variant of liposarcoma. Analysis of six cases. Am. J. Surg. Pathol. 1994, 18, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Creytens, D.; Van Gorp, J.; Savola, S.; Ferdinande, L.; Mentzel, T.; Libbrecht, L. Atypical spindle cell lipoma: A clinicopathologic, immunohistochemical, and molecular study emphasizing its relationship to classical spindle cell lipoma. Virchows Arch. 2014, 465, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Michal, M.; Agaimy, A.; Contreras, A.L.; Svajdler, M.; Kazakov, D.V.; Steiner, P.; Grossmann, P.; Martinek, P.; Hadravsky, L.; Michalova, K.; et al. Dysplastic lipoma: A distinctive atypical lipomatous neoplasm with anisocytosis, focal nuclear atypia, p53 overexpression, and a lack of MDM2 gene amplification by FISH.; A report of 66 cases demonstrating occasional multifocality and a rare association with retinoblastoma. Am. J. Surg. Pathol. 2018, 42, 1530–1540. [Google Scholar] [PubMed]
- Mentzel, T.; Palmedo, G.; Kuhnen, C. Well-differentiated spindle cell liposarcoma (‘atypical spindle cell lipomatous tumor’) does not belong to the spectrum of atypical lipomatous tumor but has a close relationship to spindle cell lipoma: Clinicopathologic, immunohistochemical, and molecular analysis of six cases. Mod. Pathol. 2010, 23, 729–736. [Google Scholar] [CrossRef] [Green Version]
- Agaimy, A. Anisometric cell lipoma: Insight from a case series and review of the literature on adipocytic neoplasms in survivors of retinoblastoma suggest a role for RB1 loss and possible relationship to fat-predominant (“fat-only”) spindle cell lipoma. Ann. Diagn. Pathol. 2017, 29, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Mariño-Enriquez, A.; Nascimento, A.F.; Ligon, A.H.; Liang, C.; Fletcher, C.D.M. Atypical spindle cell lipomatous tumor: Clinico-pathologic characterization of 232 cases demonstrating a morphologic spectrum. Am. J. Surg. Pathol. 2017, 41, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Creytens, D.; Mentzel, T.; Ferdinande, L.; Lecoutere, E.; van Gorp, J.; Atanesyan, L.; de Groot, K.; Savola, S.; Van Roy, N.; Dorpe, J.V.; et al. ‘Atypical’ pleomorphic lipomatous tumor: A clinicopathologic, immunohistochemical and molecular study of 21 cases, emphasizing its relationship to atypical spindle cell lipomatous tumor and suggesting a morphologic spectrum (atypical spindle cell/pleomorphic lipomatous tumor). Am. J. Surg. Pathol. 2017, 41, 1443–1455. [Google Scholar] [PubMed]
- Pedeutour, F.; Montgomery, E. Pleomorphic liposarcoma. In WHO Classification of Soft Tissue and Bone Tumours; IARC Press: Lyon, France, 2020. [Google Scholar]
- Alaggio, R.; Coffin, C.M.; Weiss, S.W.; Bridge, J.A.; Issakov, J.; Oliveira, A.M.; Folpe, A.L. Liposarcomas in young patients: A study of 82 cases occurring in patients younger than 22 years of age. Am. J. Surg. Pathol. 2009, 33, 645–658. [Google Scholar] [CrossRef]
- Gebhard, S.; Coindre, J.-M.; Michels, J.-J.; Terrier, P.; Bertrand, G.; Trassard, M.; Taylor, S.; Château, M.-C.; Marquès, B.; Picot, V.; et al. Pleomorphic liposarcoma: Clinicopathologic, immunohistochemical, and follow-up analysis of 63 cases: A study from the French Federation of Cancer Centers Sarcoma Group. Am. J. Surg. Pathol. 2002, 26, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Hornick, J.L.; Bosenberg, M.W.; Mentzel, T.; McMenamin, M.E.; Oliveira, A.M.; Fletcher, C.D.M. Pleomorphic liposarcoma: Clinico-pathologic analysis of 57 cases. Am. J. Surg. Pathol. 2004, 28, 1257–1267. [Google Scholar] [CrossRef] [PubMed]
- Dei Tos, A.P. Liposarcomas: Diagnostic pitfalls and new insights. Histopathology 2014, 64, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Bellver, J.L.; López, J.; Macías, E.; Alegría-Landa, V.; Gimeno, I.; Pérez-Plaza, A.; Kutzner, H.; Requena, L. Primary dermal pleomorphic lipo-sarcoma: Utility of adipophilin and MDM2/CDK4 immunostainings. J. Cutan. Pathol. 2017, 44, 283–288. [Google Scholar] [CrossRef]
- Wang, L.; Ren, W.; Zhou, X.; Sheng, W.; Wang, J. Pleomorphic liposarcoma: A clinicopathological, immunohistochemical and molecular cytogenetic study of 32 additional cases. Pathol. Int. 2013, 63, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.J.; Jo, V.Y. Pleomorphic liposarcoma: Updates and current differential diagnosis. Semin. Diagn. Pathol. 2019, 36, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Hofvander, J.; Jo, V.Y.; Ghanei, I.; Gisselsson, D.; Mårtensson, E.; Mertens, F. Comprehensive genetic analysis of a paediatric pleo-morphic myxoid liposarcoma reveals near-haploidization and loss of the RB1 gene. Histopathology 2016, 69, 141–147. [Google Scholar] [CrossRef]
- Creytens, D.; Van Gorp, J.; Ferdinande, L.; Van Roy, N.; Libbrecht, L. Array-based comparative genomic hybridisation analysis of a pleomorphic myxoid liposarcoma. J. Clin. Pathol. 2014, 67, 834–835. [Google Scholar] [CrossRef] [PubMed]
- Magro, G.; Howitt, B.; Liegl-Atzwanger, B.; McMenamin, M. Myofibroblastoma. In WHO Classification of Soft Tissue and Bone Tumours; IARC Press: Lyon, France, 2020. [Google Scholar]
- Howitt, B.E.; Fletcher, C.D.M. Mammary-type myofibroblastoma: Clinicopathologic characterization in a series of 143 cases. Am. J. Surg. Pathol. 2016, 40, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Magro, G.; Righi, A.; Casorzo, L.; Antonietta, T.; Salvatorelli, L.; Kacerovská, D.; Kazakov, D.; Michal, M. Mammary and vaginal myofibroblastomas are genetically related lesions: Fluorescence in situ hybridization analysis shows deletion of 13q14 region. Hum. Pathol. 2012, 43, 1887–1893. [Google Scholar] [CrossRef]
- Iwasa, Y.; Fletcher, C.D.M.; Flucke, U. Cellular angiofibroma. In WHO Classification of Soft Tissue and Bone Tumours; ARC Press: Lyon, France, 2020. [Google Scholar]
- Iwasa, Y.; Fletcher, C.D.M. Cellular angiofibroma: Clinicopathologic and immunohistochemical analysis of 51 cases. Am. J. Surg. Pathol. 2004, 28, 1426–1435. [Google Scholar] [CrossRef]
- Laskin, W.B.; Fetsch, J.F.; Mostofi, F.K. Angiomyofibroblastomalike tumor of the male genital tract: Analysis of 11 cases with com-parison to female angiomyofibroblastoma and spindle cell lipoma. Am. J. Surg. Pathol. 1998, 22, 6–16. [Google Scholar] [CrossRef]
- McCluggage, W.G.; Ganesan, R.; Hirschowitz, L.; Rollason, T.P. Cellular angiofibroma and related fibromatous lesions of the vulva: Report of a series of cases with a morphological spectrum wider than previously described. Histopathol. 2004, 45, 360–368. [Google Scholar] [CrossRef]
- Chen, E.; Fletcher, C.D.M. Cellular angiofibroma with atypia or sarcomatous transformation: Clinicopathologic analysis of 13 cases. Am. J. Surg. Pathol. 2010, 34, 707–714. [Google Scholar] [CrossRef]
- Flucke, U.; Van Krieken, J.H.J.M.; Mentzel, T. Cellular angiofibroma: Analysis of 25 cases emphasizing its relationship to spindle cell lipoma and mammary-type myofibroblastoma. Mod. Pathol. 2011, 24, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Brenn, T.; Agaimy, A.; Hollmann, T. Acral fibromyxoma. In WHO Classification of Soft Tissue and Bone Tumours; IARC Press: Lyon, France, 2020. [Google Scholar]
- Hollmann, T.J.; Bovée, J.V.M.G.; Fletcher, C.D.M. Digital fibromyxoma (superficial acral fibromyxoma): A detailed characterization of 124 cases. Am. J. Surg. Pathol. 2012, 36, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Fetsch, J.F.; Laskin, W.B.; Miettinen, M. Superficial acral fibromyxoma: A clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes. Hum. Pathol. 2001, 32, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Agaimy, A.; Michal, M.; Giedl, J.; Hadravsky, L. Superficial acral fibromyxoma: Clinicopathological, immunohistochemical, and molecular study of 11 cases highlighting frequent Rb1 loss/deletions. Hum. Pathol. 2017, 60, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Mentzel, T.; Schärer, L.; Kazakov, D.V.; Michal, M. Myxoid Dermatofibrosarcoma protuberans: Clinicopathologic, immunohistochemical, and molecular analysis of eight cases. Am. J. Dermatopathol. 2007, 29, 443–448. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Types | SCL/PL | ASPLT | PLS | MFB | CAF | AFM |
|---|---|---|---|---|---|---|
| Sex ratio (M:F) | 10:1 | 1.5–3:1 | 1:1 | 2:1 | 1:1 (men older than women) | 1.2–2:1 |
| Recurrence | Very rare local recurrence | 10–15% local recurrence if incompletely excised | 50% local recurrence 50% metastases 60% 5 y OS | Very rare local recurrence | Very rare local recurrence, even with sarcomatous change | 1/4th of lesion locally recur after incomplete excision; re-excision is curative. |
| Macroscopic appearance | Circumscribed, encapsulated Slow-growing Size < 5 cm | Vaguely lobular, unencapsulated, ill-defined margins Gradually enlarging Median size 5–8.5 cm (up to 28 cm) | Poorly circumscribed, infiltrative Fast growing Large (usually >5 cm) | Well circumscribed, nodular Rarely infiltrative Very slow-growing Median size 6.5 cm (up to 22 cm) | Well circumscribed, nodular, sometimes with pseudocapsule Rarely infiltrative Slow-growing Median size 2–3 cm in women and 6–7 cm in men (up to 25 cm) | Polypoid/verrucous, lobulated, sometimes infiltrative Slow-growing Recurrent lesions are always well-circumscribed. Size 1–2 cm |
| Histology Background | Variable fibromyxoid stroma, ropey collagen, mast cells | Variable fibromyxoid stroma, ropey collagen, mast cells, perivascular lymphocytic infiltrate | Highly cellular with scant fibromyxoid stroma | Fibrous stroma with variable hyalinized areas, ropey collagen, mast cells, perivascular lymphocytic infiltrate | Variably edematous and fibrous stroma, sometimes myxoid change, mast cells, perivascular lymphocytic infiltrate | Variably myxoid stroma with fibrous areas, mast cells |
| Cellularity | Low | Highly variable | High | Low to moderate | Moderate to high | Low |
| Cell type and growth pattern | Bland spindle cells, mature adipocytes, lipoblasts, scattered bizarre cells | Variably atypical spindle cells, atypical adipocytes, (pleomorphic) lipoblasts, (bizarre) floretlike giant cells | Highly atypical spindle cells, highly atypical epithelioid cells (25%), pleomorphic lipoblast (var), giant and multinucleated tumor cells | Bland spindle cells with fascicular growth, intermixed mature adipocytes, rare degenerative-type atypia, rare epithelioid cells | Short, bland spindle cells, sometimes nuclear palisading, and fascicular growth Rarely pleomorphic spindle cells, lipoblasts, and sarcomatous change | Bland spindled, ovoid or stellate cells, vague fascicular or storiform growth, dispersed small multinucleated stromal cells |
| Mitoses/necrosis | Very rare mitoses No necrosis | Often present but scarce mitoses No necrosis | High mitotic count Frequent necrosis | Very rare mitoses No necrosis | Scarce mitoses, rarely increased No necrosis | Very rare mitoses No necrosis |
| Vasculature | Low density, hyalinized | Low to moderate density, hyalinized | High vascularity, sometimes chicken wire-like vasculature | Low density, sometimes hyalinized | High vascularity, variable diameter, prominent hyalinization | Low density, inconspicuous |
| Heterologous differentiation | Cartilaginous, osseous, EMH | Smooth muscle, cartilaginous, osseous | Highly variable | Absent | Absent | Cartilaginous |
| IHC | CD34(+) S100(+) in adipocytes Desmin(-) PR rare Rb loss | CD34(+) S100(+) in adipocytes and S100 (var) in spindle cells Desmin (var) Rb loss (60–100%) | CD34 (-/var) S100 (+) in lipoblasts Desmin (-/var) Rb loss (±50%) | CD34 (+) S100 (-), except adipocytes Desmin (+) SMA (30%) Rb loss (90%) | CD34 (var) S100 (-) SMA/Desmin (rare) ER/PR (+) Rb loss | CD34 (+) S100 (-) Desmin (-) SMA (var) EMA (var) Rb loss (>90%) |
| Molecular | Heterozygous 13q14 deletion | Heterozygous 13q14 deletion (60-70%) | CNVs (loss > gain) Rearrangements Heterozygous 13q14 deletion (50%) | Heterozygous 13q14 deletion | Heterozygous 13q14 deletion | Heterozygous 13q14 deletion |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Libbrecht, S.; Van Dorpe, J.; Creytens, D. The Rapidly Expanding Group of RB1-Deleted Soft Tissue Tumors: An Updated Review. Diagnostics 2021, 11, 430. https://doi.org/10.3390/diagnostics11030430
Libbrecht S, Van Dorpe J, Creytens D. The Rapidly Expanding Group of RB1-Deleted Soft Tissue Tumors: An Updated Review. Diagnostics. 2021; 11(3):430. https://doi.org/10.3390/diagnostics11030430
Chicago/Turabian StyleLibbrecht, Sasha, Jo Van Dorpe, and David Creytens. 2021. "The Rapidly Expanding Group of RB1-Deleted Soft Tissue Tumors: An Updated Review" Diagnostics 11, no. 3: 430. https://doi.org/10.3390/diagnostics11030430
APA StyleLibbrecht, S., Van Dorpe, J., & Creytens, D. (2021). The Rapidly Expanding Group of RB1-Deleted Soft Tissue Tumors: An Updated Review. Diagnostics, 11(3), 430. https://doi.org/10.3390/diagnostics11030430