
EWSR1—The Most Common Rearranged Gene in Soft Tissue Lesions, Which Also Occurs in Different Bone Lesions: An Updated Review

 , , ,
, , ,
Abstract
1. Introduction
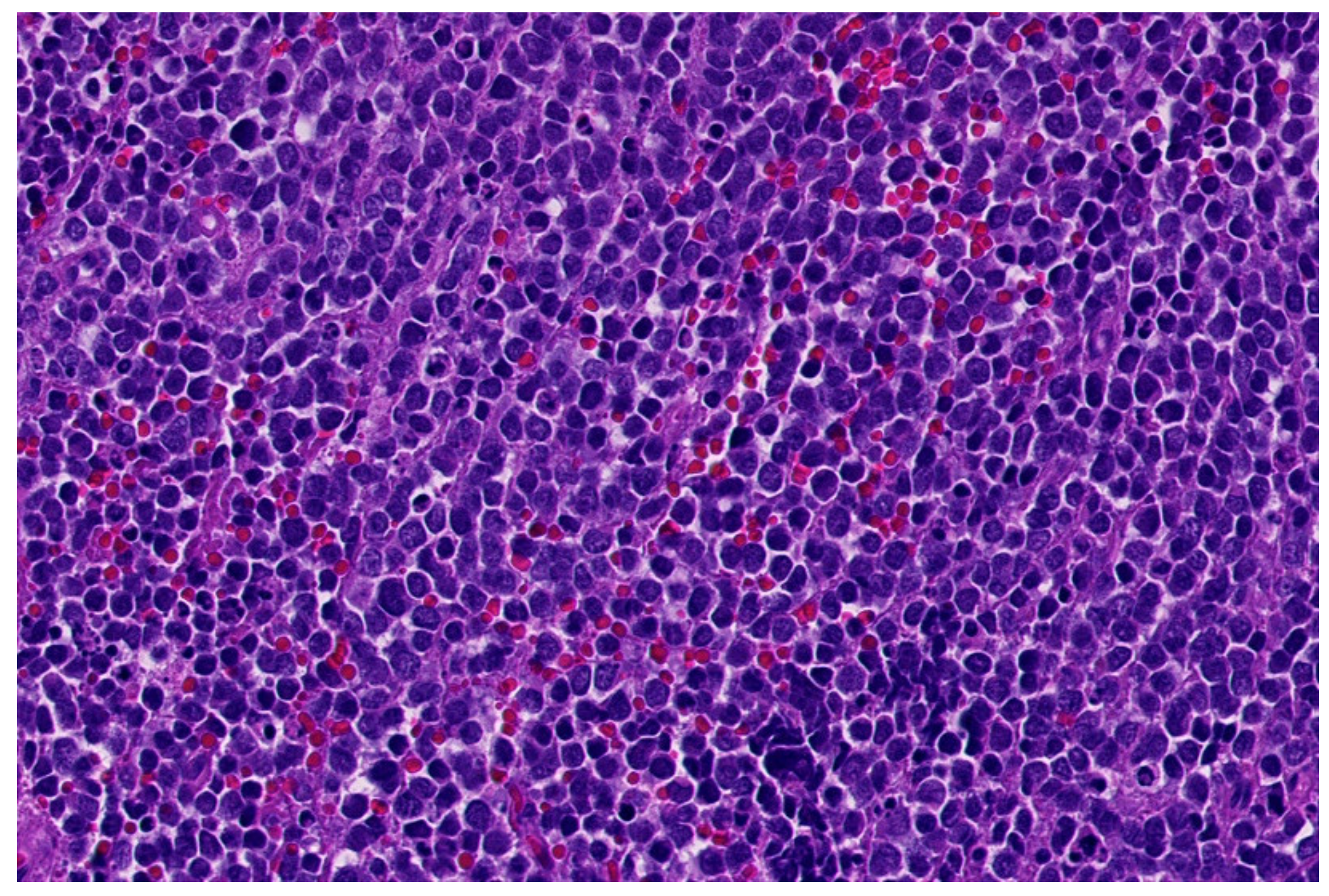
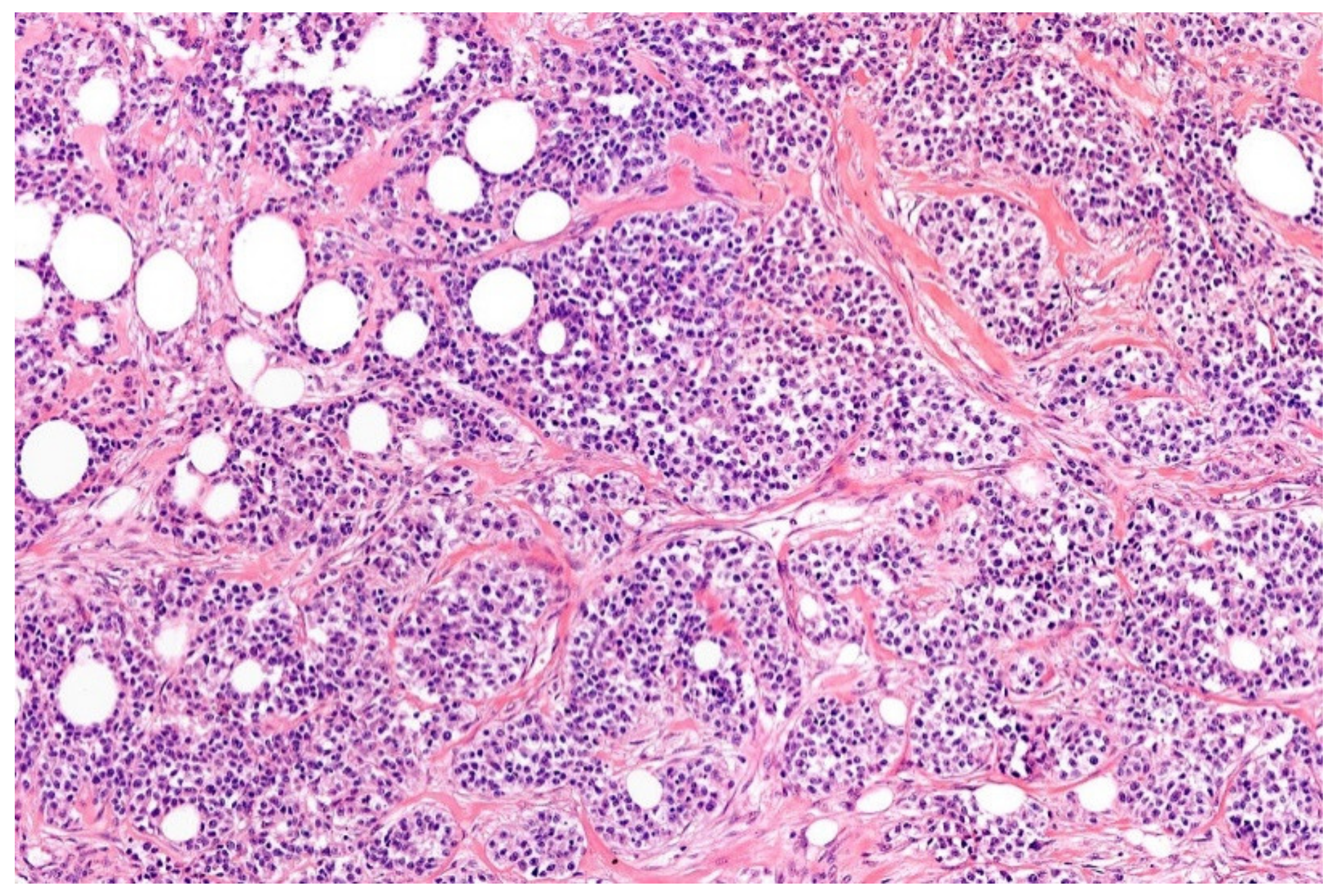
2. Ewing Sarcoma
3. Round-Cell Sarcomas with EWSR1-Non-ETS Fusions
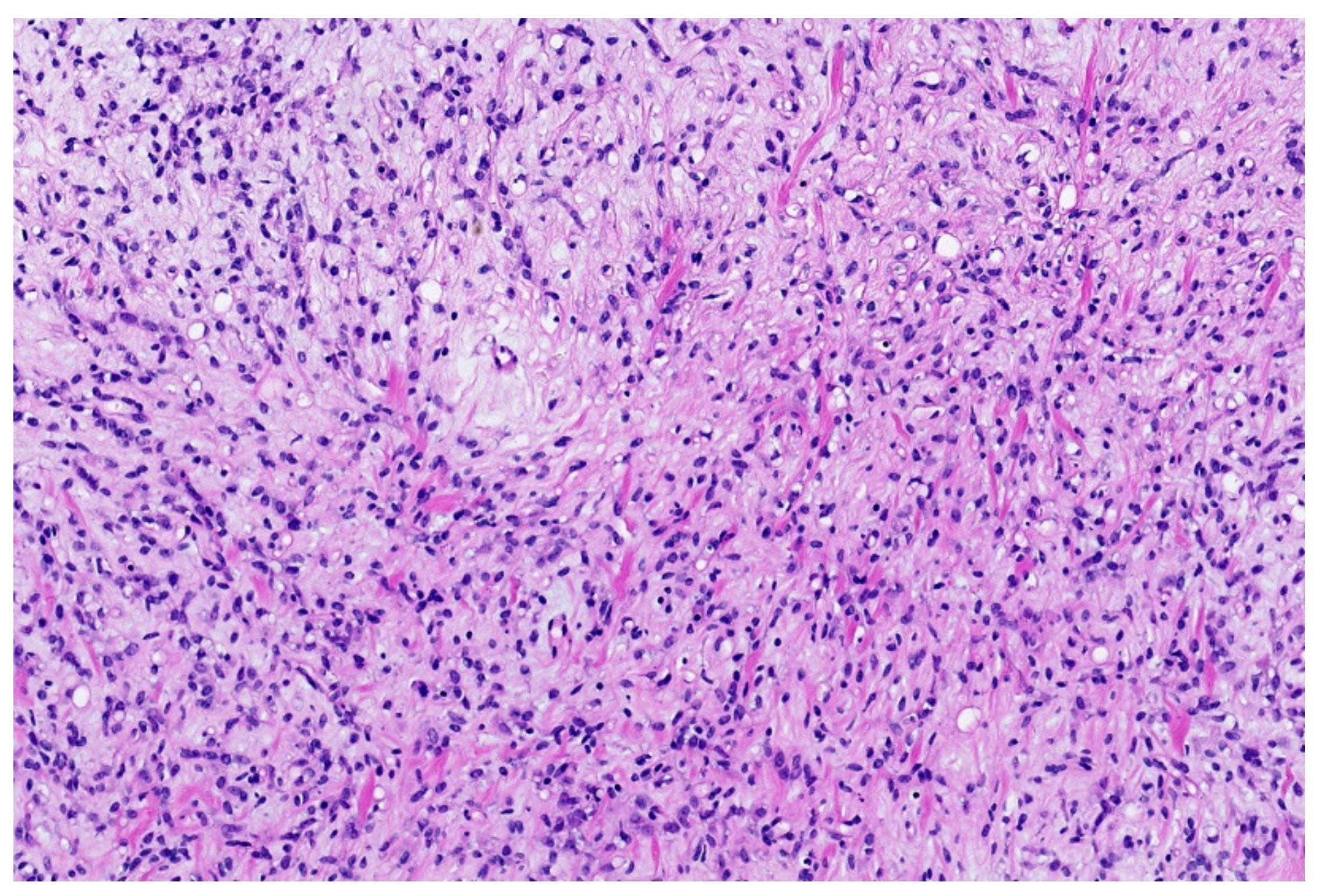
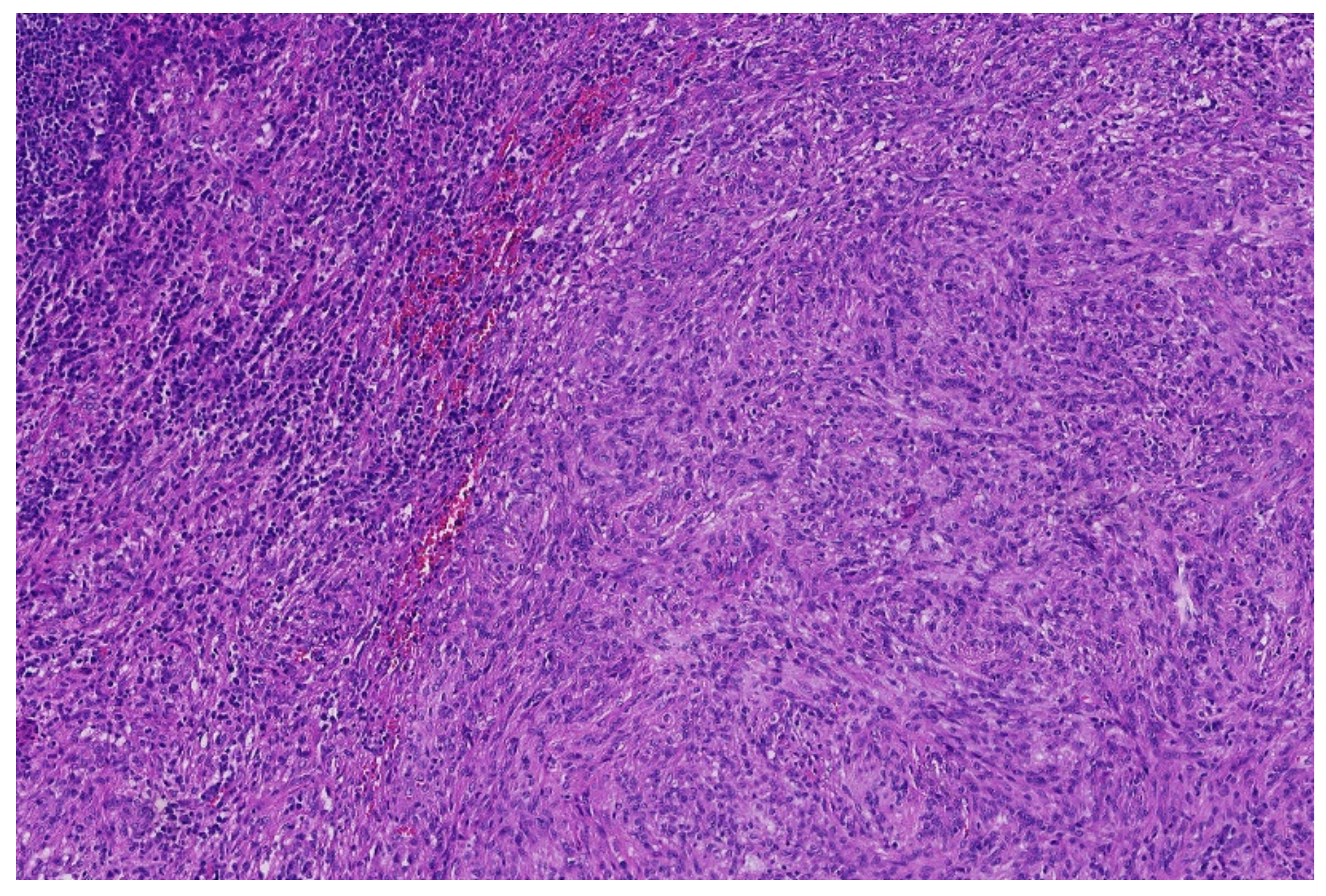
3.1. NFATc2-Rearranged Sarcoma
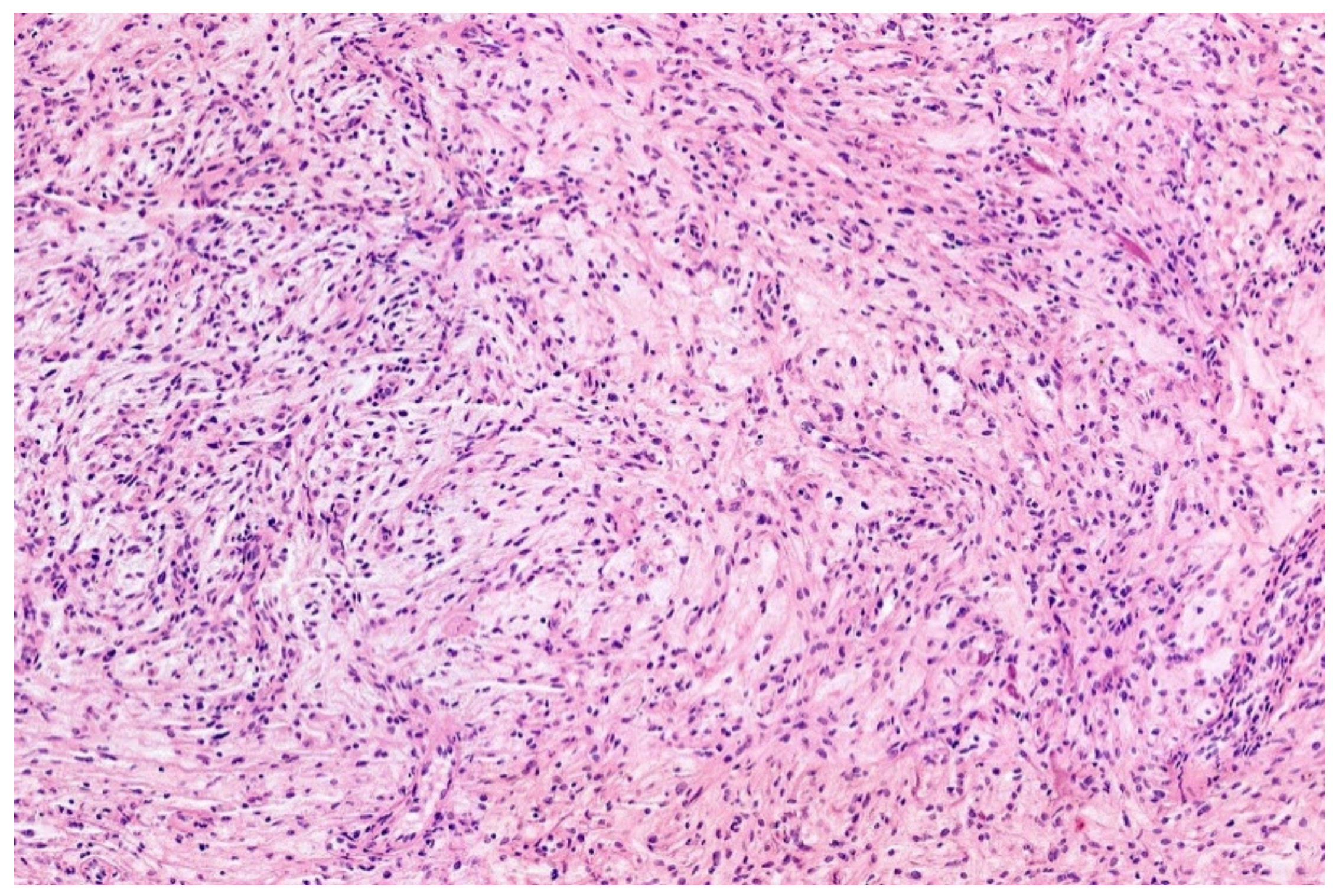
3.2. EWSR1-PATZ and EWSR1-VEZF1 Rearranged Sarcoma
- Low-grade appearing tumors: the spindled, epithelioid, ovoid, and round cells are set in a hyaline stroma reminiscent of solitary fibrous tumors and myoepithelioma.
- Intermediate and high-grade appearing neoplasms have a round and/or ovoid morphology with few spindle cells and slight stromal component. The tumors of this second subgroup resemble other small, blue round-cell tumors, e.g., ARMS, BCOR-, and CIC-rearranged sarcoma.
3.3. Small Blue Round-Cell Tumor with EWSR1-SMARCA5 Rearrangement
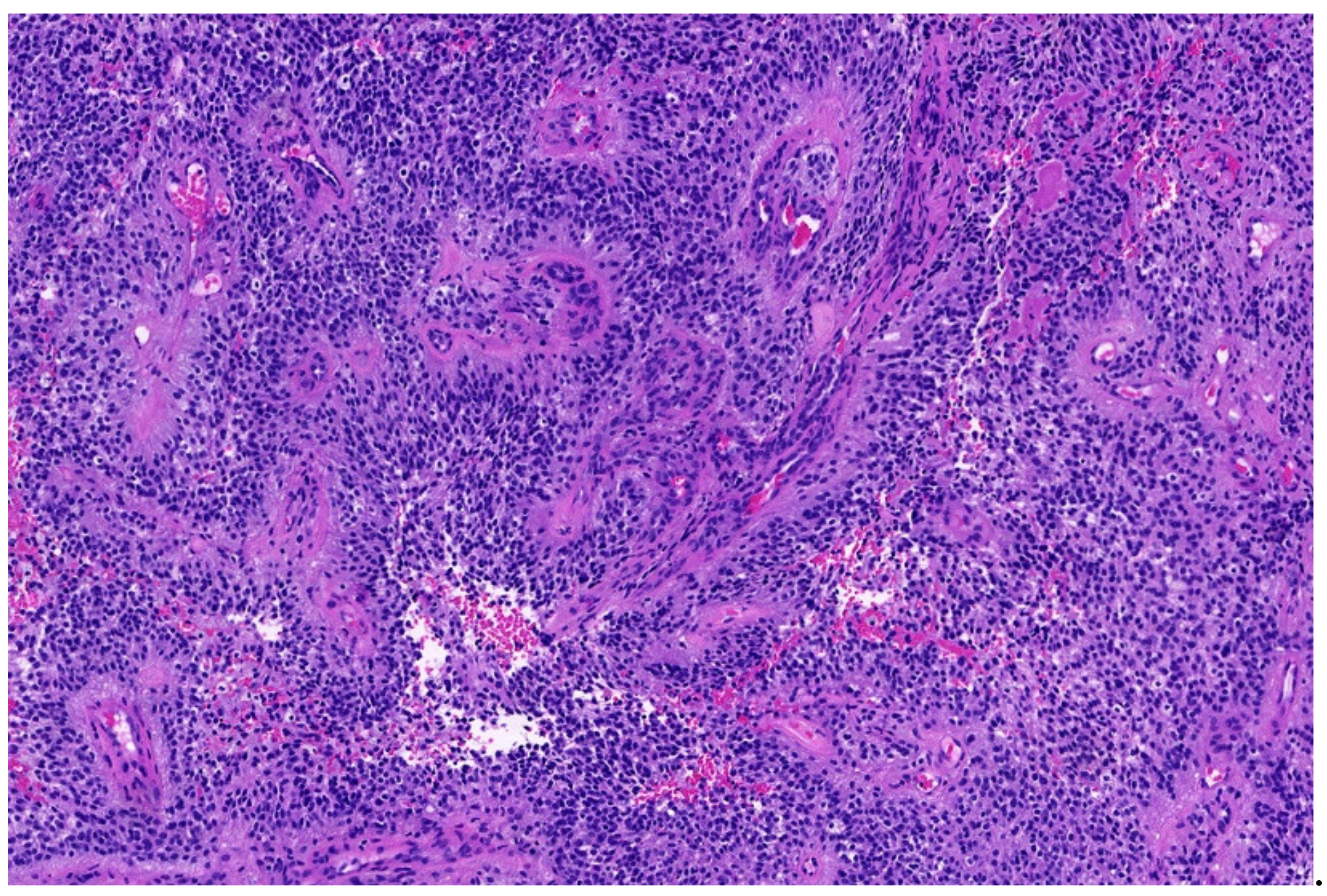
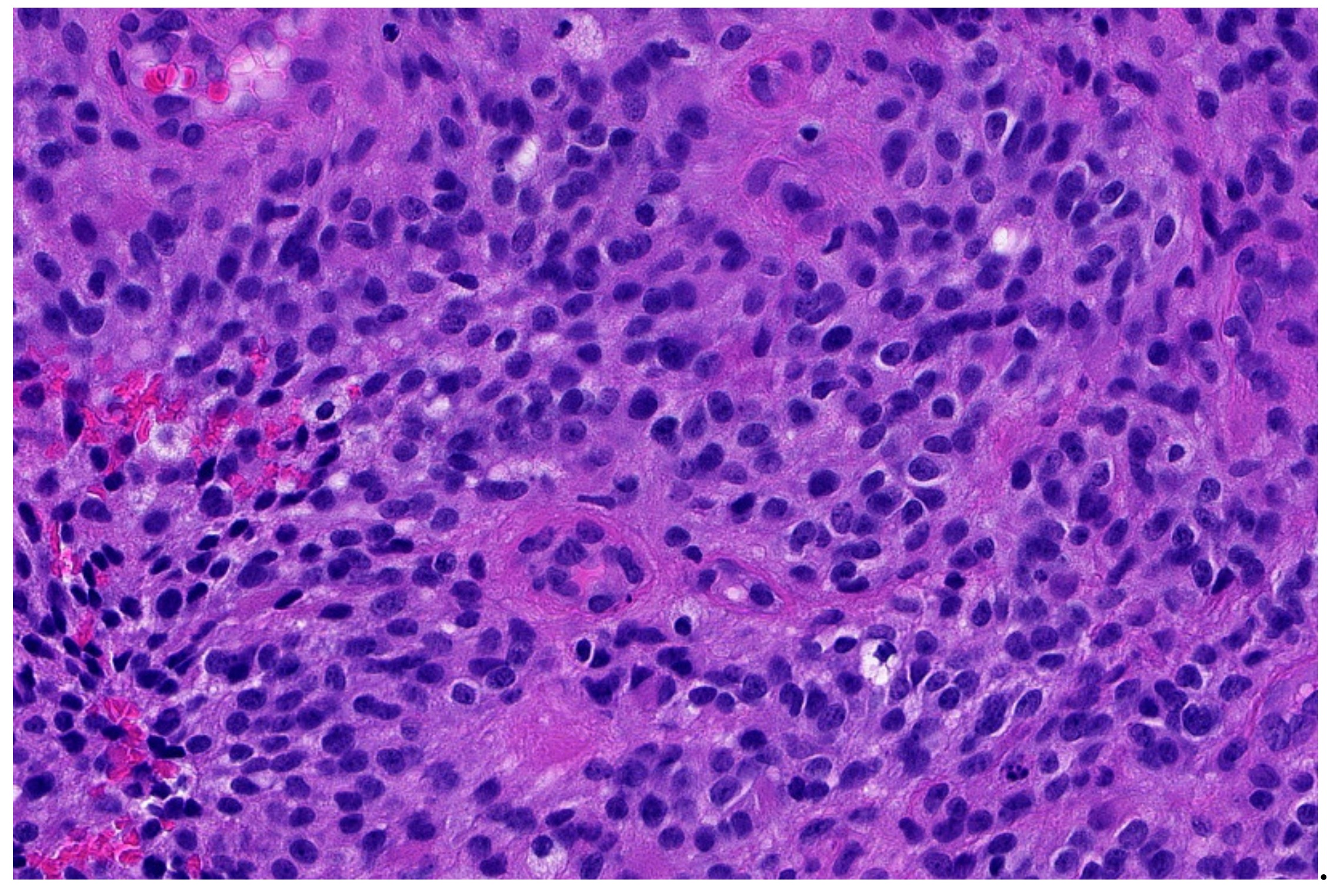
3.4. Desmoplastic Small Round-Cell Tumor (DSCRT)
4. Myxoid Liposarcoma (MLS)
5. Tumors with EWSR1/FUS Fused to the CREB-Family
5.1. Angiomatoid Fibrous Histiocytoma (AFH)
5.2. Primary Pulmonary Myxoid Sarcoma (PPMS)
5.3. EWSR1-CREM Undifferentiated Sarcoma
5.4. Clear-Cell Sarcoma (CCS)
5.5. Clear-Cell Sarcoma-Like Tumor of the Gastrointestinal Tract (Osteoclastrich Tumor of the Gastrointestinal Tract or Malignant Gastrointestinal Neuroectodermal Tumor)
5.6. Mesothelioma
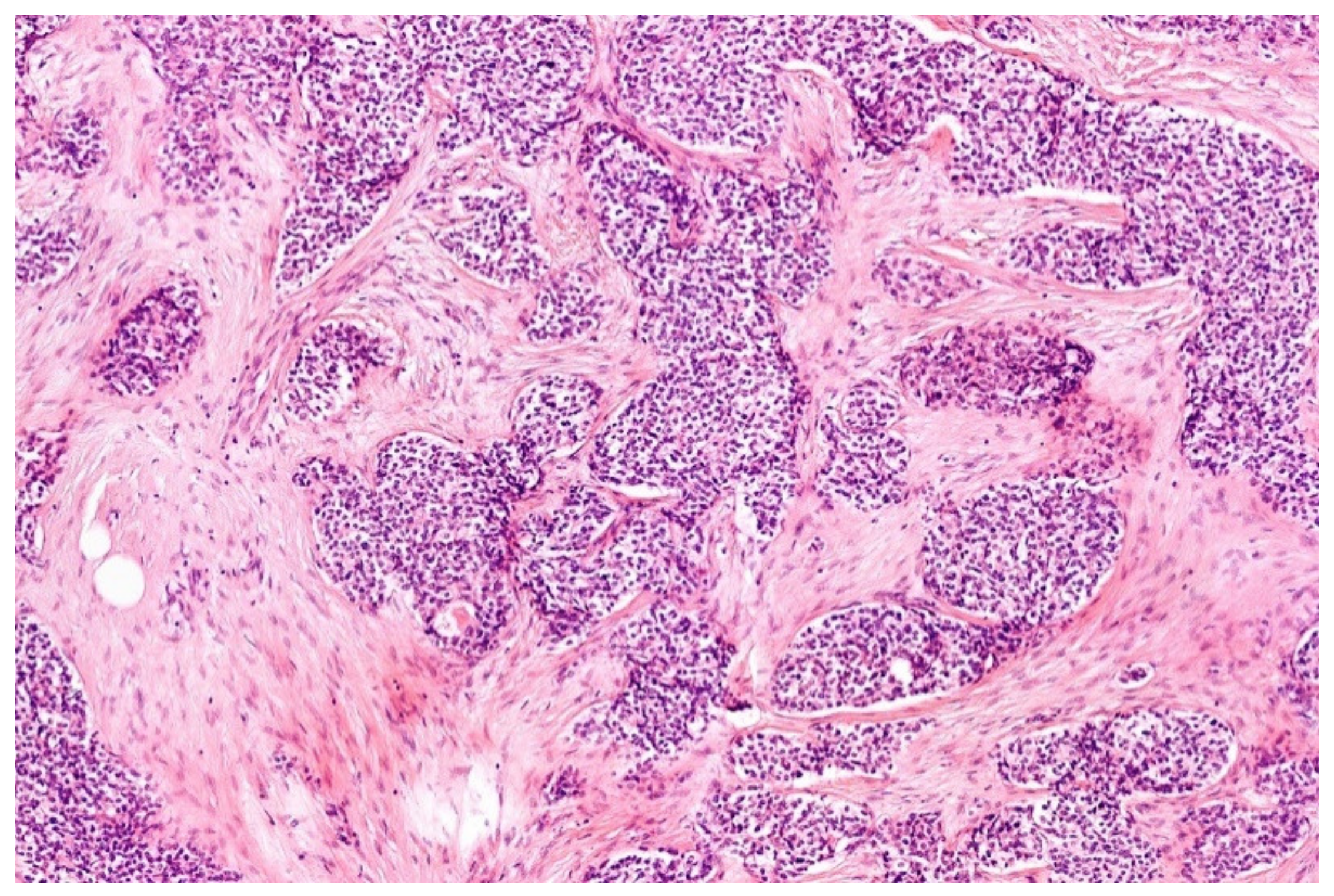
6. Myoepithelial Tumors
7. Low-Grade Fibromyxoid Sarcoma (LGFMS)/Sclerosing Epithelioid Fibrosarcoma (SEF)
8. Extraskeletal Myxoid Chondrosarcoma (EMC)
9. EWSR1-SMAD3-Positive Fibroblastic Tumor (ESFT)
10. Epithelioid and Spindle Cell Rhabdomyosarcoma with EWSR1/FUS-TFCP2 Fusion
11. Retroperitoneal Leiomyoma
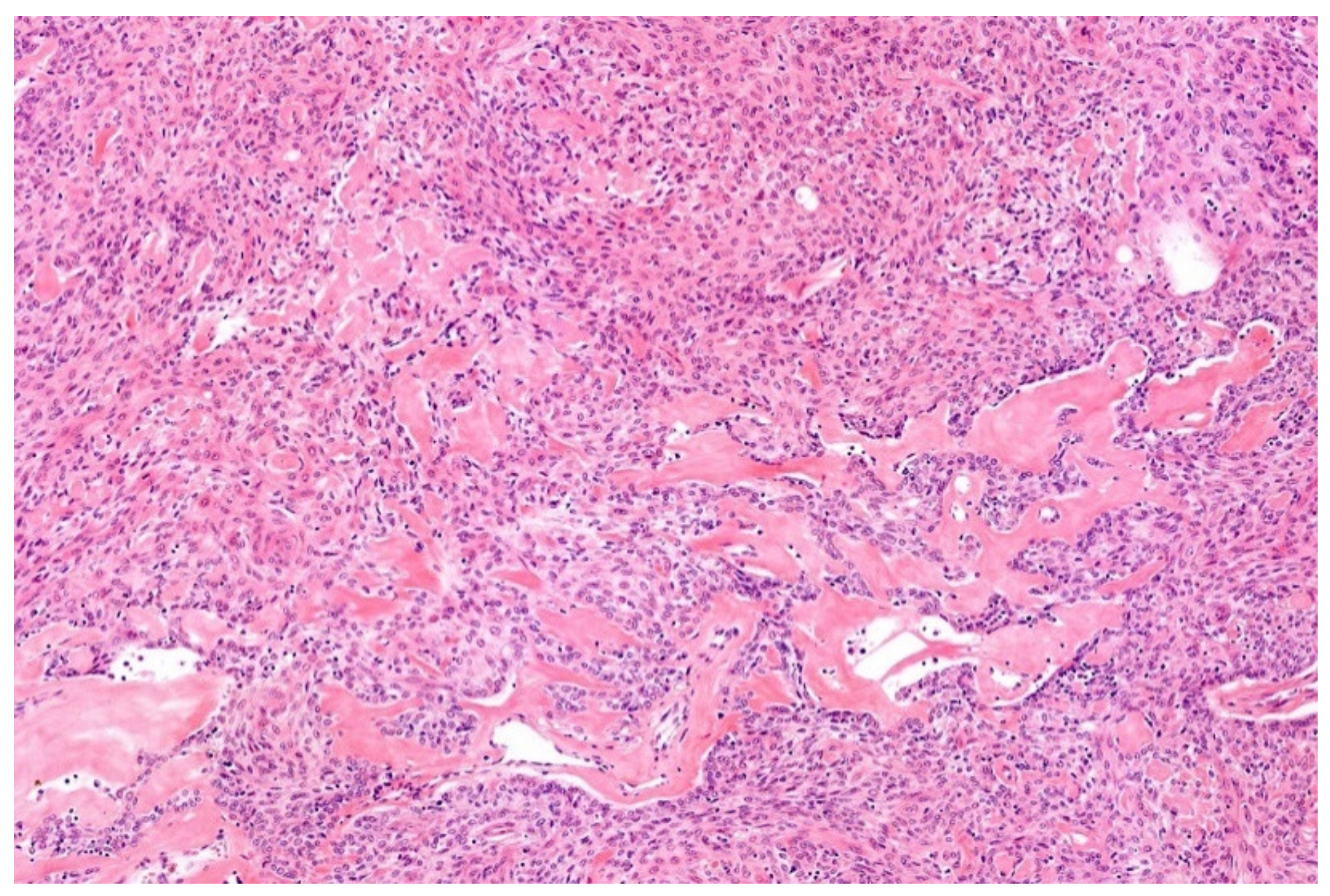
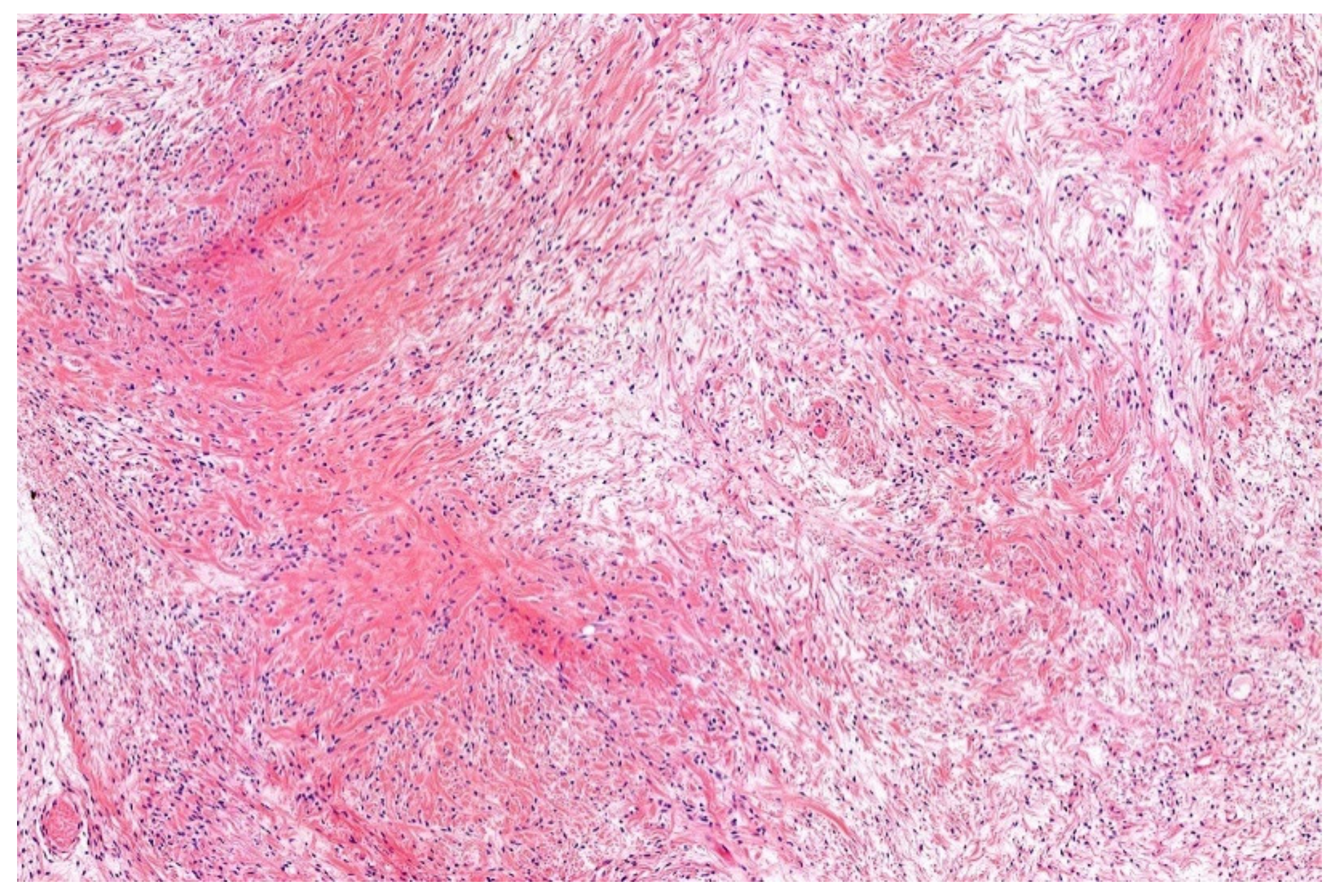
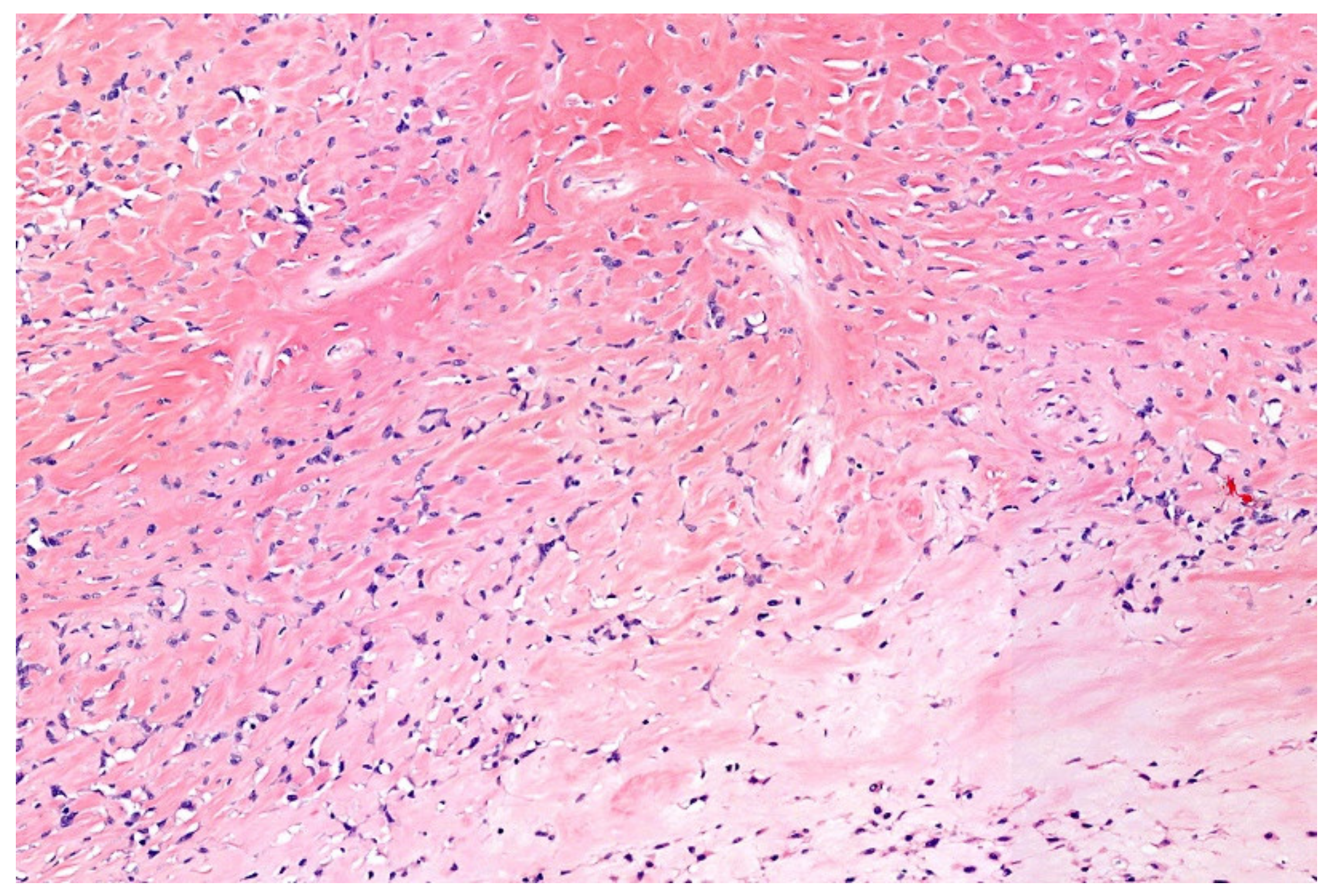
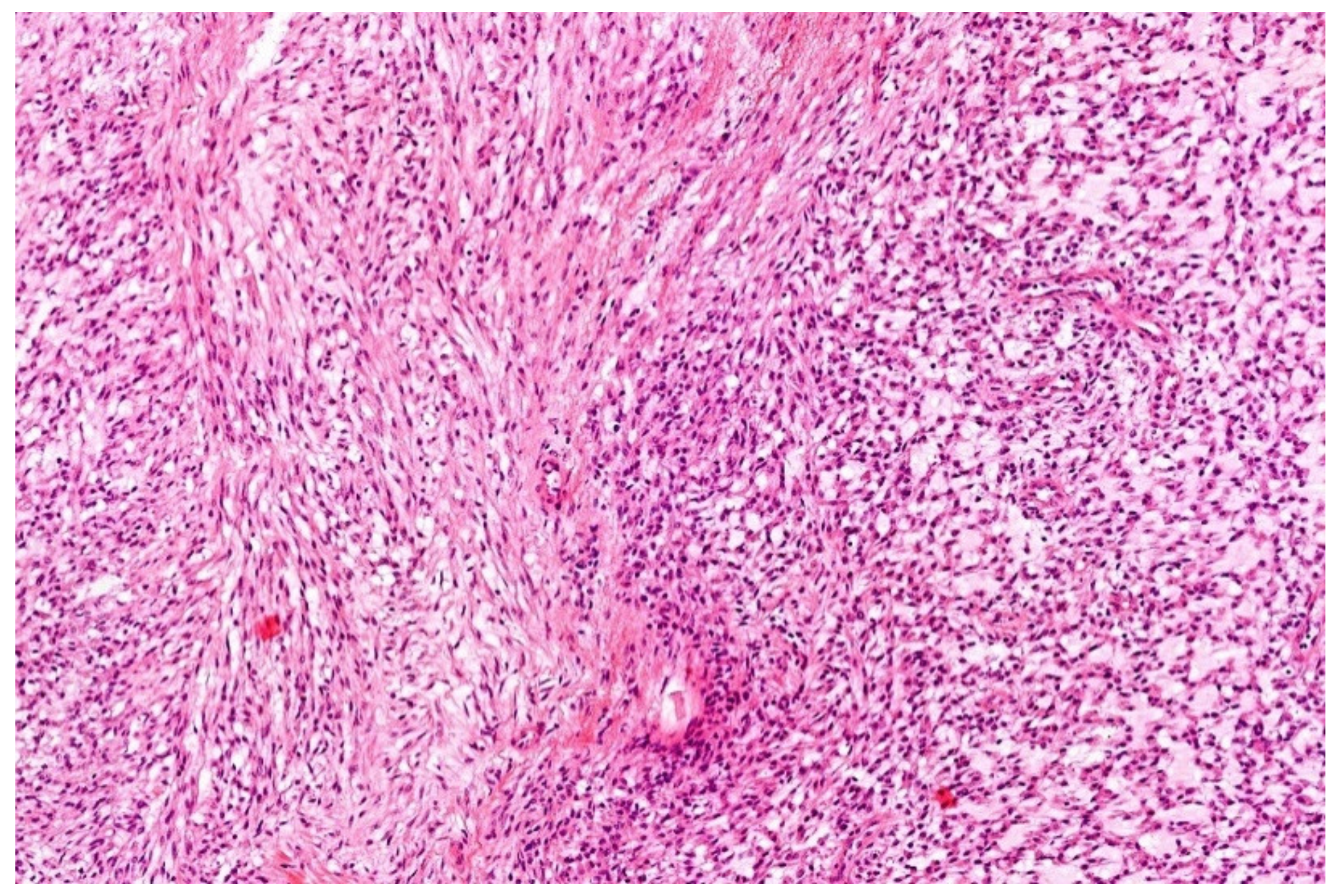
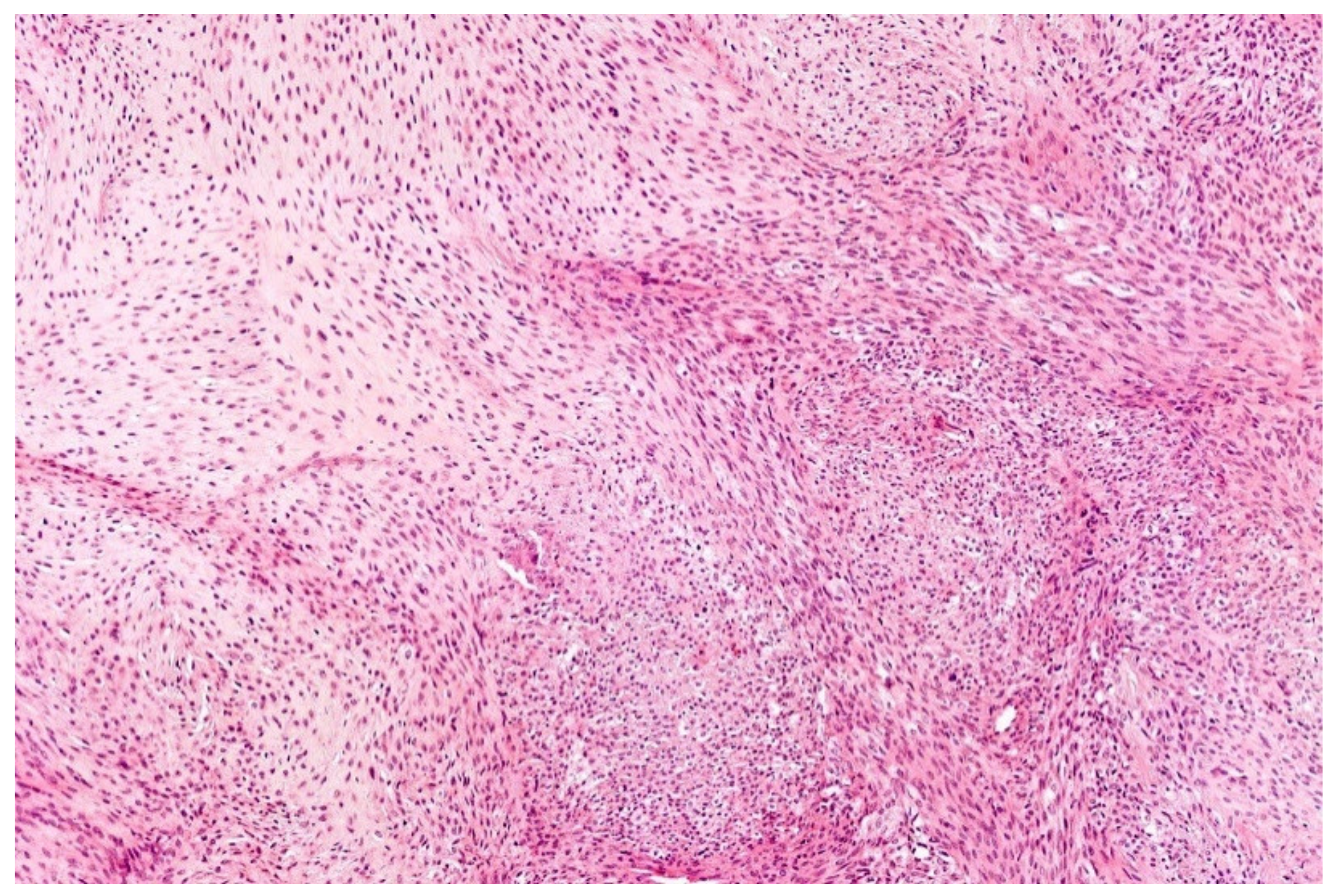
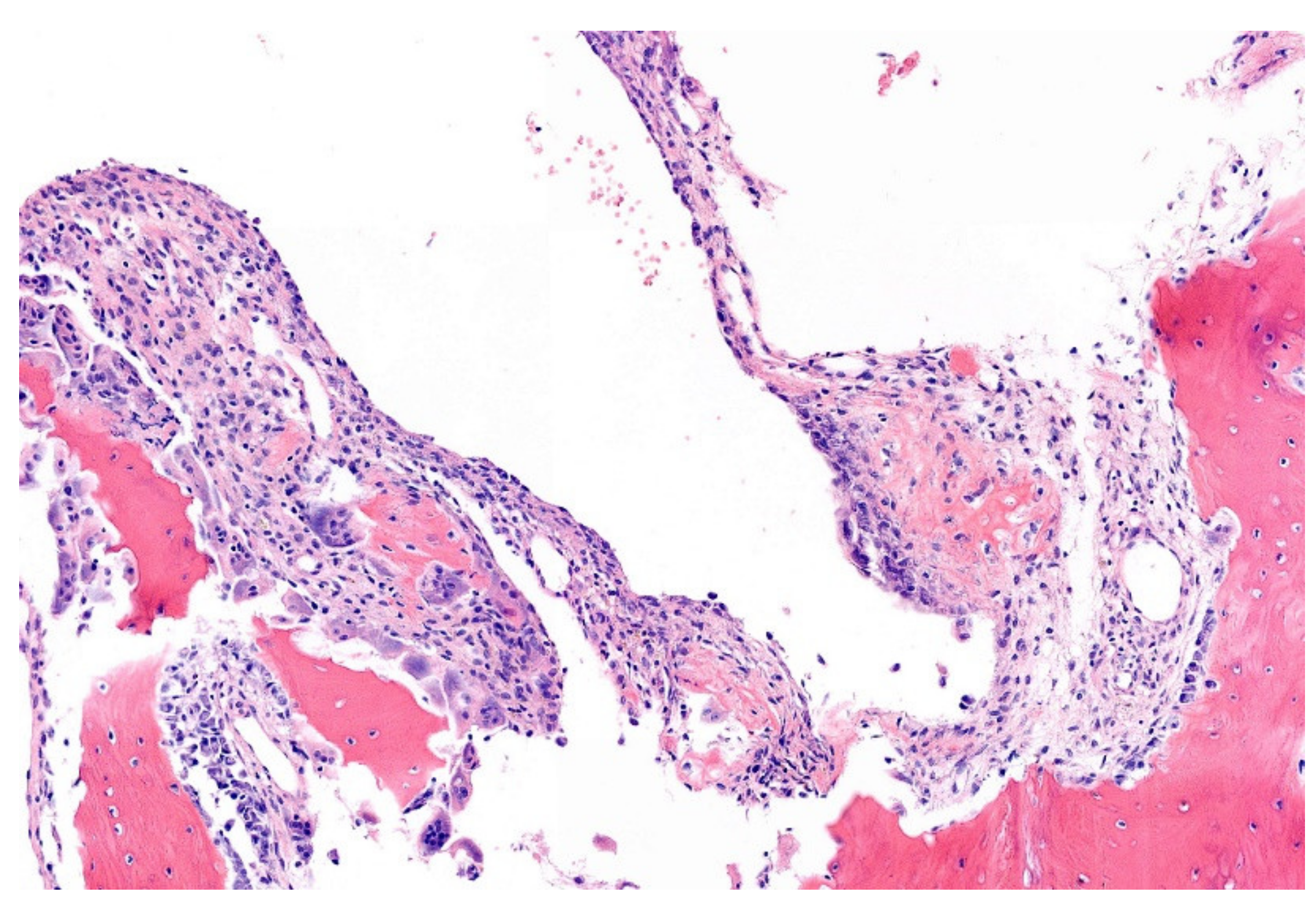
12. Simple (Unicameral) Bone Cyst (SBC)
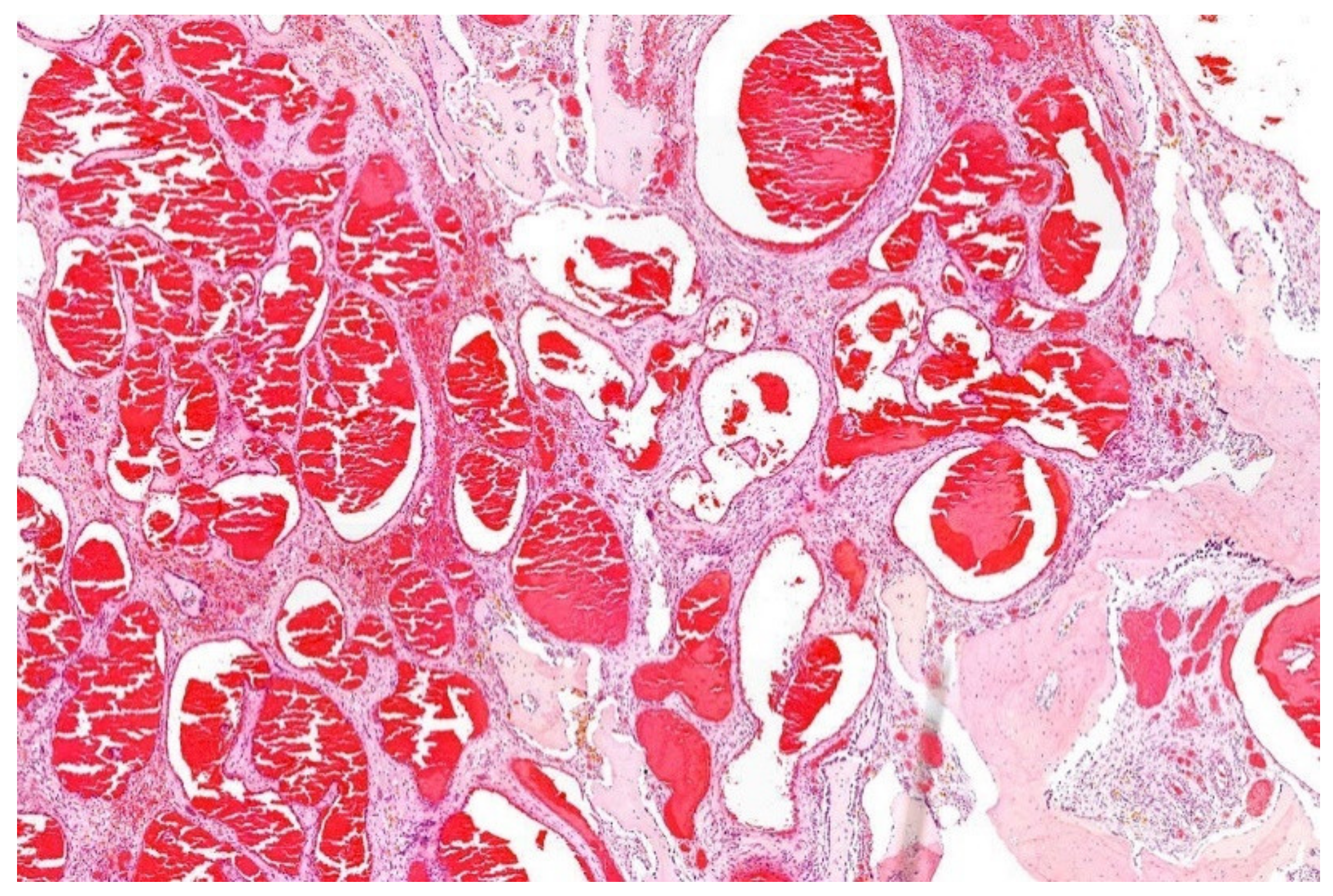
13. Hemangioma of Bone with an EWSR1-NFATC1 Fusion
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Nguyen, P.T.; Shim, H.S.; Hyeon, S.J.; Im, H.; Choi, M.H.; Chung, S.; Kowall, N.W.; Lee, S.B.; Ryu, H. EWSR1, a multifunctional protein, regulates cellular function and aging via genetic and epigenetic pathways. Biochim. Biophys. Acta Mol. Basis Dis 2019, 1865, 1938–1945. [Google Scholar] [CrossRef]
- Bertolotti, A.; Bell, B.; Tora, L. The N-terminal domain of human TAFII68 displays transactivation and oncogenic properties. Oncogene 1999, 18, 8000–8010. [Google Scholar] [CrossRef]
- Lindén, M.; Thomsen, C.; Grundevik, P.; Jonasson, E.; Andersson, D.; Runnberg, R.; Dolatabadi, S.; Vannas, C.; Luna Santamarίa, M.; Fagman, H.; et al. FET family fusion oncoproteins target the SWI/SNF chromatin remodeling complex. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- W.H.O. Classification of Soft Tissue and Bone Tumours; International Agency for Research on Cancer: Lyon, France, 2020. [Google Scholar]
- Folpe, A.L.; Goldblum, J.R.; Rubin, B.P.; Shehata, B.M.; Liu, W.; Dei Tos, A.P.; Weiss, S.W. Morphologic and immunophenotypic diversity in Ewing family tumors: A study of 66 genetically confirmed cases. Am. J. Surg. Pathol. 2005, 29, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Stout, A.P. A tumor of ulnar nerve. Proc. NY Pathol. Soc. 1918, 18, 2–12. (In Japanese) [Google Scholar]
- Ewing, J. Classics in oncology. Diffuse endothelioma of bone. James Ewing. Proceedings of the New York Pathological Society, 1921. CA Cancer J. Clin. 1972, 22, 95–98. [Google Scholar] [CrossRef]
- Seidal, T.; Mark, J.; Hagmar, B.; Angervall, L. Alveolar rhabdomyosarcoma: A cytogenetic and correlated cytological and histological study. Acta Pathol. Microbiol. Immunol. Scand. A 1982, 90, 345–354. [Google Scholar] [CrossRef]
- Aurias, A.; Rimbaut, C.; Buffe, D.; Dubousset, J.; Mazabraud, A. Translocation of chromosome 22 in Ewing’s sarcoma. C R Seances Acad Sci. III 1983, 296, 1105–1107. [Google Scholar]
- Turc-Carel, C. Chromosomal translocations in Ewing’s sarcoma. N. Engl. J. Med. 1983, 309, 496–498. [Google Scholar]
- Czerniak, B.U. Dorfman and Czerniak’s Bone Tumors; Elsevier Health Sciences: Houston, TX, USA, 2015. [Google Scholar]
- Miettinen, M.U. Modern Soft Tissue Pathology: Tumors and Non-Neoplastic Conditions; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar]
- Rooper, L.M.; Bishop, J.A. Soft Tissue Special Issue: Adamantinoma-Like Ewing Sarcoma of the Head and Neck: A Practical Review of a Challenging Emerging Entity. Head Neck Pathol. 2020, 14, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Sekine, S.; Tsuta, K.; Fukayama, M.; Furuta, K.; Tsuda, H. NKX2.2 is a useful immunohistochemical marker for Ewing sarcoma. Am. J. Surg. Pathol. 2012, 36, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, R.; Matsuyama, A.; Nakamoto, M.; Shiba, E.; Kasai, T.; Hisaoka, M. The combination of CD99 and NKX2.2, a transcriptional target of EWSR1-FLI1, is highly specific for the diagnosis of Ewing sarcoma. Virchows Arch. 2014, 465, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.P.; Fletcher, C.D.; Hornick, J.L. Evaluation of NKX2-2 expression in round cell sarcomas and other tumors with EWSR1 rearrangement: Imperfect specificity for Ewing sarcoma. Mod. Pathol. 2016, 29, 370–380. [Google Scholar] [CrossRef]
- Russell-Goldman, E.; Hornick, J.L.; Qian, X.; Jo, V.Y. NKX2.2 immunohistochemistry in the distinction of Ewing sarcoma from cytomorphologic mimics: Diagnostic utility and pitfalls. Cancer Cytopathol. 2018, 126, 942–949. [Google Scholar] [CrossRef]
- Smith, R.; Owen, L.A.; Trem, D.J.; Wong, J.S.; Whangbo, J.S.; Golub, T.R.; Lessnick, S.L. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing’s sarcoma. Cancer Cell 2006, 9, 405–416. [Google Scholar] [CrossRef]
- Barisella, M.; Collini, P.; Orsenigo, M.; Aiello, A.; Paties, C.T.; Dileo, P.; Pilotti, S. Unusual myogenic and melanocytic differentiation of soft tissue pPNETs: An immunohistochemical and molecular study of 3 cases. Am. J. Surg. Pathol. 2010, 34, 1002–1006. [Google Scholar] [CrossRef]
- Thway, K.; Fisher, C. Mesenchymal Tumors with EWSR1 Gene Rearrangements. Surg. Pathol. Clin. 2019, 12, 165–190. [Google Scholar] [CrossRef]
- Szuhai, K.; Ijszenga, M.; de Jong, D.; Karseladze, A.; Tanke, H.J.; Hogendoorn, P.C. The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin. Cancer Res. 2009, 15, 2259–2268. [Google Scholar] [CrossRef]
- Koelsche, C.; Kriegsmann, M.; Kommoss, F.K.F.; Stichel, D.; Kriegsmann, K.; Vokuhl, C.; Grünewald, T.G.P.; Romero-Pérez, L.; Kirchner, T.; de Alava, E.; et al. DNA methylation profiling distinguishes Ewing-like sarcoma with EWSR1-NFATc2 fusion from Ewing sarcoma. J. Cancer Res. Clin. Oncol. 2019, 145, 1273–1281. [Google Scholar] [CrossRef]
- Le Loarer, F.; Pissaloux, D.; Coindre, J.M.; Tirode, F.; Vince, D.R. Update on Families of Round Cell Sarcomas Other than Classical Ewing Sarcomas. Surg. Pathol. Clin. 2017, 10, 587–620. [Google Scholar] [CrossRef]
- Bode-Lesniewska, B.; Fritz, C.; Exner, G.U.; Wagner, U.; Fuchs, B. EWSR1-NFATC2 and FUS-NFATC2 Gene Fusion-Associated Mesenchymal Tumors: Clinicopathologic Correlation and Literature Review. Sarcoma 2019, 2019, 9386390. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.; Perrin, V.; Guillemot, D.; Reynaud, S.; Coindre, J.M.; Karanian, M.; Guinebretière, J.M.; Freneaux, P.; Le Loarer, F.; Bouvet, M.; et al. Transcriptomic definition of molecular subgroups of small round cell sarcomas. J. Pathol. 2018, 245, 29–40. [Google Scholar] [CrossRef]
- Perret, R.; Escuriol, J.; Velasco, V.; Mayeur, L.; Soubeyran, I.; Delfour, C.; Aubert, S.; Polivka, M.; Karanian, M.; Meurgey, A.; et al. Correction: NFATc2-rearranged sarcomas: Clinicopathologic, molecular, and cytogenetic study of 7 cases with evidence of AGGRECAN as a novel diagnostic marker. Mod. Pathol. 2020, 33, 2087. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, T.; Modena, P.; Tornielli, S.; Bullrich, F.; Testi, M.A.; Mezzelani, A.; Radice, P.; Azzarelli, A.; Pilotti, S.; Croce, C.M.; et al. A novel zinc finger gene is fused to EWS in small round cell tumor. Oncogene 2000, 19, 3799–3804. [Google Scholar] [CrossRef]
- Michal, M.; Rubin, B.P.; Agaimy, A.; Kosemehmetoglu, K.; Rudzinski, E.R.; Linos, K.; John, I.; Gatalica, Z.; Davis, J.L.; Liu, Y.J.; et al. EWSR1-PATZ1-rearranged sarcoma: A report of nine cases of spindle and round cell neoplasms with predilection for thoracoabdominal soft tissues and frequent expression of neural and skeletal muscle markers. Mod. Pathol. 2021, 34, 770–785. [Google Scholar] [CrossRef]
- Benini, S.; Gamberi, G.; Cocchi, S.; Righi, A.; Frisoni, T.; Longhi, A.; Gambarotti, M. Identification of a novel fusion transcript EWSR1-VEZF1 by anchored multiplex PCR in malignant peripheral nerve sheath tumor. Pathol. Res. Pract. 2020, 216, 152760. [Google Scholar] [CrossRef]
- Tsuda, Y.; Zhang, L.; Meyers, P.; Tap, W.D.; Healey, J.H.; Antonescu, C.R. The clinical heterogeneity of round cell sarcomas with EWSR1/FUS gene fusions: Impact of gene fusion type on clinical features and outcome. Genes Chromosomes Cancer 2020, 59, 525–534. [Google Scholar] [CrossRef]
- Siegfried, A.; Rousseau, A.; Maurage, C.A.; Pericart, S.; Nicaise, Y.; Escudie, F.; Grand, D.; Delrieu, A.; Gomez-Brouchet, A.; Le Guellec, S.; et al. EWSR1-PATZ1 gene fusion may define a new glioneuronal tumor entity. Brain Pathol. 2019, 29, 53–62. [Google Scholar] [CrossRef]
- Bridge, J.A.; Sumegi, J.; Druta, M.; Bui, M.M.; Henderson-Jackson, E.; Linos, K.; Baker, M.; Walko, C.M.; Millis, S.; Brohl, A.S. Clinical, pathological, and genomic features of EWSR1-PATZ1 fusion sarcoma. Mod. Pathol. 2019, 32, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Chougule, A.; Taylor, M.S.; Nardi, V.; Chebib, I.; Cote, G.M.; Choy, E.; Nielsen, G.P.; Deshpande, V. Spindle and Round Cell Sarcoma With EWSR1-PATZ1 Gene Fusion: A Sarcoma With Polyphenotypic Differentiation. Am. J. Surg. Pathol. 2019, 43, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Yau, D.T.W.; Wong, S.; Chow, C.; To, K.F. Round Cell Sarcoma with EWSR1-PATZ1 Fusion in the Face of a Five-Year-Old Boy: Report of a Case with Unusual Histologic Features. Head Neck Pathol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Sumegi, J.; Nishio, J.; Nelson, M.; Frayer, R.W.; Perry, D.; Bridge, J.A. A novel t(4;22)(q31;q12) produces an EWSR1-SMARCA5 fusion in extraskeletal Ewing sarcoma/primitive neuroectodermal tumor. Mod. Pathol. 2011, 24, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.R.; Tryka, A.F.; Lewis, J.M. A novel reciprocal chromosome translocation t(11;22)(p13;q12) in an intraabdominal desmoplastic small round-cell tumor. Am. J. Surg. Pathol. 1992, 16, 411–416. [Google Scholar] [CrossRef]
- Gerald, W.L.; Miller, H.K.; Battifora, H.; Miettinen, M.; Silva, E.G.; Rosai, J. Intra-abdominal desmoplastic small round-cell tumor. Report of 19 cases of a distinctive type of high-grade polyphenotypic malignancy affecting young individuals. Am. J. Surg. Pathol. 1991, 15, 499–513. [Google Scholar] [CrossRef]
- Gerald, W.L.; Rosai, J.; Ladanyi, M. Characterization of the genomic breakpoint and chimeric transcripts in the EWS-WT1 gene fusion of desmoplastic small round cell tumor. Proc. Natl. Acad. Sci. USA 1995, 92, 1028–1032. [Google Scholar] [CrossRef]
- Ladanyi, M.; Gerald, W. Fusion of the EWS and WT1 genes in the desmoplastic small round cell tumor. Cancer Res. 1994, 54, 2837–2840. [Google Scholar]
- Lae, M.E.; Roche, P.C.; Jin, L.; Lloyd, R.V.; Nascimento, A.G. Desmoplastic small round cell tumor: A clinicopathologic, immunohistochemical, and molecular study of 32 tumors. Am. J. Surg. Pathol. 2002, 26, 823–835. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Gerald, W.L.; Magid, M.S.; Ladanyi, M. Molecular variants of the EWS-WT1 gene fusion in desmoplastic small round cell tumor. Diagn. Mol. Pathol. 1998, 7, 24–28. [Google Scholar] [CrossRef]
- Gerald, W.L.; Ladanyi, M.; de Alava, E.; Cuatrecasas, M.; Kushner, B.H.; LaQuaglia, M.P.; Rosai, J. Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): Desmoplastic small round-cell tumor and its variants. J. Clin. Oncol. 1998, 16, 3028–3036. [Google Scholar] [CrossRef]
- Mora, J.; Modak, S.; Cheung, N.K.; Meyers, P.; de Alava, E.; Kushner, B.; Magnan, H.; Tirado, O.M.; Laquaglia, M.; Ladanyi, M.; et al. Desmoplastic small round cell tumor 20 years after its discovery. Future Oncol. 2015, 11, 1071–1081. [Google Scholar] [CrossRef]
- Lee, J.C.; Villanueva-Meyer, J.E.; Ferris, S.P.; Cham, E.M.; Zucker, J.; Cooney, T.; Gilani, A.; Kleinschmidt-DeMasters, B.K.; Trembath, D.; Mafra, M.; et al. Clinicopathologic and molecular features of intracranial desmoplastic small round cell tumors. Brain Pathol. 2020, 30, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Ordóñez, N.G. Desmoplastic small round cell tumor: II: An ultrastructural and immunohistochemical study with emphasis on new immunohistochemical markers. Am. J. Surg. Pathol. 1998, 22, 1314–1327. [Google Scholar] [CrossRef] [PubMed]
- Barnoud, R.; Delattre, O.; Péoc’h, M.; Pasquier, D.; Plantaz, D.; Leroux, D.; Pasquier, B. Desmoplastic small round cell tumor: RT-PCR analysis and immunohistochemical detection of the Wilm’s tumor gene WT1. Pathol. Res. Pract. 1998, 194, 693–700. [Google Scholar] [CrossRef]
- La Starza, R.; Barba, G.; Nofrini, V.; Pierini, T.; Pierini, V.; Marcomigni, L.; Perruccio, K.; Matteucci, C.; Storlazzi, C.T.; Daniele, G.; et al. Multiple EWSR1-WT1 and WT1-EWSR1 copies in two cases of desmoplastic round cell tumor. Cancer Genet. 2013, 206, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Dufresne, A.; Cassier, P.; Couraud, L.; Marec-Bérard, P.; Meeus, P.; Alberti, L.; Blay, J.Y. Desmoplastic small round cell tumor: Current management and recent findings. Sarcoma 2012, 2012, 714986. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Rosolen, A.; Sartori, F.; Leszl, A.; d’Amore, E.S.; Bisogno, G.; Carli, M.; Cecchetto, G.; Coffin, C.M.; Ninfo, V. Spindle cell tumor with EWS-WT1 transcript and a favorable clinical course: A variant of DSCT, a variant of leiomyosarcoma, or a new entity? Report of 2 pediatric cases. Am. J. Surg. Pathol. 2007, 31, 454–459. [Google Scholar] [CrossRef]
- Din, N.U.; Pekmezci, M.; Javed, G.; Horvai, A.E.; Ahmad, Z.; Faheem, M.; Navarro, A.L.; López-Terrada, D.; Perry, A. Low-grade small round cell tumor of the cauda equina with EWSR1-WT1 fusion and indolent clinical course. Hum. Pathol. 2015, 46, 153–158. [Google Scholar] [CrossRef]
- Bolen, J.W.; Thorning, D. Benign lipoblastoma and myxoid liposarcoma: A comparative light- and electron-microscopic study. Am. J. Surg. Pathol. 1980, 4, 163–174. [Google Scholar] [CrossRef]
- Haniball, J.; Sumathi, V.P.; Kindblom, L.G.; Abudu, A.; Carter, S.R.; Tillman, R.M.; Jeys, L.; Spooner, D.; Peake, D.; Grimer, R.J. Prognostic factors and metastatic patterns in primary myxoid/round-cell liposarcoma. Sarcoma 2011, 2011, 538085. [Google Scholar] [CrossRef] [PubMed]
- Bekers, E.M.; Song, W.; Suurmeijer, A.J.; Bonenkamp, J.J.; van der Geest, I.C.; Braam, P.M.; Ploegmakers, M.J.; Desar, I.M.; Tops, B.B.; van Gorp, J.M.; et al. Myxoid liposarcoma of the foot: A study of 8 cases. Ann. Diagn. Pathol. 2016, 25, 37–41. [Google Scholar] [CrossRef]
- Limon, J.; Turc-Carel, C.; Dal Cin, P.; Rao, U.; Sandberg, A.A. Recurrent chromosome translocations in liposarcoma. Cancer Genet. Cytogenet. 1986, 22, 93–94. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Höglund, M.; Mertens, F.; Mandahl, N.; Mitelman, F.; Aman, P. Fusion of the EWS and CHOP genes in myxoid liposarcoma. Oncogene 1996, 12, 489–494. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Mertens, F.; Isaksson, M.; Mandahl, N. A novel FUS/CHOP chimera in myxoid liposarcoma. Biochem. Biophys. Res. Commun. 2000, 279, 838–845. [Google Scholar] [CrossRef]
- Tallini, G.; Akerman, M.; Dal Cin, P.; De Wever, I.; Fletcher, C.D.; Mandahl, N.; Mertens, F.; Mitelman, F.; Rosai, J.; Rydholm, A.; et al. Combined morphologic and karyotypic study of 28 myxoid liposarcomas. Implications for a revised morphologic typing, (a report from the CHAMP Group). Am. J. Surg. Pathol. 1996, 20, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.C.; Renwick, P.J.; Dal Cin, P.; Van den Berghe, H.; Fletcher, C.D. Translocation t(12;16)(q13;p11) in myxoid liposarcoma and round cell liposarcoma: Molecular and cytogenetic analysis. Cancer Res. 1995, 55, 24–27. [Google Scholar] [PubMed]
- Evans, H.L. Liposarcoma: A study of 55 cases with a reassessment of its classification. Am. J. Surg. Pathol. 1979, 3, 507–523. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Coffin, C.M.; Weiss, S.W.; Bridge, J.A.; Issakov, J.; Oliveira, A.M.; Folpe, A.L. Liposarcomas in young patients: A study of 82 cases occurring in patients younger than 22 years of age. Am. J. Surg. Pathol. 2009, 33, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Fritchie, K.J.; Goldblum, J.R.; Tubbs, R.R.; Sun, Y.; Carver, P.; Billings, S.D.; Rubin, B.P. The expanded histologic spectrum of myxoid liposarcoma with an emphasis on newly described patterns: Implications for diagnosis on small biopsy specimens. Am. J. Clin. Pathol. 2012, 137, 229–239. [Google Scholar] [CrossRef]
- Dadone, B.; Refae, S.; Lemarié-Delaunay, C.; Bianchini, L.; Pedeutour, F. Molecular cytogenetics of pediatric adipocytic tumors. Cancer Genet. 2015, 208, 469–481. [Google Scholar] [CrossRef]
- Dei Tos, A.P. Liposarcomas: Diagnostic pitfalls and new insights. Histopathology 2014, 64, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Estourgie, S.H.; Nielsen, G.P.; Ott, M.J. Metastatic patterns of extremity myxoid liposarcoma and their outcome. J. Surg. Oncol. 2002, 80, 89–93. [Google Scholar] [CrossRef] [PubMed]
- De Vreeze, R.S.; de Jong, D.; Tielen, I.H.; Ruijter, H.J.; Nederlof, P.M.; Haas, R.L.; van Coevorden, F. Primary retroperitoneal myxoid/round cell liposarcoma is a nonexisting disease: An immunohistochemical and molecular biological analysis. Mod. Pathol. 2009, 22, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Scapa, J.V.; Cloutier, J.M.; Raghavan, S.S.; Peters-Schulze, G.; Varma, S.; Charville, G.W. DDIT3 Immunohistochemistry Is a Useful Tool for the Diagnosis of Myxoid Liposarcoma. Am. J. Surg. Pathol. 2021, 45, 230–239. [Google Scholar] [CrossRef]
- Baranov, E.; Black, M.A.; Fletcher, C.D.M.; Charville, G.W.; Hornick, J.L. Nuclear expression of DDIT3 distinguishes high-grade myxoid liposarcoma from other round cell sarcomas. Mod. Pathol. 2021. [Google Scholar] [CrossRef]
- Vargas, A.C.; Chan, N.L.; Wong, D.D.; Zaborowski, M.; Fuchs, T.L.; Ahadi, M.; Clarkson, A.; Sioson, L.; Sheen, A.; Maclean, F.; et al. DNA damage-inducible transcript 3 immunohistochemistry is highly sensitive for the diagnosis of myxoid liposarcoma but care is required in interpreting the significance of focal expression. Histopathology 2021. [Google Scholar] [CrossRef]
- Lansu, J.; Bovée, J.; Braam, P.; van Boven, H.; Flucke, U.; Bonenkamp, J.J.; Miah, A.B.; Zaidi, S.H.; Thway, K.; Bruland, Ø.S.; et al. Dose Reduction of Preoperative Radiotherapy in Myxoid Liposarcoma: A Nonrandomized Controlled Trial. JAMA Oncol. 2021, 7, e205865. [Google Scholar] [CrossRef]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef]
- Yoshida, A.; Wakai, S.; Ryo, E.; Miyata, K.; Miyazawa, M.; Yoshida, K.I.; Motoi, T.; Ogawa, C.; Iwata, S.; Kobayashi, E.; et al. Expanding the Phenotypic Spectrum of Mesenchymal Tumors Harboring the EWSR1-CREM Fusion. Am. J. Surg. Pathol. 2019, 43, 1622–1630. [Google Scholar] [CrossRef] [PubMed]
- Enzinger, F.M. Angiomatoid malignant fibrous histiocytoma: A distinct fibrohistiocytic tumor of children and young adults simulating a vascular neoplasm. Cancer 1979, 44, 2147–2157. [Google Scholar] [CrossRef]
- Waters, B.L.; Panagopoulos, I.; Allen, E.F. Genetic characterization of angiomatoid fibrous histiocytoma identifies fusion of the FUS and ATF-1 genes induced by a chromosomal translocation involving bands 12q13 and 16p11. Cancer Genet. Cytogenet. 2000, 121, 109–116. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Dal Cin, P.; Nafa, K.; Teot, L.A.; Surti, U.; Fletcher, C.D.; Ladanyi, M. EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 2007, 46, 1051–1060. [Google Scholar] [CrossRef]
- Tornóczky, T.; Bogner, B.; Krausz, T.; Ottóffy, G.; Szuhai, K. Angiomatoid fibrous histiocytoma: Pleomorphic variant associated with multiplication of EWSR1-CREB1 fusion gene. Pathol. Oncol. Res. 2012, 18, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Fanburg-Smith, J.C.; Miettinen, M. Angiomatoid “malignant” fibrous histiocytoma: A clinicopathologic study of 158 cases and further exploration of the myoid phenotype. Hum. Pathol. 1999, 30, 1336–1343. [Google Scholar] [CrossRef]
- Thway, K.; Fisher, C. Tumors with EWSR1-CREB1 and EWSR1-ATF1 fusions: The current status. Am. J. Surg. Pathol. 2012, 36, e1–e11. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.D. Angiomatoid “malignant fibrous histiocytoma”: An immunohistochemical study indicative of myoid differentiation. Hum. Pathol. 1991, 22, 563–568. [Google Scholar] [CrossRef]
- Chen, G.; Folpe, A.L.; Colby, T.V.; Sittampalam, K.; Patey, M.; Chen, M.G.; Chan, J.K. Angiomatoid fibrous histiocytoma: Unusual sites and unusual morphology. Mod. Pathol. 2011, 24, 1560–1570. [Google Scholar] [CrossRef] [PubMed]
- Thway, K.; Nicholson, A.G.; Wallace, W.A.; Al-Nafussi, A.; Pilling, J.; Fisher, C. Endobronchial pulmonary angiomatoid fibrous histiocytoma: Two cases with EWSR1-CREB1 and EWSR1-ATF1 fusions. Am. J. Surg. Pathol. 2012, 36, 883–888. [Google Scholar] [CrossRef]
- Thway, K.; Fisher, C. Angiomatoid fibrous histiocytoma: The current status of pathology and genetics. Arch. Pathol. Lab. Med. 2015, 139, 674–682. [Google Scholar] [CrossRef]
- Bale, T.A.; Oviedo, A.; Kozakewich, H.; Giannini, C.; Davineni, P.K.; Ligon, K.; Alexandrescu, S. Intracranial myxoid mesenchymal tumors with EWSR1-CREB family gene fusions: Myxoid variant of angiomatoid fibrous histiocytoma or novel entity? Brain Pathol. 2018, 28, 183–191. [Google Scholar] [CrossRef]
- Kao, Y.C.; Lan, J.; Tai, H.C.; Li, C.F.; Liu, K.W.; Tsai, J.W.; Fang, F.M.; Yu, S.C.; Huang, H.Y. Angiomatoid fibrous histiocytoma: Clinicopathological and molecular characterisation with emphasis on variant histomorphology. J. Clin. Pathol. 2014, 67, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Sloan, E.A.; Chiang, J.; Villanueva-Meyer, J.E.; Alexandrescu, S.; Eschbacher, J.M.; Wang, W.; Mafra, M.; Ud Din, N.; Carr-Boyd, E.; Watson, M.; et al. Intracranial mesenchymal tumor with FET-CREB fusion—A unifying diagnosis for the spectrum of intracranial myxoid mesenchymal tumors and angiomatoid fibrous histiocytoma-like neoplasms. Brain Pathol. 2020, e12918. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, I.M.; Fletcher, C.D. Myxoid variant of so-called angiomatoid “malignant fibrous histiocytoma”: Clinicopathologic characterization in a series of 21 cases. Am. J. Surg. Pathol. 2014, 38, 816–823. [Google Scholar] [CrossRef]
- Thway, K.; Nicholson, A.G.; Lawson, K.; Gonzalez, D.; Rice, A.; Balzer, B.; Swansbury, J.; Min, T.; Thompson, L.; Adu-Poku, K.; et al. Primary pulmonary myxoid sarcoma with EWSR1-CREB1 fusion: A new tumor entity. Am. J. Surg. Pathol. 2011, 35, 1722–1732. [Google Scholar] [CrossRef]
- Arbajian, E.; Puls, F.; Antonescu, C.R.; Amary, F.; Sciot, R.; Debiec-Rychter, M.; Sumathi, V.P.; Järås, M.; Magnusson, L.; Nilsson, J.; et al. In-depth Genetic Analysis of Sclerosing Epithelioid Fibrosarcoma Reveals Recurrent Genomic Alterations and Potential Treatment Targets. Clin. Cancer Res. 2017, 23, 7426–7434. [Google Scholar] [CrossRef]
- Liu, C.; Liu, Y.; Zhao, Y.; Wei, J.; Ma, Y.; Liu, Y.; Huang, J. Primary Intracranial Mesenchymal Tumor with EWSR1-CREM Gene Fusion: A Case Report and Literature Review. World Neurosurg. 2020, 142, 318–324. [Google Scholar] [CrossRef]
- Enzinger, F.M. CLEAR-CELL SARCOMA OF TENDONS AND APONEUROSES. AN ANALYSIS OF 21 CASES. Cancer 1965, 18, 1163–1174. [Google Scholar] [CrossRef]
- Chung, E.B.; Enzinger, F.M. Malignant melanoma of soft parts. A reassessment of clear cell sarcoma. Am. J. Surg. Pathol. 1983, 7, 405–413. [Google Scholar] [CrossRef]
- Bridge, J.A.; Borek, D.A.; Neff, J.R.; Huntrakoon, M. Chromosomal abnormalities in clear cell sarcoma. Implications for histogenesis. Am. J. Clin. Pathol. 1990, 93, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Peulvé, P.; Michot, C.; Vannier, J.P.; Tron, P.; Hemet, J. Clear cell sarcoma with t(12;22) (q13-14;q12). Genes Chromosomes Cancer 1991, 3, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.A. Translocation (12;22)(q13-14;q12) is a nonrandom aberration in soft-tissue clear-cell sarcoma. Genes Chromosomes Cancer 1992, 5, 184. [Google Scholar] [CrossRef]
- Zucman, J.; Delattre, O.; Desmaze, C.; Epstein, A.L.; Stenman, G.; Speleman, F.; Fletchers, C.D.; Aurias, A.; Thomas, G. EWS and ATF-1 gene fusion induced by t(12;22) translocation in malignant melanoma of soft parts. Nat. Genet. 1993, 4, 341–345. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Nafa, K.; Segal, N.H.; Dal Cin, P.; Ladanyi, M. EWS-CREB1: A recurrent variant fusion in clear cell sarcoma--association with gastrointestinal location and absence of melanocytic differentiation. Clin. Cancer Res. 2006, 12, 5356–5362. [Google Scholar] [CrossRef]
- Hisaoka, M.; Ishida, T.; Kuo, T.T.; Matsuyama, A.; Imamura, T.; Nishida, K.; Kuroda, H.; Inayama, Y.; Oshiro, H.; Kobayashi, H.; et al. Clear cell sarcoma of soft tissue: A clinicopathologic, immunohistochemical, and molecular analysis of 33 cases. Am. J. Surg. Pathol. 2008, 32, 452–460. [Google Scholar] [CrossRef]
- Coindre, J.M.; Hostein, I.; Terrier, P.; Bouvier-Labit, C.; Collin, F.; Michels, J.J.; Trassard, M.; Marques, B.; Ranchere, D.; Guillou, L. Diagnosis of clear cell sarcoma by real-time reverse transcriptase-polymerase chain reaction analysis of paraffin embedded tissues: Clinicopathologic and molecular analysis of 44 patients from the French sarcoma group. Cancer 2006, 107, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.A.; Johnson, M.B.; Thway, K.; Fisher, C.; Thomas, J.M.; Hayes, A.J. Clear cell sarcoma (melanoma of soft parts): The Royal Marsden Hospital experience. Eur. J. Surg. Oncol. 2008, 34, 800–804. [Google Scholar] [CrossRef]
- Wang, W.L.; Mayordomo, E.; Zhang, W.; Hernandez, V.S.; Tuvin, D.; Garcia, L.; Lev, D.C.; Lazar, A.J.; López-Terrada, D. Detection and characterization of EWSR1/ATF1 and EWSR1/CREB1 chimeric transcripts in clear cell sarcoma (melanoma of soft parts). Mod. Pathol. 2009, 22, 1201–1209. [Google Scholar] [CrossRef]
- Gonzaga, M.I.; Grant, L.; Curtin, C.; Gootee, J.; Silberstein, P.; Voth, E. The epidemiology and survivorship of clear cell sarcoma: A National Cancer Database (NCDB) review. J. Cancer Res. Clin. Oncol. 2018, 144, 1711–1716. [Google Scholar] [CrossRef] [PubMed]
- Lucas, D.R.; Nascimento, A.G.; Sim, F.H. Clear cell sarcoma of soft tissues. Mayo Clinic experience with 35 cases. Am. J. Surg. Pathol. 1992, 16, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Tirabosco, R.; Lang-Lazdunski, L.; Diss, T.C.; Amary, M.F.; Rodriguez-Justo, M.; Landau, D.; Lorenzi, W.; Flanagan, A.M. Clear cell sarcoma of the mediastinum. Ann. Diagn. Pathol. 2009, 13, 197–200. [Google Scholar] [CrossRef]
- Hantschke, M.; Mentzel, T.; Rütten, A.; Palmedo, G.; Calonje, E.; Lazar, A.J.; Kutzner, H. Cutaneous clear cell sarcoma: A clinicopathologic, immunohistochemical, and molecular analysis of 12 cases emphasizing its distinction from dermal melanoma. Am. J. Surg. Pathol. 2010, 34, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, T.; Menzies, A.M.; Roberts-Thomson, R. Recent advancements in melanoma management. Intern. Med. J. 2021, 51, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Ekfors, T.O.; Kujari, H.; Isomäki, M. Clear cell sarcoma of tendons and aponeuroses (malignant melanoma of soft parts) in the duodenum: The first visceral case. Histopathology 1993, 22, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Alpers, C.E.; Beckstead, J.H. Malignant neuroendocrine tumor of the jejunum with osteoclast-like giant cells. Enzyme histochemistry distinguishes tumor cells from giant cells. Am. J. Surg. Pathol. 1985, 9, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Donner, L.R.; Trompler, R.A.; Dobin, S. Clear cell sarcoma of the ileum: The crucial role of cytogenetics for the diagnosis. Am. J. Surg. Pathol. 1998, 22, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Thway, K. Clear cell sarcoma-like tumor of the gastrointestinal tract: An evolving entity. Arch. Pathol. Lab. Med. 2015, 139, 407–412. [Google Scholar] [CrossRef]
- Zambrano, E.; Reyes-Mugica, M.; Franchi, A.; Rosai, J. An osteoclast-rich tumor of the gastrointestinal tract with features resembling clear cell sarcoma of soft parts: Reports of 6 cases of a GIST simulator. Int. J. Surg. Pathol. 2003, 11, 75–81. [Google Scholar] [CrossRef]
- Stockman, D.L.; Miettinen, M.; Suster, S.; Spagnolo, D.; Dominguez-Malagon, H.; Hornick, J.L.; Adsay, V.; Chou, P.M.; Amanuel, B.; Vantuinen, P.; et al. Malignant gastrointestinal neuroectodermal tumor: Clinicopathologic, immunohistochemical, ultrastructural, and molecular analysis of 16 cases with a reappraisal of clear cell sarcoma-like tumors of the gastrointestinal tract. Am. J. Surg. Pathol. 2012, 36, 857–868. [Google Scholar] [CrossRef]
- Segawa, K.; Sugita, S.; Aoyama, T.; Kubo, T.; Asanuma, H.; Sugawara, T.; Ito, Y.; Tsujiwaki, M.; Fujita, H.; Emori, M.; et al. Detection of specific gene rearrangements by fluorescence in situ hybridization in 16 cases of clear cell sarcoma of soft tissue and 6 cases of clear cell sarcoma-like gastrointestinal tumor. Diagn. Pathol. 2018, 13, 73. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Thorsen, J.; Gorunova, L.; Micci, F.; Haugom, L.; Davidson, B.; Heim, S. RNA sequencing identifies fusion of the EWSR1 and YY1 genes in mesothelioma with t(14;22)(q32;q12). Genes Chromosomes Cancer 2013, 52, 733–740. [Google Scholar] [CrossRef]
- Desmeules, P.; Joubert, P.; Zhang, L.; Al-Ahmadie, H.A.; Fletcher, C.D.; Vakiani, E.; Delair, D.F.; Rekhtman, N.; Ladanyi, M.; Travis, W.D.; et al. A Subset of Malignant Mesotheliomas in Young Adults Are Associated With Recurrent EWSR1/FUS-ATF1 Fusions. Am. J. Surg. Pathol. 2017, 41, 980–988. [Google Scholar] [CrossRef]
- Argani, P.; Harvey, I.; Nielsen, G.P.; Takano, A.; Suurmeijer, A.J.H.; Voltaggio, L.; Zhang, L.; Sung, Y.S.; Stenzinger, A.; Mechtersheimer, G.; et al. EWSR1/FUS-CREB fusions define a distinctive malignant epithelioid neoplasm with predilection for mesothelial-lined cavities. Mod. Pathol. 2020, 33, 2233–2243. [Google Scholar] [CrossRef]
- Stout, A.P.; Gorman, J.G. Mixed tumors of the skin of the salivary gland type. Cancer 1959, 12, 537–543. [Google Scholar] [CrossRef]
- De Pinieux, G.; Beabout, J.W.; Unni, K.K.; Sim, F.H. Primary mixed tumor of bone. Skeletal Radiol 2001, 30, 534–536. [Google Scholar] [CrossRef] [PubMed]
- Dabska, M. Parachordoma: A new clinicopathologic entity. Cancer 1977, 40, 1586–1592. [Google Scholar] [CrossRef]
- Limon, J.; Babińska, M.; Denis, A.; Ryś, J.; Niezabitowski, A. Parachordoma: A rare sarcoma with clonal chromosomal changes. Cancer Genet. Cytogenet. 1998, 102, 78–80. [Google Scholar] [CrossRef]
- Tihy, F.; Scott, P.; Russo, P.; Champagne, M.; Tabet, J.C.; Lemieux, N. Cytogenetic analysis of a parachordoma. Cancer Genet. Cytogenet. 1998, 105, 14–19. [Google Scholar] [CrossRef]
- Pauwels, P.; Dal Cin, P.; Roumen, R.; van den Berghe, H.; Sciot, R. Intramuscular mixed tumour with clonal chromosomal changes. Virchows Arch. 1999, 434, 167–171. [Google Scholar] [CrossRef]
- Van den Berg, E.; Zorgdrager, H.; Hoekstra, H.J.; Suurmeijer, A.J. Cytogenetics of a soft tissue malignant myoepithelioma. Cancer Genet. Cytogenet. 2004, 151, 87–89. [Google Scholar] [CrossRef]
- Hallor, K.H.; Teixeira, M.R.; Fletcher, C.D.; Bizarro, S.; Staaf, J.; Domanski, H.A.; von Steyern, F.V.; Panagopoulos, I.; Mandahl, N.; Mertens, F. Heterogeneous genetic profiles in soft tissue myoepitheliomas. Mod. Pathol. 2008, 21, 1311–1319. [Google Scholar] [CrossRef]
- Rekhi, B.; Amare, P.; Gulia, A.; Baisane, C.; Patil, A.; Agarwal, S.; Puri, A.; Jambhekar, N.A. Primary intraosseous myoepithelioma arising in the iliac bone and displaying trisomies of 11, 15, 17 with del (16q) and del (22q11)—A rare case report with review of literature. Pathol. Res. Pract. 2011, 207, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Gleason, B.C.; Fletcher, C.D. Myoepithelial carcinoma of soft tissue in children: An aggressive neoplasm analyzed in a series of 29 cases. Am. J. Surg. Pathol. 2007, 31, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Brandal, P.; Panagopoulos, I.; Bjerkehagen, B.; Gorunova, L.; Skjeldal, S.; Micci, F.; Heim, S. Detection of a t(1;22)(q23;q12) translocation leading to an EWSR1-PBX1 fusion gene in a myoepithelioma. Genes Chromosomes Cancer 2008, 47, 558–564. [Google Scholar] [CrossRef]
- Brandal, P.; Panagopoulos, I.; Bjerkehagen, B.; Heim, S. t(19;22)(q13;q12) Translocation leading to the novel fusion gene EWSR1-ZNF444 in soft tissue myoepithelial carcinoma. Genes Chromosomes Cancer 2009, 48, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.R.; Zhang, L.; Chang, N.E.; Pawel, B.R.; Travis, W.; Katabi, N.; Edelman, M.; Rosenberg, A.E.; Nielsen, G.P.; Dal Cin, P.; et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer 2010, 49, 1114–1124. [Google Scholar] [CrossRef]
- Flucke, U.; Palmedo, G.; Blankenhorn, N.; Slootweg, P.J.; Kutzner, H.; Mentzel, T. EWSR1 gene rearrangement occurs in a subset of cutaneous myoepithelial tumors: A study of 18 cases. Mod. Pathol. 2011, 24, 1444–1450. [Google Scholar] [CrossRef]
- Puls, F.; Arbajian, E.; Magnusson, L.; Douis, H.; Kindblom, L.G.; Mertens, F. Myoepithelioma of bone with a novel FUS-POU5F1 fusion gene. Histopathology 2014, 65, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Agaram, N.P.; Chen, H.W.; Zhang, L.; Sung, Y.S.; Panicek, D.; Healey, J.H.; Nielsen, G.P.; Fletcher, C.D.; Antonescu, C.R. EWSR1-PBX3: A novel gene fusion in myoepithelial tumors. Genes Chromosomes Cancer 2015, 54, 63–71. [Google Scholar] [CrossRef]
- Huang, S.C.; Chen, H.W.; Zhang, L.; Sung, Y.S.; Agaram, N.P.; Davis, M.; Edelman, M.; Fletcher, C.D.; Antonescu, C.R. Novel FUS-KLF17 and EWSR1-KLF17 fusions in myoepithelial tumors. Genes Chromosomes Cancer 2015, 54, 267–275. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Zhang, L.; Shao, S.Y.; Mosquera, J.M.; Weinreb, I.; Katabi, N.; Fletcher, C.D. Frequent PLAG1 gene rearrangements in skin and soft tissue myoepithelioma with ductal differentiation. Genes Chromosomes Cancer 2013, 52, 675–682. [Google Scholar] [CrossRef]
- Pairet, G.; Tilman, G.; Sciot, R.; Schubert, T.; Perlepe, V.; Poirel, H.A.; Galant, C. Case Reports in Oncological Medicine Myoepithelioma: A New Rearrangement Involving the LPP Locus in a Case of Multiple Bone and Soft Tissue Lesions. Case Rep. Oncol. Med. 2018, 2018, 3512847. [Google Scholar] [CrossRef]
- Hornick, J.L.; Fletcher, C.D. Myoepithelial tumors of soft tissue: A clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am. J. Surg. Pathol. 2003, 27, 1183–1196. [Google Scholar] [CrossRef]
- Thway, K.; Fisher, C. Myoepithelial tumor of soft tissue: Histology and genetics of an evolving entity. Adv. Anat. Pathol. 2014, 21, 411–419. [Google Scholar] [CrossRef]
- Gleason, B.C.; Hornick, J.L. Myoepithelial tumours of skin and soft tissue: An update. Diagn. Histopathol. 2008, 14, 552–562. [Google Scholar] [CrossRef]
- Song, W.; Flucke, U.; Suurmeijer, A.J.H. Myoepithelial Tumors of Bone. Surg. Pathol. Clin. 2017, 10, 657–674. [Google Scholar] [CrossRef]
- Jo, V.Y.; Antonescu, C.R.; Zhang, L.; Dal Cin, P.; Hornick, J.L.; Fletcher, C.D. Cutaneous syncytial myoepithelioma: Clinicopathologic characterization in a series of 38 cases. Am. J. Surg. Pathol. 2013, 37, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Tirabosco, R.; Mangham, D.C.; Rosenberg, A.E.; Vujovic, S.; Bousdras, K.; Pizzolitto, S.; De Maglio, G.; den Bakker, M.A.; Di Francesco, L.; Kalil, R.K.; et al. Brachyury expression in extra-axial skeletal and soft tissue chordomas: A marker that distinguishes chordoma from mixed tumor/myoepithelioma/parachordoma in soft tissue. Am. J. Surg. Pathol. 2008, 32, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Guillou, L.; Benhattar, J.; Gengler, C.; Gallagher, G.; Ranchère-Vince, D.; Collin, F.; Terrier, P.; Terrier-Lacombe, M.J.; Leroux, A.; Marquès, B.; et al. Translocation-positive low-grade fibromyxoid sarcoma: Clinicopathologic and molecular analysis of a series expanding the morphologic spectrum and suggesting potential relationship to sclerosing epithelioid fibrosarcoma: A study from the French Sarcoma Group. Am. J. Surg. Pathol. 2007, 31, 1387–1402. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.R.; Rosenblum, M.K.; Pereira, P.; Nascimento, A.G.; Woodruff, J.M. Sclerosing epithelioid fibrosarcoma: A study of 16 cases and confirmation of a clinicopathologically distinct tumor. Am. J. Surg. Pathol. 2001, 25, 699–709. [Google Scholar] [CrossRef]
- Evans, H.L. Low-grade fibromyxoid sarcoma. A report of two metastasizing neoplasms having a deceptively benign appearance. Am. J. Clin. Pathol. 1987, 88, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Meis-Kindblom, J.M.; Kindblom, L.G.; Enzinger, F.M. Sclerosing epithelioid fibrosarcoma. A variant of fibrosarcoma simulating carcinoma. Am. J. Surg. Pathol. 1995, 19, 979–993. [Google Scholar] [CrossRef]
- Reid, R.; de Silva, M.V.; Paterson, L.; Ryan, E.; Fisher, C. Low-grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes share a common t(7;16)(q34;p11) translocation. Am. J. Surg. Pathol. 2003, 27, 1229–1236. [Google Scholar] [CrossRef]
- Storlazzi, C.T.; Mertens, F.; Nascimento, A.; Isaksson, M.; Wejde, J.; Brosjo, O.; Mandahl, N.; Panagopoulos, I. Fusion of the FUS and BBF2H7 genes in low grade fibromyxoid sarcoma. Hum. Mol. Genet. 2003, 12, 2349–2358. [Google Scholar] [CrossRef]
- Mertens, F.; Fletcher, C.D.; Antonescu, C.R.; Coindre, J.M.; Colecchia, M.; Domanski, H.A.; Downs-Kelly, E.; Fisher, C.; Goldblum, J.R.; Guillou, L.; et al. Clinicopathologic and molecular genetic characterization of low-grade fibromyxoid sarcoma, and cloning of a novel FUS/CREB3L1 fusion gene. Lab. Investig. 2005, 85, 408–415. [Google Scholar] [CrossRef]
- Lau, P.P.; Lui, P.C.; Lau, G.T.; Yau, D.T.; Cheung, E.T.; Chan, J.K. EWSR1-CREB3L1 gene fusion: A novel alternative molecular aberration of low-grade fibromyxoid sarcoma. Am. J. Surg. Pathol. 2013, 37, 734–738. [Google Scholar] [CrossRef]
- Arbajian, E.; Puls, F.; Magnusson, L.; Thway, K.; Fisher, C.; Sumathi, V.P.; Tayebwa, J.; Nord, K.H.; Kindblom, L.G.; Mertens, F. Recurrent EWSR1-CREB3L1 gene fusions in sclerosing epithelioid fibrosarcoma. Am. J. Surg. Pathol. 2014, 38, 801–808. [Google Scholar] [CrossRef]
- Evans, H.L. Low-grade fibromyxoid sarcoma: A clinicopathologic study of 33 cases with long-term follow-up. Am. J. Surg. Pathol. 2011, 35, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.L. Low-grade fibromyxoid sarcoma. A report of 12 cases. Am. J. Surg. Pathol. 1993, 17, 595–600. [Google Scholar] [CrossRef]
- Mok, Y.; Pang, Y.H.; Sanjeev, J.S.; Kuick, C.H.; Chang, K.T. Primary Renal Hybrid Low-grade Fibromyxoid Sarcoma-Sclerosing Epithelioid Fibrosarcoma: An Unusual Pediatric Case With EWSR1-CREB3L1 Fusion. Pediatr. Dev. Pathol. 2018, 21, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Kosemehmetoglu, K.; Ardic, F.; Kilpatrick, S.E.; Aydingoz, U.; Sumathi, V.P.; Michal, M. Sclerosing epithelioid fibrosarcoma of bone: Morphological, immunophenotypical, and molecular findings of 9 cases. Virchows Arch. 2021, 478, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.A.; Möller, E.; Dal Cin, P.; Fletcher, C.D.; Mertens, F.; Hornick, J.L. MUC4 is a highly sensitive and specific marker for low-grade fibromyxoid sarcoma. Am. J. Surg. Pathol. 2011, 35, 733–741. [Google Scholar] [CrossRef]
- Doyle, L.A.; Wang, W.L.; Dal Cin, P.; Lopez-Terrada, D.; Mertens, F.; Lazar, A.J.; Fletcher, C.D.; Hornick, J.L. MUC4 is a sensitive and extremely useful marker for sclerosing epithelioid fibrosarcoma: Association with FUS gene rearrangement. Am. J. Surg. Pathol. 2012, 36, 1444–1451. [Google Scholar] [CrossRef]
- Stout, A.P.; Verner, E.W. Chondrosarcoma of the extraskeletal soft tissues. Cancer 1953, 6, 581–590. [Google Scholar] [CrossRef]
- Enzinger, F.M.; Shiraki, M. Extraskeletal myxoid chondrosarcoma. An analysis of 34 cases. Hum. Pathol. 1972, 3, 421–435. [Google Scholar] [CrossRef]
- Labelle, Y.; Zucman, J.; Stenman, G.; Kindblom, L.G.; Knight, J.; Turc-Carel, C.; Dockhorn-Dworniczak, B.; Mandahl, N.; Desmaze, C.; Peter, M.; et al. Oncogenic conversion of a novel orphan nuclear receptor by chromosome translocation. Hum. Mol. Genet. 1995, 4, 2219–2226. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, S.H.; Jaramillo, M.A.; Gumerlock, P.H.; Gardner, M.B.; Lewis, J.P.; Freeman, A.E. Myxoid chondrosarcoma with a translocation involving chromosomes 9 and 22. Cancer Genet. Cytogenet. 1985, 14, 219–226. [Google Scholar] [CrossRef]
- Hisaoka, M.; Hashimoto, H. Extraskeletal myxoid chondrosarcoma: Updated clinicopathological and molecular genetic characteristics. Pathol. Int. 2005, 55, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Flucke, U.; Tops, B.B.; Verdijk, M.A.; van Cleef, P.J.; van Zwam, P.H.; Slootweg, P.J.; Bovée, J.V.; Riedl, R.G.; Creytens, D.H.; Suurmeijer, A.J.; et al. NR4A3 rearrangement reliably distinguishes between the clinicopathologically overlapping entities myoepithelial carcinoma of soft tissue and cellular extraskeletal myxoid chondrosarcoma. Virchows Arch. 2012, 460, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Meis-Kindblom, J.M.; Bergh, P.; Gunterberg, B.; Kindblom, L.G. Extraskeletal myxoid chondrosarcoma: A reappraisal of its morphologic spectrum and prognostic factors based on 117 cases. Am. J. Surg. Pathol. 1999, 23, 636–650. [Google Scholar] [CrossRef]
- Hachitanda, Y.; Tsuneyoshi, M.; Daimaru, Y.; Enjoji, M.; Nakagawara, A.; Ikeda, K.; Sueishi, K. Extraskeletal myxoid chondrosarcoma in young children. Cancer 1988, 61, 2521–2526. [Google Scholar] [CrossRef]
- Demicco, E.G.; Wang, W.L.; Madewell, J.E.; Huang, D.; Bui, M.M.; Bridge, J.A.; Meis, J.M. Osseous myxochondroid sarcoma: A detailed study of 5 cases of extraskeletal myxoid chondrosarcoma of the bone. Am. J. Surg. Pathol. 2013, 37, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.D.; Popat, S.; Bhuchar, G.; D’Adamo, D.R.; Keohan, M.L.; Fisher, C.; Antonescu, C.R.; Singer, S.; Brennan, M.F.; Judson, I.; et al. Extraskeletal myxoid chondrosarcoma: A retrospective review from 2 referral centers emphasizing long-term outcomes with surgery and chemotherapy. Cancer 2008, 113, 3364–3371. [Google Scholar] [CrossRef]
- Kao, Y.C.; Flucke, U.; Eijkelenboom, A.; Zhang, L.; Sung, Y.S.; Suurmeijer, A.J.H.; Antonescu, C.R. Novel EWSR1-SMAD3 Gene Fusions in a Group of Acral Fibroblastic Spindle Cell Neoplasms. Am. J. Surg. Pathol. 2018, 42, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Michal, M.; Berry, R.S.; Rubin, B.P.; Kilpatrick, S.E.; Agaimy, A.; Kazakov, D.V.; Steiner, P.; Ptakova, N.; Martinek, P.; Hadravsky, L.; et al. EWSR1-SMAD3-rearranged Fibroblastic Tumor: An Emerging Entity in an Increasingly More Complex Group of Fibroblastic/Myofibroblastic Neoplasms. Am. J. Surg. Pathol. 2018, 42, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Sun, M.; Lao, I.W.; Yu, L.; Wang, J. EWSR1-SMAD3 positive fibroblastic tumor. Exp. Mol. Pathol. 2019, 110, 104291. [Google Scholar] [CrossRef]
- Foot, O.; Hallin, M.; Jones, R.L.; Sumathi, V.P.; Thway, K. EWSR1-SMAD3-Positive Fibroblastic Tumor. Int. J. Surg. Pathol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Habeeb, O.; Korty, K.E.; Azzato, E.M.; Astbury, C.; Farkas, D.H.; Ko, J.S.; Billings, S.D. EWSR1-SMAD3 rearranged fibroblastic tumor: Case series and review. J. Cutan. Pathol. 2021, 48, 255–262. [Google Scholar] [CrossRef]
- De Noon, S.; Flanagan, A.M.; Tirabosco, R.; O’Donnell, P.; Amary, F. EWSR1-SMAD3 fibroblastic tumour of bone: Expanding the clinical spectrum. Skelet. Radiol. 2021, 50, 445–450. [Google Scholar] [CrossRef]
- Le Loarer, F.; Cleven, A.H.G.; Bouvier, C.; Castex, M.P.; Romagosa, C.; Moreau, A.; Salas, S.; Bonhomme, B.; Gomez-Brouchet, A.; Laurent, C.; et al. A subset of epithelioid and spindle cell rhabdomyosarcomas is associated with TFCP2 fusions and common ALK upregulation. Mod. Pathol. 2020, 33, 404–419. [Google Scholar] [CrossRef]
- Dashti, N.K.; Wehrs, R.N.; Thomas, B.C.; Nair, A.; Davila, J.; Buckner, J.C.; Martinez, A.P.; Sukov, W.R.; Halling, K.C.; Howe, B.M.; et al. Spindle cell rhabdomyosarcoma of bone with FUS-TFCP2 fusion: Confirmation of a very recently described rhabdomyosarcoma subtype. Histopathology 2018, 73, 514–520. [Google Scholar] [CrossRef]
- Agaram, N.P.; Zhang, L.; Sung, Y.S.; Cavalcanti, M.S.; Torrence, D.; Wexler, L.; Francis, G.; Sommerville, S.; Swanson, D.; Dickson, B.C.; et al. Expanding the Spectrum of Intraosseous Rhabdomyosarcoma: Correlation Between 2 Distinct Gene Fusions and Phenotype. Am. J. Surg. Pathol. 2019, 43, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Chrisinger, J.S.A.; Wehrli, B.; Dickson, B.C.; Fasih, S.; Hirbe, A.C.; Shultz, D.B.; Zadeh, G.; Gupta, A.A.; Demicco, E.G. Epithelioid and spindle cell rhabdomyosarcoma with FUS-TFCP2 or EWSR1-TFCP2 fusion: Report of two cases. Virchows Arch. 2020, 477, 725–732. [Google Scholar] [CrossRef]
- Tagami, Y.; Sugita, S.; Kubo, T.; Iesato, N.; Emori, M.; Takada, K.; Tsujiwaki, M.; Segawa, K.; Sugawara, T.; Kikuchi, T.; et al. Spindle cell rhabdomyosarcoma in a lumbar vertebra with FUS-TFCP2 fusion. Pathol. Res. Pract. 2019, 215, 152399. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Gorunova, L.; Bjerkehagen, B.; Heim, S. Novel KAT6B-KANSL1 fusion gene identified by RNA sequencing in retroperitoneal leiomyoma with t(10;17)(q22;q21). PLoS ONE 2015, 10, e0117010. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Gorunova, L.; Bjerkehagen, B.; Heim, S. Fusion of the genes EWSR1 and PBX3 in retroperitoneal leiomyoma with t(9;22)(q33;q12). PLoS ONE 2015, 10, e0124288. [Google Scholar] [CrossRef]
- Noordin, S.; Allana, S.; Umer, M.; Jamil, M.; Hilal, K.; Uddin, N. Unicameral bone cysts: Current concepts. Ann. Med. Surg. 2018, 34, 43–49. [Google Scholar] [CrossRef]
- Hung, Y.P.; Fisch, A.S.; Diaz-Perez, J.A.; Iafrate, A.J.; Lennerz, J.K.; Nardi, V.; Bredella, M.A.; Raskin, K.A.; Lozano-Calderon, S.A.; Rosenberg, A.E.; et al. Identification of EWSR1-NFATC2 fusion in simple bone cysts. Histopathology 2020. [Google Scholar] [CrossRef]
- Virchow, R. On the formation of bony cysts. Über die Bildung von Knochencysten 1876, 369–381. [Google Scholar]
- Pižem, J.; Šekoranja, D.; Zupan, A.; Boštjančič, E.; Matjašič, A.; Mavčič, B.; Contreras, J.A.; Gazič, B.; Martinčič, D.; Snoj, Ž.; et al. FUS-NFATC2 or EWSR1-NFATC2 Fusions Are Present in a Large Proportion of Simple Bone Cysts. Am. J. Surg. Pathol. 2020, 44, 1623–1634. [Google Scholar] [CrossRef]
- Richkind, K.E.; Mortimer, E.; Mowery-Rushton, P.; Fraire, A. Translocation (16;20)(p11.2;q13). sole cytogenetic abnormality in a unicameral bone cyst. Cancer Genet. Cytogenet. 2002, 137, 153–155. [Google Scholar] [CrossRef]
- Mulliken, J.B.; Glowacki, J. Hemangiomas and vascular malformations in infants and children: A classification based on endothelial characteristics. Plast. Reconstr. Surg. 1982, 69, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Steele, L.; Zbeidy, S.; Thomson, J.; Flohr, C. How is the term haemangioma used in the literature? An evaluation against the revised ISSVA classification. Pediatr. Dermatol. 2019, 36, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Enjolras, O.; Mulliken, J.B. Vascular tumors and vascular malformations (new issues). Adv. Dermatol. 1997, 13, 375–423. [Google Scholar] [PubMed]
- Flucke, U.; Karanian, M.; Broek, R.W.T.; Thway, K. Soft Tissue Special Issue: Perivascular and Vascular Tumors of the Head and Neck. Head Neck Pathol. 2020, 14, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Arbajian, E.; Magnusson, L.; Brosjö, O.; Wejde, J.; Folpe, A.L.; Nord, K.H.; Mertens, F. A benign vascular tumor with a new fusion gene: EWSR1-NFATC1 in hemangioma of the bone. Am. J. Surg. Pathol. 2013, 37, 613–616. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entity | Morphology | IHC | Common Genetic Alterations |
|---|---|---|---|
| CIC-sarcoma | Sheets of undifferentiated round/spindle/epitheloid cells; mild nuclear pleomorphism; and necrosis | CD99 (mostly patchy), WT1, ETV4, DUX4, and NUT (CIC-NUTM1) | CIC-DUX4/FOXO4/LEUTX/NUTM1/2A fusions |
| BCOR-sarcoma | Sheets/nests/short fascicles of uniform; bland round-oval-spindle cells; rich capillary network; and myxoid matrix (variable) | BCOR, SATB2, cyclin D1, TLE1, CCNB3 (BCOR-CCNB3), and CD99 (50%) | BCOR-CCNB3/MAML3/ZC3H7B, KMT2D); BCOR ITD*; and YWHAE-NUTM2B; *ITD, internal tandem duplication |
| EWSR1-nonETS round-cell sarcomas | Cords/nests/pseudoacinar pattern of round-spindle cells; bland-pleomorphic spectrum; and fibro-/myxohyaline stroma | CD99, NKX2.2, and CKAE1/3 (focal, dot-like) | EWSR1/FUS-NFATc2 |
| Diverse morphology: round-spindle cells; fibrous stroma | Co-expression of myogenic markers (desmin/myogenin/MyoD1), neurogenic markers (S100/SOX10/MITF/GFAP) and keratins (AE1/3) | EWSR1-PATZ1 or EWSR1-VEZF1 | |
| Desmoplastic small round-cell tumor | Sheets/nests/cords of uniform; bland round cells; and desmoplastic stroma | Desmin (dot-like), keratin, EMA, and WT1 (C-terminus) | EWSR1-WT1 |
| Lymphoblastic lymphoma | Small-medium blastoid cells; minimal cytoplasm | CD99, TdT, CD45, CD34, CD1a, and B- and T-cel markers | Diverse |
| Small-cell carcinoma | Small-medium round-oval cells; salt and pepper chromatin; indistinct nucleoli; molding; and apoptosis | Keratins, CD56, synaptophysin, chromogranin, and TTF1 | Diverse; TP53, PTEN mutations; RB1, 3p loss; and MYC amplification |
| NUT carcinoma | Poorly cohesive sheets of primitive/basaloid cells; abrupt keratinization; and coagulative necrosis | CK5/6, P40, P63, and NUT | NUT-BRD3/BRD4/NSD3/CIC/BCORL1/MGA/MXD4 |
| Myoepithelial carcinoma | Solid sheets/nests of cell with high nuclear grade or undifferentiated round-cell morphology; facultatively glandular component; necrosis; and high mitotic count | Pankeratins, S100, EMA, GFAP, SOX10, P63, SMA, calponin, desmin (focal); and INI1 loss (subset) | EWSR1 rearrangements (various fusion partners); PLAG1 rearrangements (mixed tumors) |
| ARMS | Nests with central discohesion-solid nests; monomorphic primitive round cells; and multinucleated wreath-like giant cells | Desmin, myogenin (strong, diffuse), MyoD1, keratin, neuro-endocrine markers (CD56, synaptophysin, and chromogranin) | PAX3/PAX7-FOXO1 |
| Sinonasal glomangiopericytoma | Solid-fascicular pattern; spindle-round cells with minimal atypia; arranged around staghorn vessels; and perivascular hyalinization | Beta-catenin (nuclear), SMA | CTNNB1 mutations |
| Glomus tumor | Solid-nested pattern; small, uniform round cells with round nucleus, amphophilic-slightly eosinophilic cytoplasm and sharply defined cell borders; and variable vascular pattern | SMA with membranous accentuation, caldesmon, and collagen IV | MIR143-NOTCH1/2/3, and BRAF/KRAS mutations |
| Rhabdoid tumor | Solid pattern; rounded-polygonal cells with vesicular nuclei and prominent nucleoli; and eosinophil hyaline-like cytoplasmic inclusions | Diverse; keratins, EMA, CD99, synaptophysin, SALL4, glypican-3, and INI1 loss | SMARCB1 biallelic loss, SMARCB1 or SMARCA4 (germline) mutations |
| Mesenchymal chondrosarcoma | Biphasic: poorly differentiated round cells and islands of hyaline cartilage; staghorn-like vessels | S100, CD99, SOX9, EMA, desmin, myogenin, and MyoD1 | HEY1-NCOA2 |
| Synovial sarcoma with round-cell features | Fascicles or sheets of small round hyperchromatic cells; high N/C ratio; staghorn vessels; necrosis; and thin fibrovascular septa | CD99, BCL2, CD56, TLE1, S100 (focal), EMA, and keratins (variable) | SS18-SSX1/2/4 |
| Diagnosis | Morphology | IHC | Common Genetic Alterations |
|---|---|---|---|
| Solitary fibrous tumor | Patternless pattern of spindle cells or round cells; hyalinized stroma; collagen bundles; staghorn vessels; and possibly fat component | CD34, BCL2, CD99, and STAT6 | NAB2-STAT6 |
| Myoepithelial tumor | Divers spectrum; reticular/trabecular pattern; variable spindle/epithelioid/plasmocytoid/clear cells; rarely ductal component (mixed tumors); and myxoid stroma | Pankeratins, S100, SOX10, EMA, GFAP, P63, SMA, calponin, and desmin (focal) | EWSR1 rearrangements (various fusion partners); PLAG1 rearrangements (mixed tumors) |
| DSRCT | Sheets/nests/cords of uniform, bland, round cells; and desmoplastic stroma | Desmin (dot-like), keratin, EMA, and WT1 (C-terminus) | EWSR1-WT1 |
| CIC-sarcoma | Sheets of undifferentiated round/spindle/epitheloid cells; mild nuclear pleomorphism; and necrosis | CD99 (mostly patchy), WT1, ETV4, DUX4, and NUT (CIC-NUTM1) | CIC-DUX4/FOXO4/LEUTX/NUTM1/2A |
| BCOR-sarcoma | Sheets/nests/short fascicles of uniform, bland round-oval-spindle cells; rich capillary network; and myxoid matrix (variable) | BCOR, SATB2, cyclin D1, TLE1, CCNB3 (BCOR-CCNB3), and CD99 (50%) | BCOR-CCNB3/MAML3/ZC3H7B, KMT2D); BCOR ITD; and YWHAE-NUTM2B |
| ARMS | Nests/sheets with central discohesion (pseudoalveolar) or solid nests; monomorphic, primitive round cells; and multinucleated wreath-like giant cells | Desmin, myogenin (strong, diffuse), MyoD1, keratin, and neuro-endocrine markers (CD56, synaptophysin, chromogranin) | PAX3/PAX7-FOXO1 |
| Malignant peripheral nerve sheath tumor/Triton | Fascicles of spindle cells and/or sheets epithelioid cells with perivascular accentuation and alternating cellularity; staghorn vessels; geographic necrosis; and heterologous differentiation (rhabdomyoblasts, glandular structures) | S100, SOX10 (focally), and loss of H3K27me3 | Inactivating mutations of NF1, CDKN2A/B, EED, and SUZ2 |
| Synovial sarcoma | Sheets-fascicles; cellular, monomorphic spindle cells; high N/C ratio; variable epithelial differentiation; staghorn vessels; variable amount of collagen; mast cells; and calcification/ossification; poorly differentiated areas may show round-epithelioid cells | CD99, BCL2, CD56, TLE1, S100 (focal), EMA, and keratins (variable) | SS18-SSX1/2/4 |
| Diagnosis | Morphology | IHC | Common Genetic Alterations |
|---|---|---|---|
| Neuroblastoma | Primitive cells; rosettes; neuropil; and ganglion cells | CD56, synaptophysin, and chromogranin | nMYC amplification; ATRK, ALK mutations; and chromosomal aberrations (1p, 17q, and 11q) |
| Lymphoma | Variably sized hyperchromatic-blastoid cells with variable atypia; minimal cytoplasm | CD45, B/T-cell markers | Diverse |
| Blastemic Wilms | Primitive, undifferentiated round-to-spindled cells | WT1 (N-terminus), CD56 | WT1, TP53 mutations; 11p15.5 deletion |
| Small cell/neuroendocrine carcinoma | Small-medium round-oval cells; salt and pepper chromatin; indistinct nucleoli; molding; and apoptosis | Keratins CD56, synaptophysin, chromogranin, and TTF1 | Diverse; TP53, PTEN mutations; RB1, 3p loss; and MYC amplification |
| Metastatic Merkel cell carcinoma | Round-oval nuclei; high N/C-ratio; salt and pepper chromatin; indistinct nucleoli; molding; and apoptosis | Broad spectrum keratins; CK20 (dot-like), CD56, chromogranin, and synaptophysin | Diverse; depends on polyomavirus (PyV) status; PyV- tumors: Rb1, TP53 mutations |
| Small cell mesothelioma | Solid nests; high N/C-ratio; well defined membrane; fine chromatin; and indistinct nucleoli | Calretinin, CK5/6, WT1 (N-terminus), and D2-40 | Diverse; P16 loss; BAP1 mutation; NF2 deletion; and ALK/EWSR1/FUS rearrangments |
| Diagnosis | Morphology | IHC | Common Genetic Alterations |
|---|---|---|---|
| Lipoblastoma | Lobulated architecture with fibrous septa with often prominent vasculature; possibly myxoid stroma with possibly plexiform vasculature; resembling fetal fat with prelipoblasts, lipoblasts, and mature fat in variable portions | Not specific | PLAG1 rearrangements (various fusion partners) |
| Myxoid pleomorphic liposarcoma | Progressive transition between areas resembling myxoid liposarcoma and pleomorphic liposarcoma; pleomorphic cells; and myxoid matrix | Not specific | RB1 deletion, TP53 mutations |
| Chondroid lipoma | Myxohyaline chondroid matrix; lipoblasts intermingled with mature adipocytes and chondroid cells; and vascularized septa | S100 (mature adipocytes and lipoblasts); keratins (rare) | C110r95-MRTFB |
| Soft tissue angiofibroma | Myxoid-collagenous stroma; prominent, branching vasculature; and bland spindle cells, possibly adipocytes | CD34, EMA, desmine (dendritic cells) | NCOA2 rearrangements (various fusion partners) |
| Small, blue round-cell tumors (when round-cell liposarcoma) | Cells with small round-oval-spindle cells with little cytoplasm | See Table Ewing and Ewing-like sarcomas | See Table Ewing and Ewing-like sarcomas |
| Diagnosis | Morphology | IHC | Common Genetic Alterations |
|---|---|---|---|
| Histiocytic lesions | Diverse; histiocytes; and multinucleated giant cells | Diverse (depending on entity); CD68, CD163, and FXIIIA | Diverse (depending on entity); activating MAPK signaling mutations (BRAF, NRAS, KRAS, ARAF, and MAP2K1) |
| Follicular dentritic sarcoma | Fascicles/whorls/storiform pattern; oval-spindle cells with small nucleoli and syncytial borders; nuclear pseudoinclusions; binucleate (often) or multinucleate (rare) forms; and admixed lymphocytic infiltrate with perivascular lymphocytic cuffs | CD21, CD23, CD35, and D2-40 | Highly diverse mutational profile |
| Small, blue round-cell tumors See Table 1 and Table 2 | Cells with small round-oval-spindle cells with scant cytoplasm | Diverse (depending on entity) | Diverse (depending on entity) |
| Epithelioid fibrous histiocytoma | Polypoid; epidermal collarette; epithelioid cells with vesicular nuclei, small nucleoli, and amphophilic cytoplasm | FXIIIA, EMA, and ALK | ALK rearrangements (various fusion partners) |
| Aneurysmatic fibrous histiocytoma | Epidermal hyperplasia and basal layer pigmentation; circumscribed, dermal based proliferation; haphazard arrangement of ovoid spindle cells; admixed foam and giant cell; central blood-filled cystic space; and abundant hemosiderin deposition | FXIIIA, SMA | Not specific |
| Rhabdomyosarcoma | Monomorphic primitive round cells with variable rhabdomyoblastic differentiation (depending on subtype) | Desmin, myogenin, and MyoD1 | Diverse (depending on subtype); PAX3/PAX7-FOXO1 fusions (ARMS); alterations of RAS signaling pathway (embryonal RMS) |
| Rhabdoid tumor | Solid pattern; rounded-polygonal cells with vesicular nuclei and prominent nucleoli; and eosinophil hyaline-like cytoplasmic inclusions | Diverse; keratins, EMA, CD99, synaptophysin, SALL4, glypican-3, and INI1 loss | SMARCB1 biallelic loss |
| Inflammatory myofibroblastic tumor | Fascicular pattern (variable); plump-spindle cells with vesicular nuclei and small nucleoli; amphophilic cytoplasm; oedematous-myxoid-fibrous stroma; and mixed inflammatory infiltrate | SMA, calponin, desmin, keratin (focal), ALK, and ROS1 | ALK rearrangements (various fusion partners); ROS1, NTRK3, RET, or PDGFRB rearrangements |
| Carcinoma | Sheets/nests/trabecula; round-oval-epithelioid cells with nuclear atypia and variable amount of cytoplasm | Pankeratins, lineage specific markers (depending on site of origine) | Diverse (depending on site of origin) |
| Meningeoma | Highly diverse (according to subtype and grade): lobulated; whorled, fascicular pattern; spindle or plump syncytial cells; intranuclear pseudoinclusions; and psammoma bodies | EMA, S100, and PR | Monosomy 22; NF2 deletions |
| Extraskeletal myxoid chondrosarcoma | Multinodular; lace-like/reticular pattern; round-spindle monomorphic cells; eosinophilic cytoplasm; and myxoid matrix | Non-specific; S100 (focal) | NR4A3-EWSR1-/TAF15/TCF12/TFG |
| Myoepithelioma (syncytial) | Cutaneous; poorly marginated; syncytial growth; sheets of uniform ovoid-histiocytoid-epithelioid cells; and minimal stroma | S100, EMA, GFAP, SMA, and calponin | EWSR1-PBX3 |
| Myxoid liposarcoma | Lobulated; primitive uniform round-ovoid cells; variable number of lipoblasts; myxoid stroma; and plexiform vasculature (chicken wire) | DDIT3 | FUS/EWSR1-DDIT3 |
| Diagnosis | Morphology | IHC | Common Genetic Alterations |
|---|---|---|---|
| Salivary gland myoepithelial tumors | Strands/nests/ductular structures of epithelial/myoepithelial cells; and chondromyxoid/hyalinised stroma | Epithelial cells: EMA, cytokeratins; Myoepithelial cells: GFAP, S100, SOX10, p40, p63, and SMA | PLAG1 or HMGA2 fusions |
| Angiomatoid fibrous histiocytoma | Syncytial/whorling pattern (classic AFH); reticular/lace-like pattern (myxoid AFH); uniform histiocytoid cells; blood-filled pseudocysts; and inflammatory/lymphocytic reaction (lymph node-like) | Desmin, EMA, and ALK | EWSR1-ATF1 or EWSR1-CREB1 |
| Extraskeletal myxoid chondrosarcoma | Multinodular; lace-like/reticular pattern; round-spindle monomorphic cells with eosinophilic cytoplasm; and myxoid stroma | Non-specific; S100 (focal) | NR4A3-EWSR1/TAF15/TCF12/TFG |
| Diagnosis | Morphology | IHC | Common Genetic Alterations |
|---|---|---|---|
| Melanoma | Diverse growth patterns; large, atypical spindle-epithelioid-bizarre cells with vesicular nuclei and prominent, eosinophilic nucleoli; nuclear pseudo-inclusions; abundant eosinophilic-clear cytoplasm; and melanin pigment | S100, SOX10, Melan-A, HMB45, and MITF | Diverse: ARID2, BAP1, BRAF, GNAQ, HRAS, KIT, NF1, NRAS, PTEN mutations; and chromosomal gains/losses |
| Epithelioid Schwannoma | Multilobulated growth; encapsulated; nests or single cells; variableschwannoma epithelioid cells; and myxoid-hyalinized stroma | S100, SOX10, Loss of INI1 (~40%) | Loss of SMARCB1 (~40%) |
| Myoepithelial tumors | Divers spectrum; reticular/trabecular pattern; variable spindle/epithelioid/clear/plasmocytoid/rhabdoid cells; rarely ductal component (mixed tumors); fibromyxoid stroma; and hyalinization | Pankeratins, S100, SOX10, EMA, GFAP, P63, SMA, calponin, and desmin (focal) | EWSR1 rearrangements (various fusion partners); PLAG1 rearrangements (mixed tumors) |
| Diagnosis | Morphology | IHC | Common Genetic Alterations |
|---|---|---|---|
| Clear-cell sarcoma | Nested-fascicular pattern; epithelioid-plump spindle cells with vesicular nuclei and macronucleoli; fibrous septa; and scattered wreath-like multinucleated giant cells | Melanocytic markers (S100, SOX10, Melan-A, HMB45, and MITF) | EWSR1-ATF1/CREB (most often); EWSR1-CREM (rare) |
| Desmoplastic small round-cell tumor | Sheets/nests/cords of uniform, small round cells; and variable desmoplastic stroma | Desmin (dot-like), keratin, EMA, and WT1 (C-terminus) | EWSR1-WT1 |
| Carcinoma | Sheets/nests/trabecules; round-oval-epithelioid cells with nuclear atypia and variable cytoplasm | Pankeratins, lineage-specific markers (depending on site of origin) | Diverse (depending on site of origin) |
| Diagnosis | Morphology | IHC | Common Genetic Alterations |
|---|---|---|---|
| Extraskeletal myxoid chondrosarcoma | Multinodular; lace-like/reticular pattern; round-spindle monomorphic cells with eosinophilic cytoplasm; and myxoid stroma | Non-specific; S100 (focal) | NR4A3-EWSR1/TAF15/TCF12/TFG |
| Chordoma | Lobulated; fibrous septa; cords/nests of large epithelioid/polygonal cells, physaliphorous cells (bubbly cytoplasm); and variable myxoid stroma | Cytokeratin, EMA, S100, Brachyury | Germline tandem duplication of TBXT (rare); germline loss-of-function mutations of TSC1/2 (rare) |
| Sclerosing epitheloid fibrosarcoma | Infiltrative; cords/nests of monomorphic epithelioid cells; and hyalinized/sclerotic/collagenous stroma; | MUC4, SMA, and EMA | EWSR1/FUS/PAX5-CREB3L1/CREB3L2/CREB3L3/CREM |
| (Adeno)carcinoma | Sheets/nests/trabeculae, glands; round-oval-epithelioid cells with nuclear atypia and variable cytoplasm; and ductular structures (adenocarcinoma) | Pankeratins, lineage specific markers (depending on site of origin) | Diverse (depending on site of origin) |
| Small, blue, round cell tumors | Cells with small round-oval-spindle cells with little cytoplasm | Diverse (depending on entity) | See Table 1, Table 2 and Table 3 |
| Epitheloid sarcoma | Nodules of uniform epithelioid-spindled cells with eosinophilic cytoplasm; central geographic necrosis (classic type); multinodular/sheet-like growth; large-slightly pleomorphic epithelioid cells with eosinophilic cytoplasm (proximal type) | CD34, keratins, EMA, and loss of INI1 | Loss of SMARCB1 |
| Melanoma | Diverse growth patterns; large, atypical spindle-epithelioid-bizarre cells with vesicular nuclei and prominent, eosinophilic nucleoli; nuclear pseudo-inclusions; abundant eosinophilic-clear cytoplasm; and melanin pigment | S100, SOX10, Melan-A, HMB45, and MITF | Diverse: ARID2, BAP1, BRAF, GNAQ, HRAS, KIT, NF1, NRAS, PTEN mutations; and chromosomal gains/losses |
| Epithelioid schwannoma | Multilobulated growth; capsule; nests or singly cells; variable epithelioid cells; and myxoid-hyalinized stroma | S100, SOX10, and loss of INI1 (~40%) | Loss of SMARCB1 (~40%) |
| Epitheloid malignant peripheral nerve sheath tumor | Lobulated growth; atypical epithelioid cells with enlarged nuclei; and prominent nucleoli and eosinophilic cytoplasm | S100, SOX10 (strong and diffuse), and loss in INI1 (~75%) | Loss of SMARCB1 (~75%) |
| Ossifying fibromyxoid tumor (mostly benign, rarely malignant) | Multilobulated; nests/cords of uniform round-spindle cells; indistinct cytoplasm; no atypia (rarely high nuclear grade in malignant lesions); fibromyxoid stroma; partial rim of mature bone; and atypical osteoid in malignant tumors | S100, desmin, GFAP (focal), and pankeratin (rare) | PHF1 rearrangements (diverse fusion partners) |
| Clear-cell sarcoma | Nested-fascicular pattern; epithelioid-plump spindle cells with vesicular nuclei and macronucleoli; fibrous septa; and scattered wreath-like multinucleated giant cells | Melanocytic markers (S100, SOX10, Melan-A, HMB45, and MITF) | EWSR1-ATF1/CREB (most often); EWSR1-CREM (rare) |
| Malignant rhabdoid tumors | Solid pattern; uniform rounded-polygonal cells with vesicular nuclei and prominent nucleoli; and eosinophilic hyaline-like cytoplasmic inclusions | Diverse; keratins, EMA, CD99, synaptophysin, SALL4, glypican-3, and INI1 loss | SMARCB1 biallelic loss |
| Epithelioid hemangio-endothelioma | Infiltrative, sometimes angiocentric growth; cords/nests of bland looking epithelioid and spindle cells; glassy cytoplasm; intracytoplasmic vacuoles (blister cells); and myxohyaline stroma | CD34, CD31, ERG, D2-40, keratins (subset), SMA, CAMTA1, and TFE3 | WWTR1-CAMTA1 (>90%); YAP1-TFE3 |
| Pseudomyogenic (epitheloid sarcoma-like) hemangio-endothelioma | Multiple discontinuous nodules; possibly involvement of different tissue planes; sheets/fascicles of plump-spindle-epithelioid cells with abundant, brightly eosinophilic cytoplasm; vesicular nuclei with small nucleoli; mild nuclear atypia; not obvious vascular; and prominent stromal neutrophils (50%) | Keratins (AE1/AE3 but not MNF116), FLI, ERG, CD31 (50%), SMA (focal), and FOSB | SERPINE1/ACTB-FOSB |
| Diagnosis | Morphology | IHC | Common Genetic Alterations |
|---|---|---|---|
| Desmoid-type fibromatosis | Long, sweeping fascicles; slender uniform spindle cells; pale cytoplasm; and parallel to fascicles thin-walled blood vessels with perivascular edema | Beta-catenin (nuclear), SMA, and desmin (focal) | CTNNB1 or APC mutations |
| Nodular fasciitis | Plump spindle cells; tissue-culture aspect; extravasated erythrocytes; lymphocytes; and sometimes osteoclast-like giant cells | Non-specific: SMA, CD68, and desmin (focal) | USP6 rearrangements (diverse fusion partners) |
| Ossifying fibromyxoid tumor (mostly benign, rarely malignant) | Multilobulated; nests/cords of uniform round-spindle cells; indistinct cytoplasm; no atypia (rarely high nuclear grade in malignant lesions); fibromyxoid stroma; partial rim of mature bone; and atypical osteoid in malignant tumors | S100, desmin, GFAP (focal), and pankeratin (rare) | PHF1 rearrangements (diverse fusion partners) |
| Neurofibroma | Nodular or diffuse growth; variable admixture of perineurial cells, schwann cells and fibroblasts; hyperchromasia; usually no atypia or mitoses; and myxoid-collagenous stroma with “shredded-carrot” collagen | S100, SOX10, CD34, and EMA | NF1 deletions |
| Perineurioma | Nodular; non-encapsulated; circumscribed or infiltrative; whorled/storiform/fascicular pattern; and slender spindle cells with bipolar cytoplasmic extensions and uniform oval or elongated nuclei | EMA, GLUT1, CD34, and Claudin 1 | TRAF7 mutations (intraneural perineurioma); NF1/2 deletions |
| Desmoplastic fibroblastoma | Paucicellular; bland stellate-spindle cells; and abundant collagenous-myxocollagenous stroma | FOSL1, SMA (focal) | t(2;11) |
| Malignant peripheral nerve sheath tumor | Fascicles of monomorphic atypical spindle cells with perivascular accentuation and alternating cellularity; pleomorphism is possible; staghorn vessels; geographic necrosis; and heterologous differentiation | S100, SOX10 (focal), and loss of H3K27me3 | Inactivating mutations of NF1, CDKN2A/B, EED, and SUZ2 |
| Fibroma nuchae | Paucicellular; bland spindle cells; thick collagen bundles; and entrapped adipose tissue and/or small nerves | CD34 | Not relevant |
| Intramuscular (cellular) myxoma | Myxoid stroma; hypocellular; uniform spindle-stellate cells; inconspicuous vessels; and infiltration into skeletal muscle | CD34 | GNAS mutations |
| Dermatofibrosarcoma protuberans | Dermal based; cellular, storiform pattern of uniform spindle cells; encasement of skin appendages; and infiltration in subcutaneous fat with honeycombing | CD34 | COL1A1-PDGFB (most often); COL6A3-PDGFD or EMILIN2-PDGFD (rare) |
| NTRK-rearranged spindle cell neoplasm (emerging) | Wide spectrum of morphologies and histological grades; most often haphazardly arranged monomorphic spindle cells; variable stromal/perivascular hyalinization; and infiltrative growth into fat | S100, CD34 (co-expression), and NTRK | NTRK1-3 rearrangements (diverse fusion partners); RAF1 or BRAF fusions (rare) |
| Diagnosis | Morphology | IHC | Common Genetic Alterations |
|---|---|---|---|
| Ossifying fibromyxoid tumor | Multilobulated; nests/cords of uniform round-spindle cells; indistinct cytoplasm; no atypia (rarely high nuclear grade in malignant lesions); fibromyxoid stroma; partial rim of mature bone; and atypical osteoid in malignant tumors | S100, desmin, GFAP (focal), and pankeratin (rare) | PHF1 rearrangements (diverse fusion partners) |
| Carcinoma (lobular, signet ring cell) | Files-small nests; round-oval cells with variable cytoplasm and nuclear atypia; and intracytoplasmic mucin vacuole | Pankeratins, lineage specific markers (depending on site of origine) | Diverse (depending on site of origin) |
| Sclerosing lymphoma | Variably sized hyperchromatic-blastoid cells with variable atypia; scant cytoplasm; and sclerotic stroma | CD45, B/T-cell markers | Diverse |
| Synovial sarcoma | Sheets-fascicles; cellular, monomorphic spindle cells; high N/C ratio, variable epithelial differentiation; staghorn vessels; variable amount of collagen; mast cells; and calcification/ossification; poorly differentiated areas may show round-epithelioid cells | CD99, BCL2, CD56, TLE1, S100 (focal), EMA, and keratins (variable) | SS18-SSX1/2/4 |
| Clear-cell sarcoma | Nested-fascicular pattern; epithelioid-plump spindle cells with vesicular nuclei and macronucleoli; fibrous septa; and scattered wreath-like multinucleated giant cells | Melanocytic markers (S100, SOX10, Melan-A, HMB45, and MITF) | EWSR1-ATF1/CREB (most often); EWSR1-CREM (rare) |
| Osteosarcoma | Highly diverse; infiltrative growth; severely anaplastic and pleiomorphic cells; monomorphic small cell appearance is rare; and atypical neoplastic bone formation (essential) | SATB2, osteocalcin (BGLAP), osteonectin (SPARC), osteoprotegerin (TNFRSF11B), RUNX2, S100, actins, CD99, keratin, and EMA | Highly complex chromosomal aneuploidy |
| Myoepithelioma | Divers spectrum; reticular/trabecular pattern; variable spindle/epithelioid/clear/plasmocytoid/rhabdoid cells; rarely ductal component (mixed tumors); fibromyxoid stroma; and hyalinization | Pankeratins, S100, EMA, GFAP, SOX10, P63, SMA, calponin, and desmin (focal) | EWSR1 rearrangements (various fusion partners); PLAG1 rearrangements (mixed tumors) |
| Diagnosis | Morphology | IHC | Common Genetic Alterations |
|---|---|---|---|
| Cellular schwannoma | Encapsulation; predominantly Antoni A areas; variable neuroid spindle cells; possibly hyperchromasia and frequent mitoses; rarely Verocay bodies or schwannian whorls; and hyalinized vessels subcapsular lymphocytes | S100, SOX10 | NF2 deletion; LATS1/2, ARID1A/1B, and DDR1 mutations; SH3PXD2A-HTRA1 |
| Perineurioma | Whorled/storiform pattern; slender spindle cell with bipolar cytoplasmic extensions and oval/elongated uniform nuclei | EMA, GLUT1, CD34, Claudin 1 | TRAF7 mutations (intraneural perineurioma); NF/2 deletions |
| Myofibroma/myopericytoma | Myofibroma: nodular; biphasic pattern: immature plump-spindle cells around staghorn vessels; and nodules/fascicles of hyalinized-myoid-chondroid appearing cells. Myopericytoma: lobular; variably cellular; bland oval-spindle-myoid cells; and perivascular growth | SMA (myofibroma/myopericytoma), caldesmon (myopericytoma) | PDGFRB, NOTCH3 mutations; SRF-RELA |
| Angioleiomyoma | Bundles of bland leiomyocytes around numerous vessels | SMA, calponin, caldesmon, and desmin (variable) | Not relevant |
| Cellular digital fibroma | Whorls/short fascicles; bland spindle cells with slightly eosinophilic cytoplasm; pale pink-red, paranuclear inclusions; and collagenous stroma | Actin, calponin, and caldesmon | Not relevant |
| Calcifying aponeurotic fibroma | Fibromatosis-like, infiltrative component of bland spindle cells; nodular calcified component with hyalinized-chondroid matrix encircled by rounded-chondrocyte like cells; and osteoclast-type giant cells | SMA, CD99, S100 (chondroid areas) | FN1-EGF |
| Acral fibromyxoma | Nodular or diffuse; infiltrative growth of bland spindle-stellate cells; and variably myxoid-collagenous stroma | CD34, EMA, SMA, and RB1 (loss) | RB1 deletions |
| Superficial fibromatosis | Plexiform or multinodular; moderately cellular bland spindle cells; and collagenous stroma | SMA, desmin, beta-catenin (nuclear) positive in a subset of cases despite absence of CTNNB1 or APC mutation | Not relevant |
| Cellular dermatofibroma | Radiar configuration; storiform/short fascicular pattern; cellular center of fibrohistiocytic cells; and peripheral collagen entrapment | SMA, FXIIA, and CD68 | PRKCB/PRKCD-PDPN/CD63/LAMTOR1 |
| Lipofibromatosis | Admixture of mature fat, fascicles of bland myofibroblastic spindle cells (fibromatosis-like) and lipoblast-like cells at the interface | CD34, SMA | EGFR/HER1/ROS/RET/PDGFRB-EGF/HBEGF/TGF-α |
| Smooth muscle neoplams | Intersecting fascicles of smooth muscle cells; blunt-ended, cigar-shaped nuclei; and eosinophilic cytoplasm | SMA, desmin, and caldesmon | Not relevant; |
| Synovial sarcoma | Sheets-fascicles; cellular, monomorphic spindle cells; high N/C ratio; variable epithelial differentiation; staghorn vessels; variable amount of collagen; mast cells; calcification/ossification; and poorly differentiated areas may show round-epithelioid cells | CD99, BCL2, CD56, TLE1, S100 (focal), EMA, and keratins (variable) | SS18-SSX1/2/4 fusion |
| Malignant peripheral nerve sheath tumor | Fascicles of spindle cells with perivascular accentuation and alternating cellularity; staghorn vessels; georgraphic necrosis; and heterologous differentiation | S100, SOX10 (focal), and loss of H3K27me3 | Inactivating mutations of NF1, CDKN2A/B, EED, and SUZ2 |
| Acral dermatofibrosarcoma protuberans | Dermal-based; cellular, storiform pattern of uniform spindle cells; encasement of skin appendages; and infiltration in subcutaneous fat with honeycombing | CD34 | COL1A1-PDGFB fusion (most often); COL6A3-PDGFD or EMILIN2-PDGFD (rare) |
| Low-grade fibromyxoid sarcoma | Alternating myxoid-fibrous areas; whorling/fascicular pattern of bland spindle cells; arcades of small vessels; and sometimes hyaline rosettes, which sometimes overlap with sclerosing epitheloid fibrosarcoma | MUC4 (highly sensitive and specific); EMA, S100, CD34, and SMA (variable) | EWSR1/FUS-CREB3L1/2 |
| Pseudomyogenic (epitheloid sarcoma-like) hemangioendothelioma | Multiple discontinuous nodules; possible involvement of different tissue planes; sheets/fascicles of plump-spindle-epithelioid cells with abundant, brightly eosinophilic cytoplasm; vesicular nuclei with small nucleoli; mild nuclear atypia; not obvious vascular; and prominent stromal neutrophils (50%) | Keratins (AE1/AE3 but not MNF116), FLI, ERG, CD31 (50%), SMA (focal), and FOSB | SERPINE1/ACTB-FOSB |
| NTRK-rearranged spindle cell neoplasms | Wide spectrum of morphologies and histological grades; most often haphazardly arranged monomorphic spindle cells; variable stromal/perivascular hyalinization; and infiltrative growth into fat | S100, CD34 (co-expression), and NTRK | NTRK1-3 rearrangements (diverse fusion partners); RAF1 or BRAF fusions (rare) |
| Entity | Morphology | IHC | Common Genetic Alterations |
|---|---|---|---|
| Inflammatory myofibroblastic tumor | Fascicular pattern (variable); plump-spindle cells with vesicular nuclei and small nucleoli and amphophilic cytoplasm; oedematous-myxoid-fibrous stroma; and mixed inflammatory infiltrate | SMA, calponin, desmin, keratin (focal), ALK, and ROS1 | ALK rearrangements (various fusion partners); ROS1, NTRK3, RET, or PDGFRB rearrangements |
| Carcinoma | Sheets/nests/trabecules; round-oval-epithelioid cells with variable cytoplasm and nuclear atypia | Pankeratins, lineage-specific markers (depending on site of origine) | Diverse (depending on site of origin) |
| Myoepithelial carcinoma | Solid sheets/nests of variable myoepithelial cells (epitheloid, spindled, plasmocytoid, rhabdoid, and clear) with high nuclear grade or undifferentiated round-cell morphology; necrosis; and high mitotic count | Pankeratins, EMA, S100, SOX10, GFAP, P63, SMA, calponin, desmin (focal); and INI1 loss (subset) | EWSR1 rearrangements (various fusion partners); PLAG1 rearrangements (mixed tumors) |
| EWSR1-PATZ1 sarcoma | Diverse morphology: round-spindle cells; fibrous stroma | Co-expression of myogenic markers (desmin/myogenin/MyoD1) and neurogenic markers (S100/SOX10/MITF/GFAP) | EWSR1-PATZ1 |
| Dedifferentiated chondrosarcoma | Conventional chondrosarcoma with abrupt transition to a high-grade non-cartilaginous sarcoma (undifferentiated sarcoma, osteosarcoma, angiosarcoma, leiomyosarcoma, and rhabdomyosarcoma) | Diverse (according to line of differentiation); loss of H3K27me3, MDM2 overexpression, p53 overexpression, and PDL1 | Complex karyotype; IDH1/2, TP53 mutations |
| Pseudomyogenic (epitheloid sarcoma-like) hemangioendothelioma | Multiple discontinuous nodules; possibly involvement of different tissue planes; sheets/fascicles of plump-spindle-epithelioid cells with abundant, brightly eosinophilic cytoplasm; vesicular nuclei with small nucleoli; mild nuclear atypia; not obvious vascular; and prominent stromal neutrophils (50%) | Keratins (AE1/AE3 but not MNF116), FLI, ERG, CD31 (50%), SMA (focal), and FOSB | SERPINE1/ACTB-FOSB |
| Rhabdomyosarcoma (spindle cell) | Cellular fascicles with intersecting/herringbone pattern; atypical uniform spindle cells with pale eosinophilic cytoplasm; primitive round cells may be present; and tadpole/strap cells (sometimes) | Desmin, MyoD1 (focal or diffuse), myogenin (focal) | SRF/VGLL2/TEAD1-NCOA2, VGLL2-CITED2 (congenital spindle cell RMS); and MYOD1 mutation |
| Leiomyosarcoma | Intersecting fascicles of smooth muscle cells; blunt-ended, cigar-shaped nuclei; variable atypia and pleomorphism (depending on grade); eosinophilic cytoplasm; mitoses; necrosis | SMA, desmin, and caldesmon | Extensive genomic instability (leiomyosarcoma); diverse gene involvement with p53 mutations; deleterious ATRX alterations; ALK rearrangement (small subset); and NF1 mutations (subset of inflammatory leiomyosarcoma); |
| Melanoma | Diverse growth patterns; large, atypical spindle-epithelioid-bizarre cells with vesicular nuclei and prominent, eosinophilic nucleoli; nuclear pseudo-inclusions; abundant eosinophilic-clear cytoplasm; and melanin pigment | S100, SOX10, Melan-A, HMB45, and MITF | Diverse: ARID2, BAP1, BRAF, GNAQ, HRAS, KIT, NF1, NRAS, and PTEN mutations; chromosomal gains/losses |
| Malignant peripheral nerve sheath tumor | Fascicles of spindle cells with perivascular accentuation and alternating cellularity; staghorn vessels; necrosis; and heterologous differentiation | S100, SOX10 (focal), and loss of H3K27me3 | Inactivating mutations of NF1, CDKN2A/B, EED, and SUZ2 |
| Diagnosis | Morphology | Common Genetic Alterations |
|---|---|---|
| Fibrous dysplasia | Irregular, curvilinear trabeculae of woven (or rarely lamellar) bone without osteoblast rimming; inconspicuous osteoblasts; sharpey fibers; and osteoclasts bland fibroblastic stroma | GNAS mutations (50–70%) |
| Intraosseous ganglion | Cavity without lining; filled with mucoid viscous material | None (probably degenerative) |
| Lipoma | Nodules of mature adipose tissue, often fibrotic | Unknown |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flucke, U.; van Noesel, M.M.; Siozopoulou, V.; Creytens, D.; Tops, B.B.J.; van Gorp, J.M.; Hiemcke-Jiwa, L.S. EWSR1—The Most Common Rearranged Gene in Soft Tissue Lesions, Which Also Occurs in Different Bone Lesions: An Updated Review. Diagnostics 2021, 11, 1093. https://doi.org/10.3390/diagnostics11061093
Flucke U, van Noesel MM, Siozopoulou V, Creytens D, Tops BBJ, van Gorp JM, Hiemcke-Jiwa LS. EWSR1—The Most Common Rearranged Gene in Soft Tissue Lesions, Which Also Occurs in Different Bone Lesions: An Updated Review. Diagnostics. 2021; 11(6):1093. https://doi.org/10.3390/diagnostics11061093
Chicago/Turabian StyleFlucke, Uta, Max M. van Noesel, Vasiliki Siozopoulou, David Creytens, Bastiaan B. J. Tops, Joost M. van Gorp, and Laura S. Hiemcke-Jiwa. 2021. "EWSR1—The Most Common Rearranged Gene in Soft Tissue Lesions, Which Also Occurs in Different Bone Lesions: An Updated Review" Diagnostics 11, no. 6: 1093. https://doi.org/10.3390/diagnostics11061093
APA StyleFlucke, U., van Noesel, M. M., Siozopoulou, V., Creytens, D., Tops, B. B. J., van Gorp, J. M., & Hiemcke-Jiwa, L. S. (2021). EWSR1—The Most Common Rearranged Gene in Soft Tissue Lesions, Which Also Occurs in Different Bone Lesions: An Updated Review. Diagnostics, 11(6), 1093. https://doi.org/10.3390/diagnostics11061093