
Progress in Diagnosing Primary Ciliary Dyskinesia: The North American Perspective

Abstract
:1. Summary
2. Recognition of the Clinical Phenotype
3. PCD Diagnostic Testing
3.1. Nasal NO Testing
3.2. PCD Genetic Testing
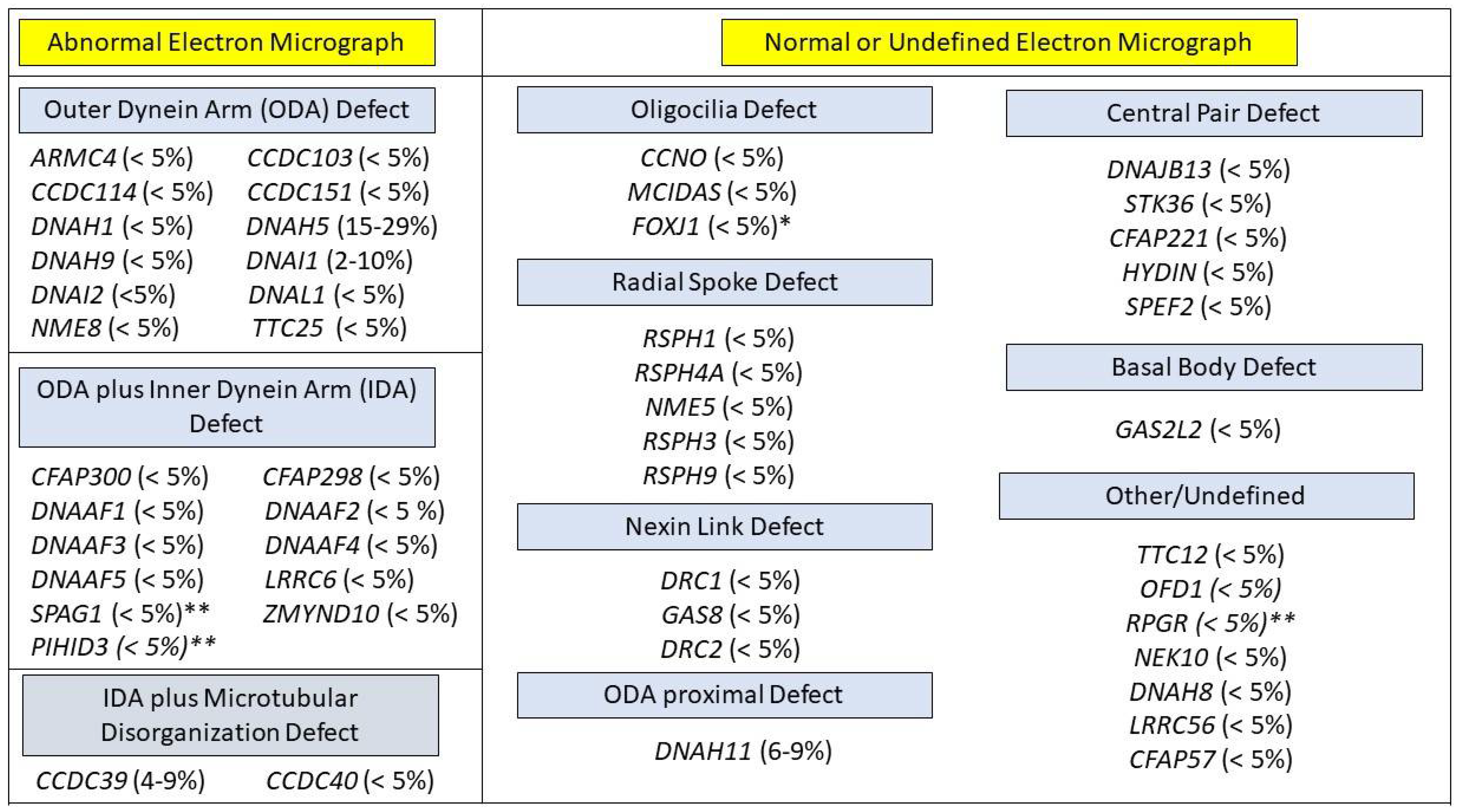
3.3. Cilia Ultrastructure Evaluation
3.4. Ciliary Motility and Highspeed Video Microscopy
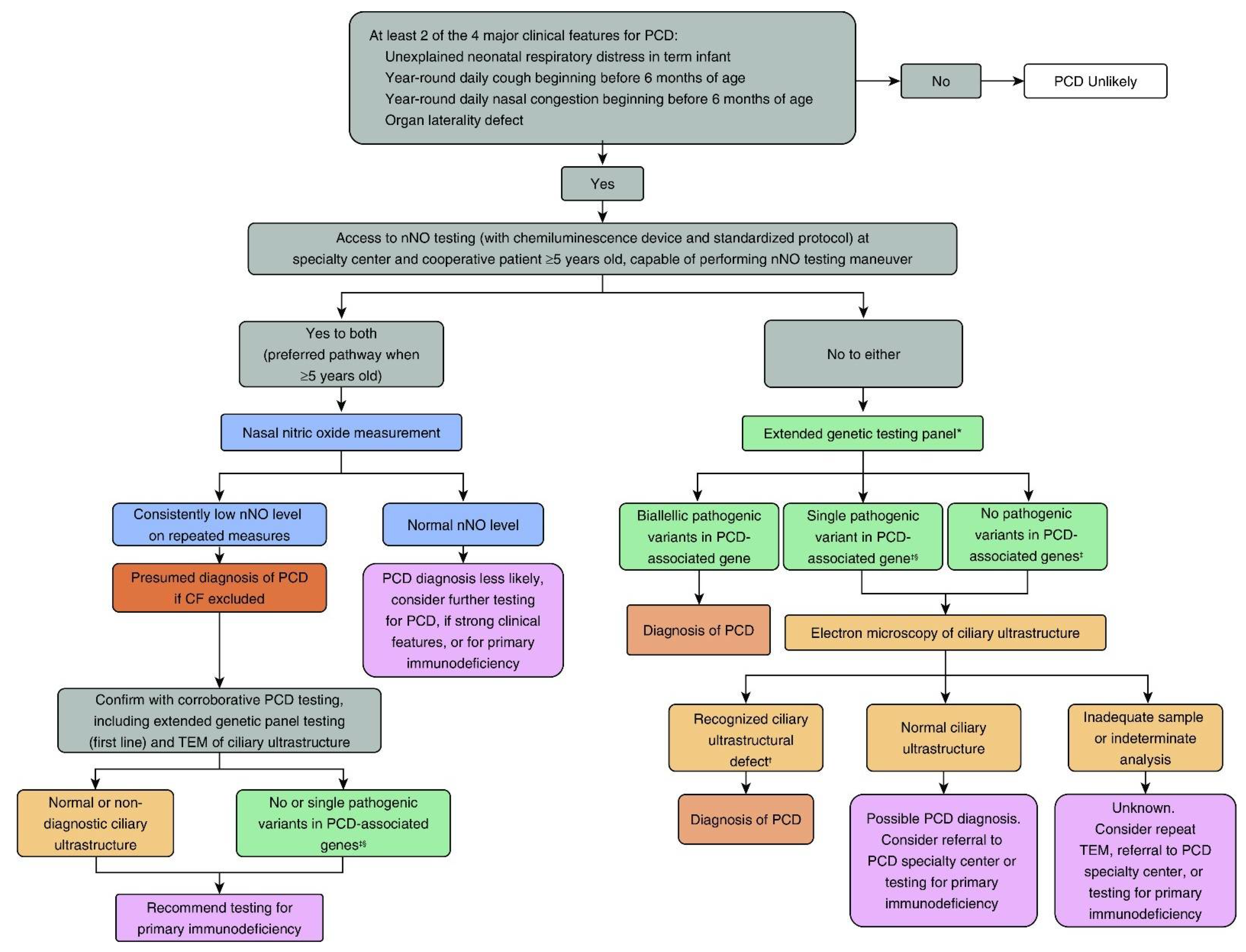
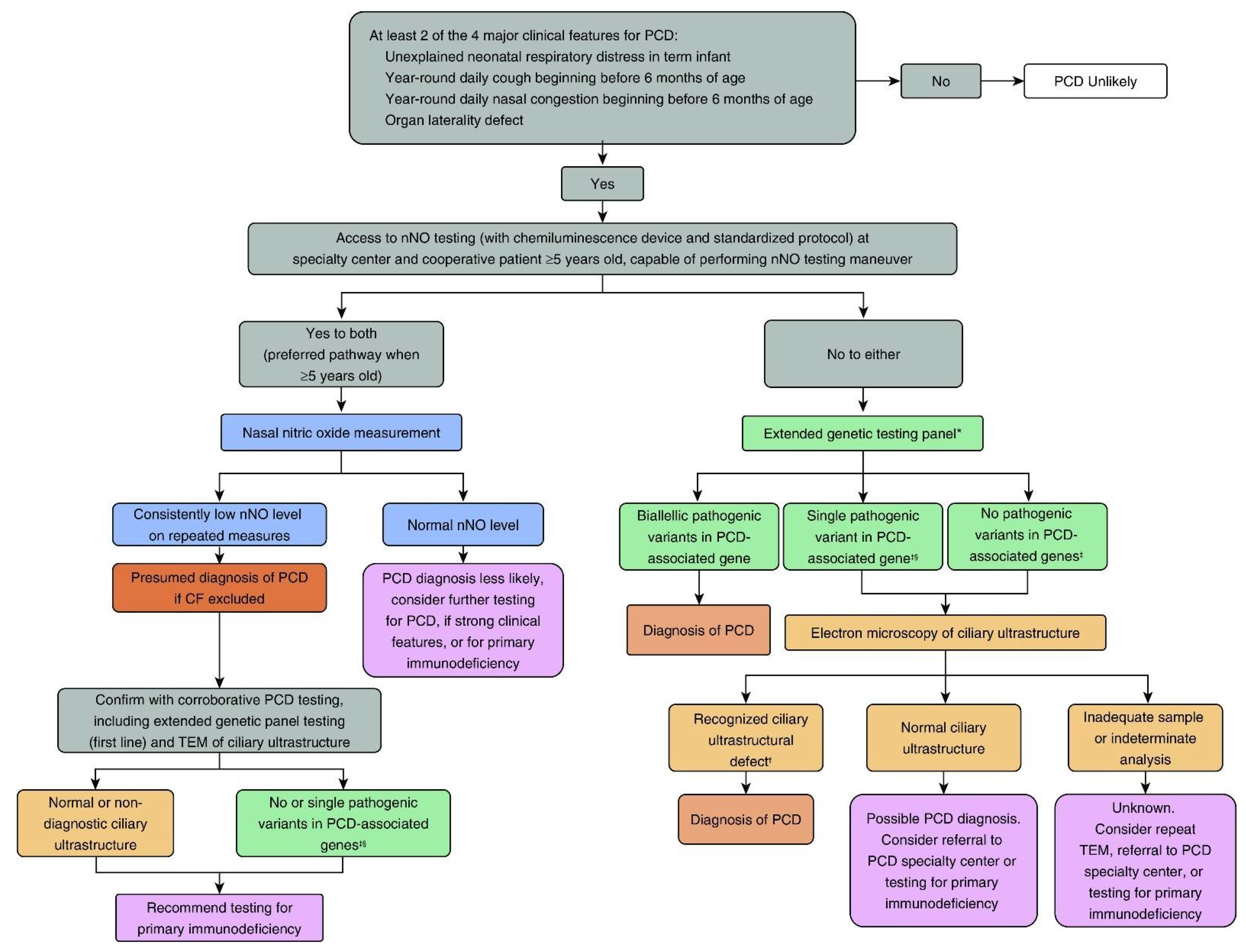
4. Summary Considerations for PCD Diagnostic Testing in North America
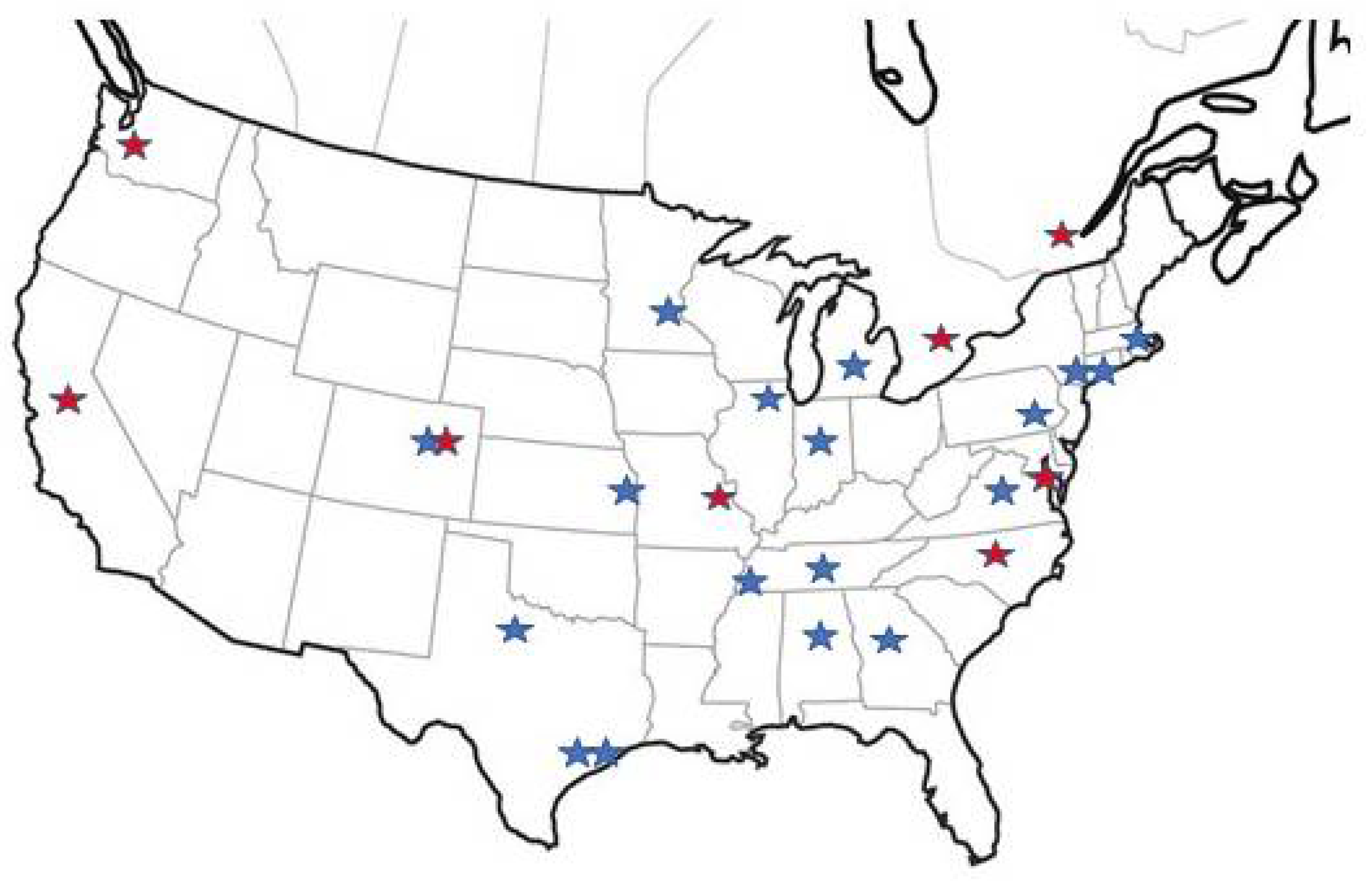
5. PCD Foundation CRCN Overview
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Knowles, M.R.; Daniels, L.A.; Davis, S.D.; Zariwala, M.A.; Leigh, M.W. Primary Ciliary Dyskinesia. Recent Advances in Diagnostics, Genetics, and Characterization of Clinical Disease. Am. J. Respir. Crit. Care Med. 2013, 188, 913–922. [Google Scholar] [CrossRef] [Green Version]
- Leigh, M.W.; Horani, A.; Kinghorn, B.; O’Connor, M.G.; Zariwala, M.A.; Knowles, M.R. Primary ciliary dyskinesia (PCD): A genetic disorder of motile cilia. Transl. Sci. Rare Dis. 2019, 4, 51–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, A.J.; Davis, S.D.; Polineni, D.; Manion, M.; Rosenfeld, M.; Dell, S.D.; Chilvers, M.A.; Ferkol, T.W.; Zariwala, M.A.; Sagel, S.D.; et al. Diagnosis of Primary Ciliary Dyskinesia. An Official American Thoracic Society Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 197, e24–e39. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.S.; Barbato, A.; Collins, S.A.; Goutaki, M.; Behan, L.; Caudri, D.; Dell, S.; Eber, E.; Escudier, E.; Hirst, R.A.; et al. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur. Respir. J. 2016, 49, 1601090. [Google Scholar] [CrossRef] [PubMed]
- Shoemark, A.; Dell, S.; Shapiro, A.; Lucas, J.S. ERS and ATS diagnostic guidelines for primary ciliary dyskinesia: Similarities and differences in approach to diagnosis. Eur. Respir. J. 2019, 54, 1901066. [Google Scholar] [CrossRef] [PubMed]
- Rubbo, B.; Shoemark, A.; Jackson, C.L.; Hirst, R.; Thompson, J.; Hayes, J.; Frost, E.; Copeland, F.; Hogg, C.; O’Callaghan, C.; et al. Accuracy of High-Speed Video Analysis to Diagnose Primary Ciliary Dyskinesia. Chest 2019, 155, 1008–1017. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, M.G.; Griffiths, A.; Iyer, N.P.; Shapiro, A.; Wilson, K.C.; Thomson, C.C. Summary for Clinicians: Diagnosis of Primary Ciliary Dyskinesia. Ann. Am. Thorac. Soc. 2019, 16, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Leigh, M.W.; Ferkol, T.W.; Davis, S.D.; Lee, H.-S.; Rosenfeld, M.; Dell, S.; Sagel, S.D.; Milla, C.; Olivier, K.N.; Sullivan, K.M.; et al. Clinical Features and Associated Likelihood of Primary Ciliary Dyskinesia in Children and Adolescents. Ann. Am. Thorac. Soc. 2016, 13, 1305–1313. [Google Scholar] [CrossRef]
- Mullowney, T.; Manson, D.; Kim, R.; Stephens, D.; Shah, V.; Dell, S. Primary Ciliary Dyskinesia and Neonatal Respiratory Distress. Pediatrics 2014, 134, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Goutaki, M.; Halbeisen, F.; Barbato, A.; Crowley, S.; Harris, A.; Hirst, R.; Karadag, B.; Martinu, V.; Morgan, L.; O’Callaghan, C.; et al. Late Diagnosis of Infants with PCD and Neonatal Respiratory Distress. J. Clin. Med. 2020, 9, 2871. [Google Scholar] [CrossRef]
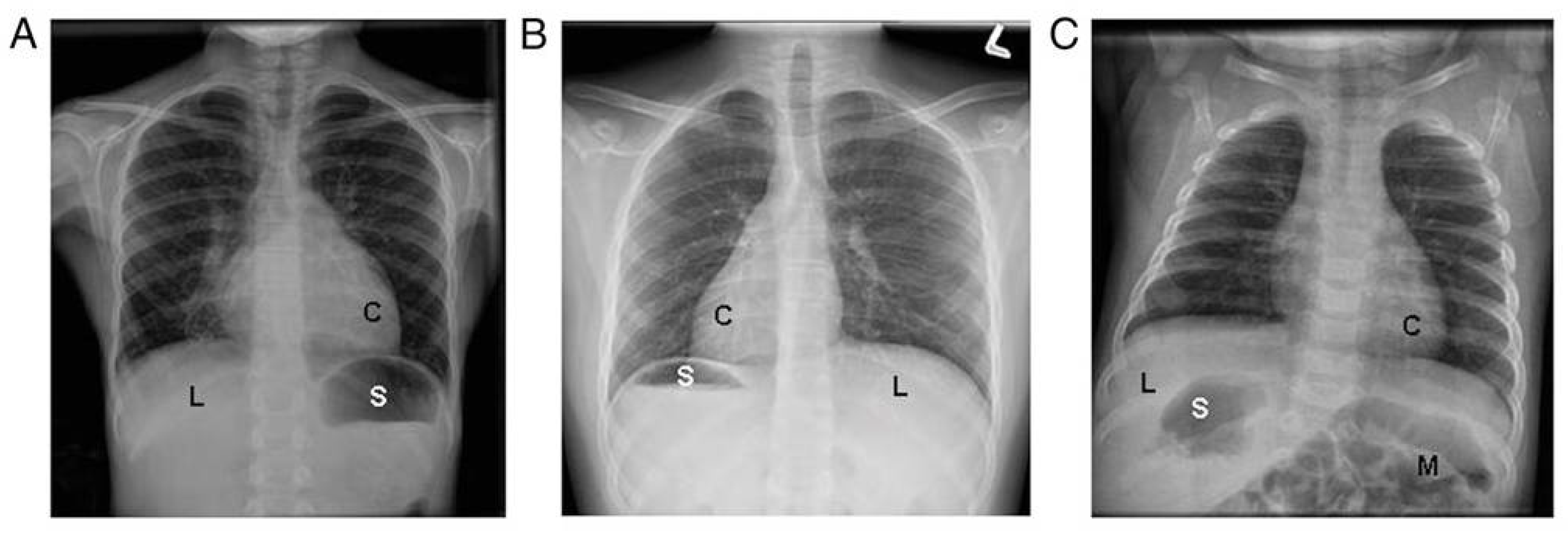
- Shapiro, A.J.; Davis, S.D.; Ferkol, T.; Dell, S.D.; Rosenfeld, M.; Olivier, K.N.; Sagel, S.D.; Milla, C.; Zariwala, M.A.; Wolf, W.; et al. Laterality defects other than situs inversus totalis in primary ciliary dyskinesia: Insights into situs ambiguus and heterotaxy. Chest 2014, 146, 1176–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frostell, C.; Fratacci, M.D.; Wain, J.C.; Jones, R.; Zapol, W.M.; Frostell, C.; Fratacci, M.D.; Wain, J.C.; Jones, R.; Zapol, W.M. Inhaled nitric oxide. A selective pulmonary vasodilator reversing hypoxic pulmonary vasoconstriction. Circulation 1991, 83, 2038–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albina, J.E.; Reichner, J. Role of nitric oxide in mediation of macrophage cytotoxicity and apoptosis. Cancer Metastasis Rev. 1998, 17, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.D.; Ferkol, T.W.; Rosenfeld, M.; Lee, H.-S.; Dell, S.; Sagel, S.D.; Milla, C.; Zariwala, M.A.; Pittman, J.E.; Shapiro, A.; et al. Clinical Features of Childhood Primary Ciliary Dyskinesia by Genotype and Ultrastructural Phenotype. Am. J. Respir. Crit. Care Med. 2015, 191, 316–324. [Google Scholar] [CrossRef] [Green Version]
- Arnal, J.-F.; Flores, P.; Rami, J.; Murris-Espin, M.; Bremont, F.; I Aguilla, M.P.; Serrano, E.; Didier, A. Nasal nitric oxide concentration in paranasal sinus inflammatory diseases. Eur. Respir. J. 1999, 13, 307–312. [Google Scholar] [CrossRef] [Green Version]
- Rybnikar, T.; Senkerik, M.; Chladek, J.; Chladkova, J.; Kalfert, D.; Skoloudik, L. Adenoid hypertrophy affects screening for primary ciliary dyskinesia using nasal nitric oxide. Int. J. Pediatric Otorhinolaryngol. 2018, 115, 6–9. [Google Scholar] [CrossRef]
- Walker, W.T.; Jackson, C.L.; Lackie, P.M.; Hogg, C.; Lucas, J.S. Nitric oxide in primary ciliary dyskinesia. Eur. Respir. J. 2012, 40, 1024–1032. [Google Scholar] [CrossRef] [Green Version]
- Leigh, M.W.; Hazucha, M.J.; Chawla, K.K.; Baker, B.R.; Shapiro, A.; Brown, D.E.; LaVange, L.M.; Horton, B.J.; Qaqish, B.; Carson, J.L.; et al. Standardizing Nasal Nitric Oxide Measurement as a Test for Primary Ciliary Dyskinesia. Ann. Am. Thorac. Soc. 2013, 10, 574–581. [Google Scholar] [CrossRef] [Green Version]
- Groot, K.M.D.W.; Noman, S.V.H.; Speleman, L.; Schilder, A.G.M.; Van Der Ent, C.K. Nasal Nitric Oxide Levels and Nasal Polyposis in Children and Adolescents With Cystic Fibrosis. JAMA Otolaryngol. Neck Surg. 2013, 139, 931. [Google Scholar] [CrossRef] [Green Version]
- Zysman-Colman, Z.N.; Kaspy, K.R.; Alizadehfar, R.; Nykamp, K.R.; Zariwala, M.A.; Knowles, M.R.; Vinh, D.C.; Shapiro, A.J. Nasal Nitric Oxide in Primary Immunodeficiency and Primary Ciliary Dyskinesia: Helping to Distinguish Between Clinically Similar Diseases. J. Clin. Immunol. 2019, 39, 216–224. [Google Scholar] [CrossRef]
- Harris, A.; Bhullar, E.; Gove, K.; Joslin, R.; Pelling, J.; Evans, H.J.; Walker, W.T.; Lucas, J.S. Validation of a portable nitric oxide analyzer for screening in primary ciliary dyskinesias. BMC Pulm. Med. 2014, 14, 18. [Google Scholar] [CrossRef] [Green Version]
- Marthin, J.K.; Nielsen, K.G. Hand-Held Tidal Breathing Nasal Nitric Oxide Measurement—A Promising Targeted Case-Finding Tool for the Diagnosis of Primary Ciliary Dyskinesia. PLoS ONE 2013, 8, e57262. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, A.J.; Josephson, M.; Rosenfeld, M.; Yilmaz, O.; Davis, S.D.; Polineni, D.; Guadagno, E.; Leigh, M.W.; Lavergne, V. Accuracy of Nasal Nitric Oxide Measurement as a Diagnostic Test for Primary Ciliary Dyskinesia. A Systematic Review and Meta-analysis. Ann. Am. Thorac. Soc. 2017, 14, 1184–1196. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, A.J.; Davis, S.D.; Leigh, M.W.; Knowles, M.R.; Lavergne, V.; Ferkol, T. Limitations of Nasal Nitric Oxide Testing in Primary Ciliary Dyskinesia. Am. J. Respir. Crit. Care Med. 2020, 202, 476–477. [Google Scholar] [CrossRef] [PubMed]
- Loges, N.T.; Antony, D.; Maver, A.; Deardorff, M.A.; Güleç, E.Y.; Gezdirici, A.; Nöthe-Menchen, T.; Höben, I.M.; Jelten, L.; Frank, D.; et al. Recessive DNAH9 Loss-of-Function Mutations Cause Laterality Defects and Subtle Respiratory Ciliary-Beating Defects. Am. J. Hum. Genet. 2018, 103, 995–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassad, M.; Shoemark, A.; le Borgne, P.; Koll, F.; Patel, M.; Dixon, M.; Hayward, J.; Richardson, C.; Frost, E.; Jenkins, L.; et al. C11orf70 Mutations Disrupting the Intraflagellar Transport-Dependent Assembly of Multiple Axonemal Dyneins Cause Primary Ciliary Dyskinesia. Am. J. Hum. Genet. 2018, 102, 956–972. [Google Scholar] [CrossRef] [Green Version]
- Shoemark, A.; Moya, E.; Hirst, R.A.; Patel, M.P.; Robson, E.A.; Hayward, J.; Scully, J.; Fassad, M.; Lamb, W.; Schmidts, M.; et al. High prevalence of CCDC103 p.His154Pro mutation causing primary ciliary dyskinesia disrupts protein oligomerisation and is associated with normal diagnostic investigations. Thorax 2017, 73, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, L.; Bouhouche, K.; Whitfield, M.; Thouvenin, G.; Coste, A.; Louis, B.; Szymanski, C.; Bequignon, E.; Papon, J.-F.; Castelli, M.; et al. TTC12 Loss-of-Function Mutations Cause Primary Ciliary Dyskinesia and Unveil Distinct Dynein Assembly Mechanisms in Motile Cilia Versus Flagella. Am. J. Hum. Genet. 2020, 106, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Ostrowski, L.E.; Leigh, M.W.; Sears, P.R.; Davis, S.D.; Wolf, W.E.; Hazucha, M.J.; Carson, J.L.; Olivier, K.N.; Sagel, S.D.; et al. Mutations inRSPH1Cause Primary Ciliary Dyskinesia with a Unique Clinical and Ciliary Phenotype. Am. J. Respir. Crit. Care Med. 2014, 189, 707–717. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.; Escudier, E.; Roger, G.; Tamalet, A.; Pelosse, B.; Marlin, S.; Clément, A.; Geremek, M.; Delaisi, B.; Bridoux, A.-M.; et al. RPGR is mutated in patients with a complex X linked phenotype combining primary ciliary dyskinesia and retinitis pigmentosa. J. Med. Genet. 2005, 43, 326–333. [Google Scholar] [CrossRef] [Green Version]
- Bukowy-Bieryllo, Z.; Zietkiewicz, E.; Loges, N.T.; Wittmer, M.; Geremek, M.; Olbrich, H.; Fliegauf, M.; Voelkel, K.; Rutkiewicz, E.; Rutland, J.; et al. RPGR mutations might cause reduced orientation of respiratory cilia. Pediatric pulmonology 2013, 48, 352–363. [Google Scholar] [CrossRef]
- Wallmeier, J.; Al-Mutairi, D.A.; Chen, C.-T.; Loges, N.T.; Pennekamp, P.; Menchen, T.; Ma, L.; E Shamseldin, H.; Olbrich, H.; Dougherty, G.W.; et al. Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat. Genet. 2014, 46, 646–651. [Google Scholar] [CrossRef]
- Wallmeier, J.; Frank, D.; Shoemark, A.; Nöthe-Menchen, T.; Cindric, S.; Olbrich, H.; Loges, N.T.; Aprea, I.; Dougherty, G.W.; Pennekamp, P.; et al. De Novo Mutations in FOXJ1 Result in a Motile Ciliopathy with Hydrocephalus and Randomization of Left/Right Body Asymmetry. Am. J. Hum. Genet. 2019, 105, 1030–1039. [Google Scholar] [CrossRef]
- Chivukula, R.R.; Montoro, D.; Leung, H.M.; Yang, J.; Shamseldin, H.E.; Taylor, M.; Dougherty, G.W.; Zariwala, M.A.; Carson, J.; Daniels, M.L.A.; et al. A human ciliopathy reveals essential functions for NEK10 in airway mucociliary clearance. Nat. Med. 2020, 26, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Al Mutairi, F.; Alkhalaf, R.; Alkhorayyef, A.; Alroqi, F.; Yusra, A.; Umair, M.; Nouf, F.; Khan, A.; Meshael, A.; Hamad, A.; et al. Homozygous truncating NEK10 mutation, associated with primary ciliary dyskinesia: A case report. BMC Pulm. Med. 2020, 20, 1–5. [Google Scholar] [CrossRef]
- Marin, X.B.; Yin, W.-N.; Sears, P.R.; Werner, M.E.; Brotslaw, E.; Mitchell, B.J.; Jania, C.M.; Zeman, K.L.; Rogers, T.D.; Herring, L.E.; et al. Lack of GAS2L2 Causes PCD by Impairing Cilia Orientation and Mucociliary Clearance. Am. J. Hum. Genet. 2019, 104, 229–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olbrich, H.; Cremers, C.; Loges, N.T.; Werner, C.; Nielsen, K.G.; Marthin, J.K.; Philipsen, M.; Wallmeier, J.; Pennekamp, P.; Menchen, T.; et al. Loss-of-Function GAS8 Mutations Cause Primary Ciliary Dyskinesia and Disrupt the Nexin-Dynein Regulatory Complex. Am. J. Hum. Genet. 2015, 97, 546–554. [Google Scholar] [CrossRef] [Green Version]
- Edelbusch, C.; Cindrić, S.; Dougherty, G.W.; Loges, N.T.; Olbrich, H.; Rivlin, J.; Wallmeier, J.; Pennekamp, P.; Amirav, I.; Omran, H. Mutation of serine/threonine protein kinase 36 ( STK36 ) causes primary ciliary dyskinesia with a central pair defect. Hum. Mutat. 2017, 38, 964–969. [Google Scholar] [CrossRef]
- Bustamante-Marin, X.M.; Shapiro, A.; Sears, P.R.; Charng, W.L.; Conrad, D.F.; Leigh, M.W.; Knowles, M.R.; Ostrowski, L.E.; Zariwala, M.A. Identification of genetic variants in CFAP221 as a cause of primary ciliary dyskinesia. J. Hum. Genet. 2020, 65, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Sha, Y.; Li, Y.; Mei, L.; Lin, S.; Huang, X.; Lu, J.; Ding, L.; Kong, S.; Lu, Z. Loss-of-function mutations in SPEF2 cause multiple morphological abnormalities of the sperm flagella (MMAF). J. Med. Genet. 2019, 56, 678–684. [Google Scholar] [CrossRef]
- Liu, C.; Lv, M.; He, X.; Zhu, Y.-J.; Amiri-Yekta, A.; Li, W.; Wu, H.; Kherraf, Z.-E.; Liu, W.; Zhang, J.; et al. Homozygous mutations in SPEF2 induce multiple morphological abnormalities of the sperm flagella and male infertility. J. Med. Genet. 2019, 57, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Cindrić, S.; Dougherty, G.W.; Olbrich, H.; Hjeij, R.; Loges, N.T.; Amirav, I.; Philipsen, M.C.; Marthin, J.K.; Nielsen, K.G.; Sutharsan, S.; et al. SPEF2- and HYDIN-Mutant Cilia Lack the Central Pair–associated Protein SPEF2, Aiding Primary Ciliary Dyskinesia Diagnostics. Am. J. Respir. Cell Mol. Biol. 2020, 62, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, S.; Watson, C.M.; Kernohan, K.D.; Lemos, M.; Hutchinson, S.; Poulter, J.; Crinnion, L.A.; Berry, I.; Simmonds, J.; Vasudevan, P.; et al. Biallelic Mutations in LRRC56, Encoding a Protein Associated with Intraflagellar Transport, Cause Mucociliary Clearance and Laterality Defects. Am. J. Hum. Genet. 2018, 103, 727–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, A.J.; Dell, S.D.; Gaston, B.; O’Connor, M.; Marozkina, N.; Manion, M.; Hazucha, M.J.; Leigh, M.W. Nasal Nitric Oxide Measurement in Primary Ciliary Dyskinesia. A Technical Paper on Standardized Testing Protocols. Ann. Am. Thorac. Soc. 2020, 17, e1–e12. [Google Scholar] [CrossRef] [PubMed]
- Olbrich, H.; Häffner, K.; Kispert, A.; Völkel, A.; Volz, A.; Sasmaz, G.; Reinhardt, R.; Hennig, S.; Lehrach, H.; Konietzko, N.; et al. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left–right asymmetry. Nat. Genet. 2002, 30, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Harricane, M.-C.; Lafitte, J.-J.; Godard, P.; Zaegel, M.; Tack, V.; Lalau, G.; Bouvagnet, P. Axonemal Dynein Intermediate-Chain Gene (DNAI1) Mutations Result in Situs Inversus and Primary Ciliary Dyskinesia (Kartagener Syndrome). Am. J. Hum. Genet. 2001, 68, 1030–1035. [Google Scholar] [CrossRef] [Green Version]
- Zariwala, M.A.; Omran, H.; Ferkol, T.W. The Emerging Genetics of Primary Ciliary Dyskinesia. Proc. Am. Thorac. Soc. 2011, 8, 430–433. [Google Scholar] [CrossRef] [Green Version]
- Zariwala, M.A.; Knowles, M.R.; Omran, H. Genetic Defects in Ciliary Structure and Function. Annu. Rev. Physiol. 2007, 69, 423–450. [Google Scholar] [CrossRef]
- Zariwala, M.A.; Knowles, M.R.; Leigh, M.W. Primary Ciliary Dyskinesia: GeneReviews. 2019. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1122/ (accessed on 1 April 2021).
- Marshall, C.R.; Scherer, S.W.; Zariwala, M.A.; Lau, L.; Paton, T.A.; Stockley, T.; Jobling, R.K.; Ray, P.N.; Knowles, M.R.; Hall, D.A.; et al. Whole-Exome Sequencing and Targeted Copy Number Analysis in Primary Ciliary Dyskinesia. G3 Genes Genomes Genet. 2015, 5, 1775–1781. [Google Scholar] [CrossRef] [Green Version]
- Gileles-Hillel, A.; Mor-Shaked, H.; Shoseyov, D.; Reiter, J.; Tsabari, R.; Hevroni, A.; Cohen-Cymberknoh, M.; Amirav, I.; Brammli-Greenberg, S.; Horani, A.; et al. Whole-exome sequencing accuracy in the diagnosis of primary ciliary dyskinesia. ERJ Open Res. 2020, 6. [Google Scholar] [CrossRef]
- A Afzelius, B. A human syndrome caused by immotile cilia. Science 1976, 193, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Shoemark, A.; Boon, M.; Brochhausen, C.; Bukowy-Bieryllo, Z.; De Santi, M.M.; Goggin, P.; Griffin, P.; Hegele, R.; Hirst, R.A.; Leigh, M.W.; et al. International consensus guideline for reporting transmission electron microscopy results in the diagnosis of primary ciliary dyskinesia (BEAT PCD TEM Criteria). Eur. Respir. J. 2020, 55, 1900725. [Google Scholar] [CrossRef] [PubMed]
- Bustamante-Marin, X.M.; Horani, A.; Stoyanova, M.; Charng, W.-L.; Bottier, M.; Sears, P.R.; Yin, W.-N.; Daniels, L.A.; Bowen, H.; Conrad, D.F.; et al. Mutation of CFAP57, a protein required for the asymmetric targeting of a subset of inner dynein arms in Chlamydomonas, causes primary ciliary dyskinesia. PLoS Genet. 2020, 16, e1008691. [Google Scholar] [CrossRef] [PubMed]
- Shoemark, A.; Frost, E.; Dixon, M.; Ollosson, S.; Kilpin, K.; Patel, M.; Scully, J.; Rogers, A.V.; Mitchison, H.M.; Bush, A.; et al. Accuracy of Immunofluorescence in the Diagnosis of Primary Ciliary Dyskinesia. Am. J. Respir. Crit. Care Med. 2017, 196, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Rubbo, B.; Best, S.; Hirst, R.A.; Shoemark, A.; Goggin, P.; Carr, S.B.; Chetcuti, P.; Hogg, C.; Kenia, P.; Lucas, J.S.; et al. Clinical features and management of children with primary ciliary dyskinesia in England. Arch. Dis. Child. 2020, 105, 724–729. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, A.J.; Leigh, M.W.; Omran, H.; Lavergne, V.; Knowles, M.R. Errors in Methodology Affect Diagnostic Accuracy of High-Speed Videomicroscopy Analysis in Primary Ciliary Dyskinesia. Chest 2019, 156, 1032–1033. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, A.J.; Ferkol, T.W.; Manion, M.; Leigh, M.W.; Davis, S.D.; Knowles, M.R. High-Speed Videomicroscopy Analysis Presents Limitations in Diagnosis of Primary Ciliary Dyskinesia. Am. J. Respir. Crit. Care Med. 2020, 201, 122–123. [Google Scholar] [CrossRef]
- Simoneau, T.; Zandieh, S.O.; Rao, D.R.; Vo, P.; Palm, K.E.; McCown, M.; Kopel, L.S.; Dias, A.; Casey, A.; Perez-Atayde, A.R.; et al. Impact of Cilia Ultrastructural Examination on the Diagnosis of Primary Ciliary Dyskinesia. Pediatr. Dev. Pathol. 2013, 16, 321–326. [Google Scholar] [CrossRef]
- Shoemark, A.; Dixon, M.; Corrin, B.; Dewar, A. Twenty-year review of quantitative transmission electron microscopy for the diagnosis of primary ciliary dyskinesia. J. Clin. Pathol. 2011, 65, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Yin, W.; Smith, M.C.; Song, K.; Leigh, M.W.; Zariwala, M.A.; Knowles, M.R.; Ostrowski, L.E.; Nicastro, D. Cryo-electron tomography reveals ciliary defects underlying human RSPH1 primary ciliary dyskinesia. Nat. Commun. 2014, 5, 5727. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| nNO Routinely ≤ 77 nL/min | Limited Cases with nNO > 77 nL/min | nNO Routinely > 77 nL/min | |
|---|---|---|---|
| ARMC4 | DNAI2 | DNAH9 (9 cases) [25,26] | CCDC103 [27] |
| CCDC39 | DNAJB13 | TTC12 (2 cases) [28] | RSPH1 [29] |
| CCDC40 | DNAL1 | RPGR (<10 cases) [30,31] | |
| CCDC114 | DRC1 | CCNO (2 cases) [32] | |
| CCDC151 | DRC2 | FOXJ1 (4 cases) [33] | |
| CFAP57 | HYDIN | NEK10 (1 case) [34,35] | |
| CFAP298 | LRRC6 | GAS2L2 (2 cases) [36] | |
| CFAP300 | MCIDAS | GAS8 (1 cases) [37] | |
| DNAAF1 | NME5 | STK36 (1 case) [38] | |
| DNAAF2 | NME8 | CFAP221 (3 cases) [39] | |
| DNAAF3 | OFD1 | SPEF2 (1 cases) [40,41,42] | |
| DNAAF4 | PIHID3 | LRRC56 (1 case) [43] | |
| DNAAF5 | RSPH3 | ||
| DNAH1 | RSPH4A | ||
| DNAH5 | RSPH9 | ||
| DNAH8 | SPAG1 | ||
| DNAH11 | TTC25 | ||
| DNAI1 | ZMYND10 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Connor, M.G.; Horani, A.; Shapiro, A.J. Progress in Diagnosing Primary Ciliary Dyskinesia: The North American Perspective. Diagnostics 2021, 11, 1278. https://doi.org/10.3390/diagnostics11071278
O’Connor MG, Horani A, Shapiro AJ. Progress in Diagnosing Primary Ciliary Dyskinesia: The North American Perspective. Diagnostics. 2021; 11(7):1278. https://doi.org/10.3390/diagnostics11071278
Chicago/Turabian StyleO’Connor, Michael Glenn, Amjad Horani, and Adam J. Shapiro. 2021. "Progress in Diagnosing Primary Ciliary Dyskinesia: The North American Perspective" Diagnostics 11, no. 7: 1278. https://doi.org/10.3390/diagnostics11071278
APA StyleO’Connor, M. G., Horani, A., & Shapiro, A. J. (2021). Progress in Diagnosing Primary Ciliary Dyskinesia: The North American Perspective. Diagnostics, 11(7), 1278. https://doi.org/10.3390/diagnostics11071278