
3D Printed Personalized External Aortic Root Model in Marfan Syndrome with Isolated Sinus of Valsalva Aneurysm Caused by a Novel Pathogenic FBN1 p.Gly1127Cys Variant


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
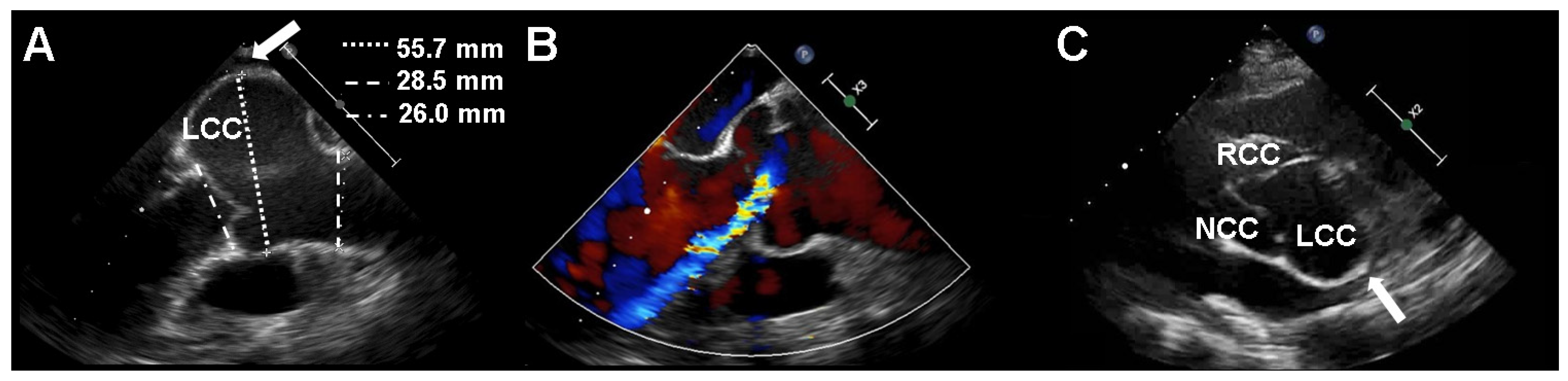
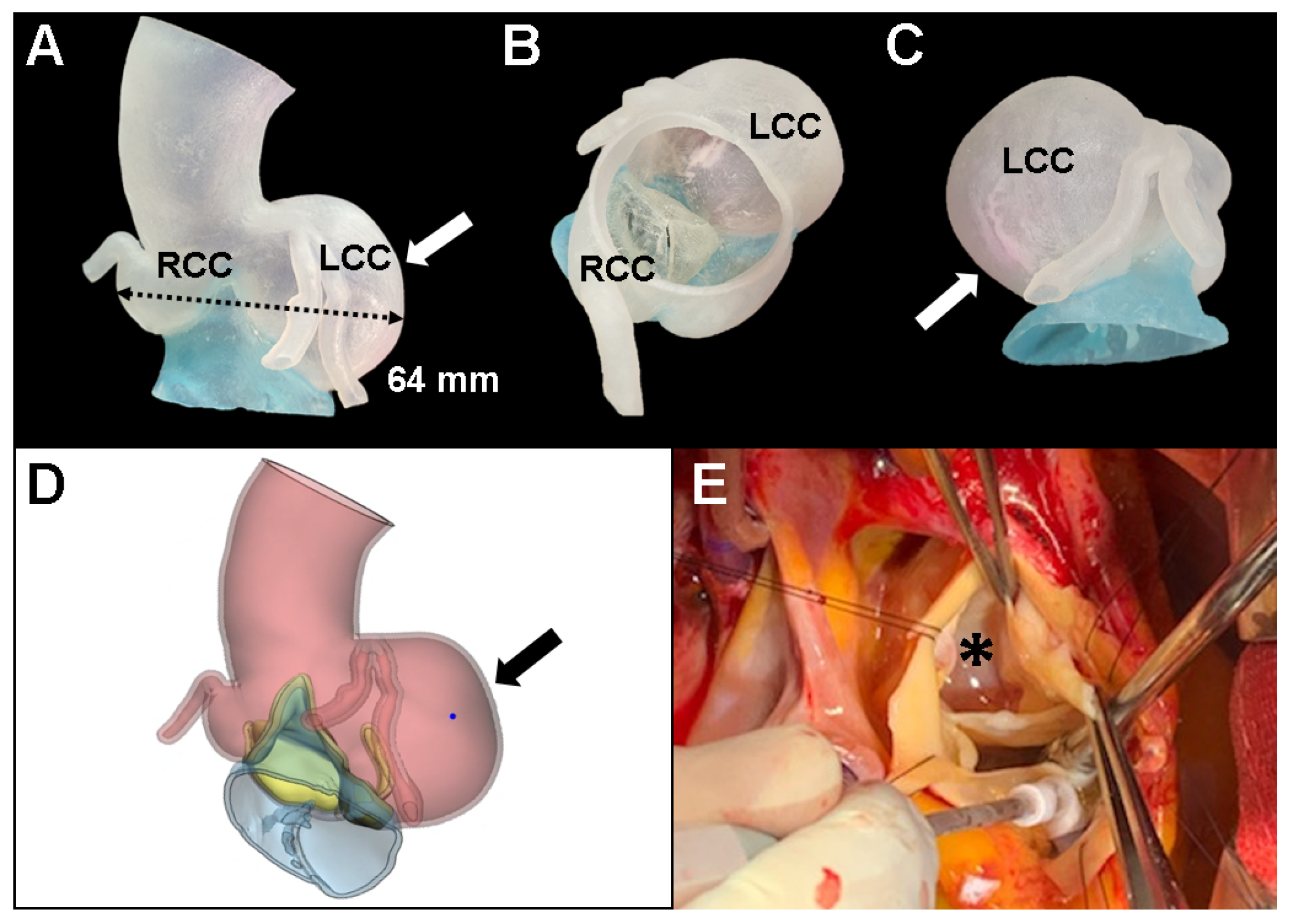
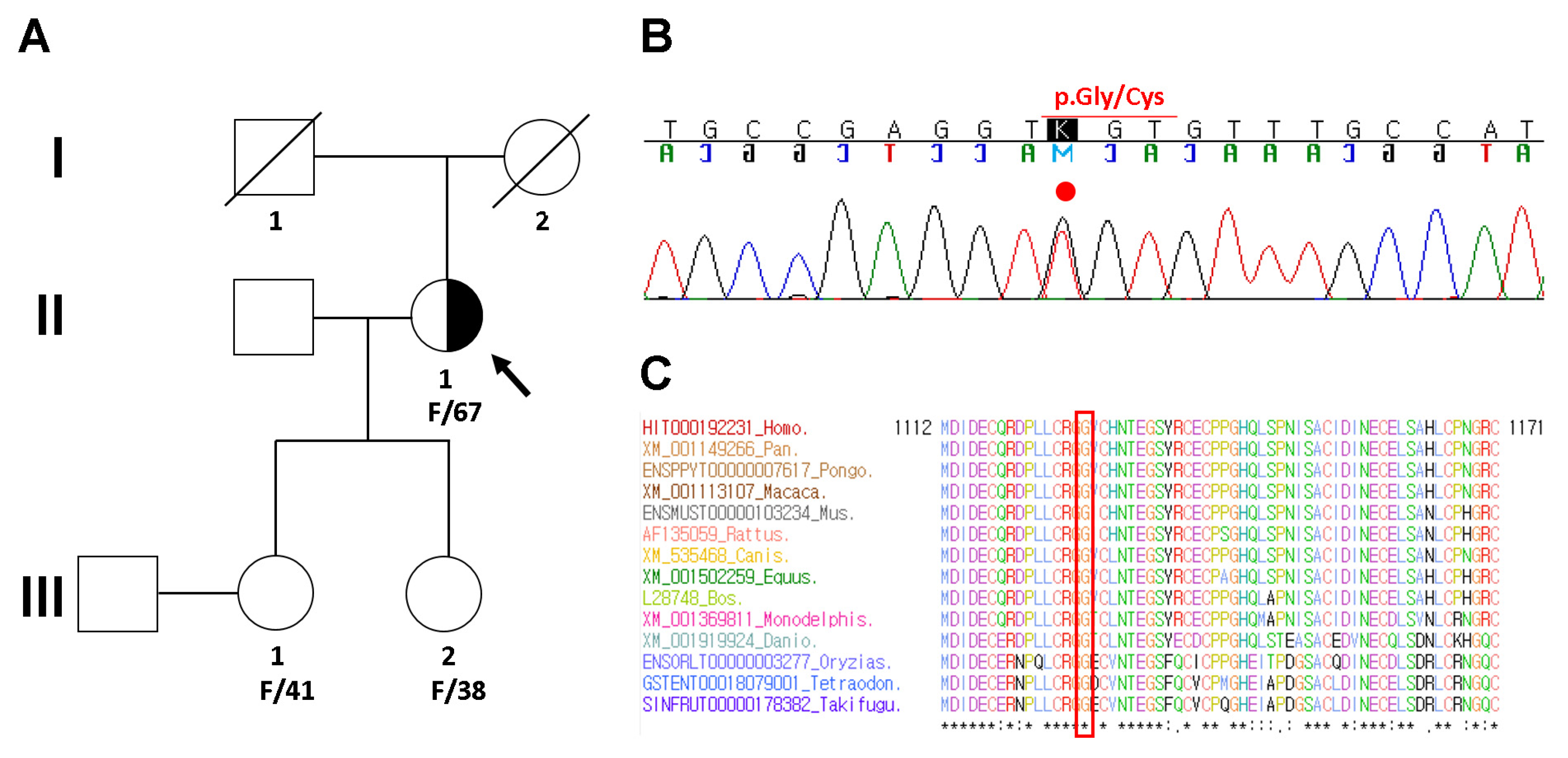
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Groth, K.A.; Hove, H.; Kyhl, K.; Folkestad, L.; Gaustadnes, M.; Vejlstrup, N.; Stochholm, K.; Østergaard, J.R.; Andersen, N.H.; Gravholt, C.H. Prevalence, incidence, and age at diagnosis in Marfan Syndrome. Orphanet. J. Rare Dis. 2015, 10, 153. [Google Scholar] [CrossRef] [Green Version]
- Sakai, L.Y.; Keene, D.R.; Renard, M.; De Backer, J. FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders. Gene 2016, 591, 279–291. [Google Scholar] [CrossRef]
- Takeda, N.; Inuzuka, R.; Maemura, S.; Morita, H.; Nawata, K.; Fujita, D.; Taniguchi, Y.; Yamauchi, H.; Yagi, H.; Kato, M.; et al. Impact of Pathogenic FBN1 Variant Types on the Progression of Aortic Disease in Patients With Marfan Syndrome. Circ. Genom. Precis Med. 2018, 11, e002058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Kodolitsch, Y.; De Backer, J.; Schüler, H.; Bannas, P.; Behzadi, C.; Bernhardt, A.M.; Hillebrand, M.; Fuisting, B.; Sheikhzadeh, S.; Rybczynski, M.; et al. Perspectives on the revised Ghent criteria for the diagnosis of Marfan syndrome. Appl. Clin. Genet. 2015, 8, 137–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franken, R.; Groenink, M.; de Waard, V.; Feenstra, H.M.; Scholte, A.J.; van den Berg, M.P.; Pals, G.; Zwinderman, A.H.; Timmermans, J.; Mulder, B.J. Genotype impacts survival in Marfan syndrome. Eur. Heart J. 2016, 37, 3285–3290. [Google Scholar] [CrossRef]
- Groth, K.A.; Stochholm, K.; Hove, H.; Andersen, N.H.; Gravholt, C.H. Causes of Mortality in the Marfan Syndrome(from a Nationwide Register Study). Am. J. Cardiol. 2018, 122, 1231–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, M.J.; Devereux, R.B.; Preiss, L.R.; Asch, F.M.; Eagle, K.A.; Holmes, K.W.; LeMaire, S.A.; Maslen, C.L.; Milewicz, D.M.; Morris, S.A.; et al. Associations of Age and Sex With Marfan Phenotype: The National Heart, Lung, and Blood Institute GenTAC (Genetically Triggered Thoracic Aortic Aneurysms and Cardiovascular Conditions) Registry. Circ. Cardiovasc. Genet. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Treasure, T.; Takkenberg, J.J.; Golesworthy, T.; Rega, F.; Petrou, M.; Rosendahl, U.; Mohiaddin, R.; Rubens, M.; Thornton, W.; Lees, B.; et al. Personalised external aortic root support (PEARS) in Marfan syndrome: Analysis of 1-9 year outcomes by intention-to-treat in a cohort of the first 30 consecutive patients to receive a novel tissue and valve-conserving procedure, compared with the published results of aortic root replacement. Heart 2014, 100, 969–975. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Pan, X.; Zhao, K.; Zhao, C. Two novel mutations of fibrillin-1 gene correlate with different phenotypes of Marfan syndrome in Chinese families. Mol. Vis. 2013, 19, 751–758. [Google Scholar] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Francke, U.; Berg, M.A.; Tynan, K.; Brenn, T.; Liu, W.; Aoyama, T.; Gasner, C.; Miller, D.C.; Furthmayr, H. A Gly1127Ser mutation in an EGF-like domain of the fibrillin-1 gene is a risk factor for ascending aortic aneurysm and dissection. Am. J. Hum. Genet. 1995, 56, 1287–1296. [Google Scholar]
- von Kodolitsch, Y.; Robinson, P.N. Marfan syndrome: An update of genetics, medical and surgical management. Heart 2007, 93, 755–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, N.; Hara, H.; Fujiwara, T.; Kanaya, T.; Maemura, S.; Komuro, I. TGF-β Signaling-Related Genes and Thoracic Aortic Aneurysms and Dissections. Int J. Mol. Sci 2018, 19, 2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, F.; Caescu, C.; Wondimu, E.; Galatioto, J. Marfan syndrome; A connective tissue disease at the crossroads of mechanotransduction, TGFβ signaling and cell stemness. Matrix Biol. 2018, 71–72, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Faivre, L.; Collod-Beroud, G.; Loeys, B.L.; Child, A.; Binquet, C.; Gautier, E.; Callewaert, B.; Arbustini, E.; Mayer, K.; Arslan-Kirchner, M.; et al. Effect of mutation type and location on clinical outcome in 1013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: An international study. Am. J. Hum. Genet. 2007, 81, 454–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franken, R.; Teixido-Tura, G.; Brion, M.; Forteza, A.; Rodriguez-Palomares, J.; Gutierrez, L.; Garcia Dorado, D.; Pals, G.; Mulder, B.J.; Evangelista, A. Relationship between fibrillin-1 genotype and severity of cardiovascular involvement in Marfan syndrome. Heart 2017, 103, 1795–1799. [Google Scholar] [CrossRef]
- Schrijver, I.; Liu, W.; Odom, R.; Brenn, T.; Oefner, P.; Furthmayr, H.; Francke, U. Premature termination mutations in FBN1: Distinct effects on differential allelic expression and on protein and clinical phenotypes. Am. J. Hum. Genet. 2002, 71, 223–237. [Google Scholar] [CrossRef] [Green Version]
- Hernándiz, A.; Zúñiga, A.; Valera, F.; Domingo, D.; Ontoria-Oviedo, I.; Marí, J.F.; Román, J.A.; Calvo, I.; Insa, B.; Gómez, R.; et al. Genotype FBN1/phenotype relationship in a cohort of patients with Marfan syndrome. Clin. Genet. 2021, 99, 269–280. [Google Scholar] [CrossRef]
- Grange, T.; Aubart, M.; Langeois, M.; Benarroch, L.; Arnaud, P.; Milleron, O.; Eliahou, L.; Gross, M.S.; Hanna, N.; Boileau, C.; et al. Quantifying the Genetic Basis of Marfan Syndrome Clinical Variability. Genes (Basel) 2020, 11, 574. [Google Scholar] [CrossRef]
- Vanem, T.T.; Geiran, O.R.; Krohg-Sørensen, K.; Røe, C.; Paus, B.; Rand-Hendriksen, S. Survival, causes of death, and cardiovascular events in patients with Marfan syndrome. Mol. Genet. Genom. Med. 2018, 6, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- Demolder, A.; von Kodolitsch, Y.; Muiño-Mosquera, L.; De Backer, J. Myocardial Function, Heart Failure and Arrhythmia in Marfan Syndrome: A Systematic Literature Review. Diagnostics (Basel) 2020, 10, 751. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, C.; Bulut, H.; van Gemert, W.; Staal, A.H.; Cortenbach, K.; Snoeren, M.; Nijveldt, R.; Duijnhouwer, A.; Loeys, B.L.; van Royen, N.; et al. Progressive Pulmonary Artery Dilatation is Associated with Type B Aortic Dissection in Patients with Marfan Syndrome. J. Clin. Med. 2019, 8, 1848. [Google Scholar] [CrossRef] [Green Version]
- Treasure, T.; Pepper, J. Personalised External Aortic Root Support (PEARS) Compared with Alternatives for People with Life-Threatening Genetically Determined Aneurysms of the Aortic Root. Diseases 2015, 3, 2–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Zhou, Y.; Peng, Y.; Jin, L. Two rare missense mutations in the fibrillin-1 gene associated with atypical cardiovascular manifestations in a Chinese patient affected by Marfan syndrome. Mol. Med. Rep. 2018, 18, 877–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamsi-Pasha, M.A.; Lawrie, G.M. Aneurysmal left sinus of Valsalva in Marfan’s syndrome. Eur. Heart J. 2018, 39, 285. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Santos, M.; Oliveira-Santos, E.; Gonçalves, L.; Silva Marques, J. Cardiovascular Three-Dimensional Printing in Non-Congenital Percutaneous Interventions. Heart Lung Circ. 2019, 28, 1525–1534. [Google Scholar] [CrossRef]
- Haghiashtiani, G.; Qiu, K.; Zhingre Sanchez, J.D.; Fuenning, Z.J.; Nair, P.; Ahlberg, S.E.; Iaizzo, P.A.; McAlpine, M.C. 3D printed patient-specific aortic root models with internal sensors for minimally invasive applications. Sci. Adv. 2020, 6, eabb4641. [Google Scholar] [CrossRef] [PubMed]
- Koolbergen, D.R.; Manshanden, J.S.; Bouma, B.J.; Blom, N.A.; Mulder, B.J.; de Mol, B.A.; Hazekamp, M.G. Valve-sparing aortic root replacement†. Eur. J. Cardiothorac. Surg. 2015, 47, 348–354; discussion 354. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, J.S.; Park, J.; Kwon, J.B.; Kim, D.-W.; Park, M.-W. 3D Printed Personalized External Aortic Root Model in Marfan Syndrome with Isolated Sinus of Valsalva Aneurysm Caused by a Novel Pathogenic FBN1 p.Gly1127Cys Variant. Diagnostics 2021, 11, 1057. https://doi.org/10.3390/diagnostics11061057
Cho JS, Park J, Kwon JB, Kim D-W, Park M-W. 3D Printed Personalized External Aortic Root Model in Marfan Syndrome with Isolated Sinus of Valsalva Aneurysm Caused by a Novel Pathogenic FBN1 p.Gly1127Cys Variant. Diagnostics. 2021; 11(6):1057. https://doi.org/10.3390/diagnostics11061057
Chicago/Turabian StyleCho, Jung Sun, Joonhong Park, Jong Bum Kwon, Dae-Won Kim, and Mahn-Won Park. 2021. "3D Printed Personalized External Aortic Root Model in Marfan Syndrome with Isolated Sinus of Valsalva Aneurysm Caused by a Novel Pathogenic FBN1 p.Gly1127Cys Variant" Diagnostics 11, no. 6: 1057. https://doi.org/10.3390/diagnostics11061057
APA StyleCho, J. S., Park, J., Kwon, J. B., Kim, D.-W., & Park, M.-W. (2021). 3D Printed Personalized External Aortic Root Model in Marfan Syndrome with Isolated Sinus of Valsalva Aneurysm Caused by a Novel Pathogenic FBN1 p.Gly1127Cys Variant. Diagnostics, 11(6), 1057. https://doi.org/10.3390/diagnostics11061057