Abstract
Gardner syndrome is a neoplasic disease that associates intestinal polyposis and colorectal adenocarcinoma with osteomas and soft tissue tumors determined by germline mutations in the APC gene. The early diagnosis and identification of high-risk individuals are important because patients have a 100% risk of colon cancer. We present the case of a family with Gardner syndrome. Cephalometric, panoramic X-rays and CBCT of the proband and her brother showed multiple osteomas affecting the skull bones, mandible and paranasal sinuses. The detailed family history showed an autosomal dominant transmission with the presence of the disease in the mother and maternal grandfather of the proband. Both had the typical signs of disease and died in the fourth decade of life. Based on these aspects the clinical diagnosis was Gardner syndrome. By gene sequencing, a novel pathogenic variant c.4609dup (p.Thr1537Asnfs*7) in heterozygous status was identified in the APC gene in both siblings. We reviewed literature data concerning the correlation between the localization of mutations in the APC gene and the extracolonic manifestations of familial adenomatous polyposis as well as their importance in early diagnosis and adequate oncological survey of patients and families based on abnormal genomic variants.
1. Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide (approximately 1.8 million new cancer diagnoses yearly) and the second most common cause of cancer death (over 880,000 deaths in 2018) [1]. A special entity is represented by CRC with early onset that has a different clinical, pathological and molecular profile compared to CRC with a late onset. Thus, distinction between the two forms is important for effective prevention, detection and the therapeutic plan [2].
Usually, colorectal cancer in young people is discovered at an advanced stage due to the lack of screening programs in this age group. Exceptions are cases of hereditary syndromes predisposed to CRC (10% of all colorectal cancers), where, after the discovery of an index case, a large screening of the family follows [3,4].
Most early-onset cancers are monogenic (16–35% of total) [2,4,5,6,7]. The most common forms are Lynch Syndrome and familial adenomatous polyposis (FAP). The high percentage of monogenic cancers with early onset indicates and justifies genetic counselling and multigene panel testing [4]. However, nearly half of patients with a genetic form of CRC do not have a positive family history [6]. Thus, genetic diagnosis is important for patients, but also for the identification and management of relatives at risk of inheriting the mutation.
Familial adenomatous polyposis accounts for approximately 1% of all colorectal cancers [8]. FAP (OMIM #175100) is caused by germline mutations in a tumor suppressor gene—APC regulator of WNT signaling pathway (APC). Mutations of the APC gene have an autosomal dominant inheritance pattern. FAP can be divided in several forms: classic FAP (characterized by the presence of a large number >100 of colorectal adenomatous polyps), attenuated FAP (AFAP—characterized by a lower number of polyps, usually under 30), gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS), Gardner syndrome (associates FAP with osteomas and soft tissue tumors) and Turcot syndrome (association of FAP with central nervous system tumors) [9].
The presence of extracolonic non-malignant manifestations in Gardner syndrome (including osteomas, dental abnormalities, congenital hypertrophy of the retinal pigment epithelium, benign cutaneous lesions, desmoid tumors and adrenal masses) and their early appearance, even before the development of polyps/cancer, is an important factor for diagnosis and prevention.
We present two cases, siblings diagnosed with a new pathogenic variant in the APC gene to highlight the importance of extracolonic manifestations and family history in the early diagnosis of FAP. We also reviewed the extracolonic manifestations in FAP and their correlations with the location of mutations in the APC gene.
2. Materials and Methods
2.1. Patients
Two patients were included in the study: the proband and her brother. The proband is a young Caucasian female (16 year old at the time of diagnosis). She was referred to an oral surgeon because of a mandibular osteoma discovered during a routine consultation. The proband’s brother (12 year old at the time) had supernumerary teeth and a mandibular tumor on palpation. Family history showed that the proband’s mother was diagnosed with mandibular osteoma, supernumerary teeth and died at 34 years old because of intra-abdominal and abdominal wall tumors. The maternal grandfather had supernumerary teeth and soft tissue tumors and died around the age of 35 (Figure 1).
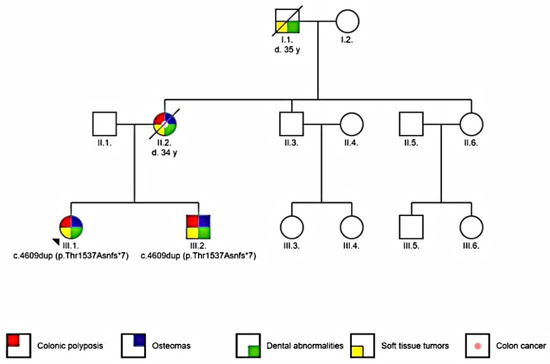
Figure 1.
Pedigree of the analyzed family (arrow indicates the proband).
The association of multiple osteomas, dental abnormalities, and family history of colon cancer with dominant transmission has led to clinical diagnosis of familial adenomatous polyposis Gardner variant.
2.2. Methods
Patients were evaluated by a multidisciplinary team consisting of an oral surgeon, radiologist, geneticist, gastroenterologist, endocrinologist and an ophthalmologist.
A complete evaluation was performed by anterior-posterior cephalometric and panoramic X-rays, Cone-beam computed tomography (CBCT), digestive endoscopic evaluation, molecular testing, ophthalmological and endocrinological evaluation. At the age of 18, the proband underwent surgical removal of the most prominent osteoma of the left angle of the mandible and a pathological examination was performed.
The patients underwent Invitae Multi-Cancer panel (Invitae, San Francisco, CA, USA). Full-gene sequencing, deletion/duplication analysis and variant interpretation were performed at Invitae (Table S1).
3. Results
3.1. Radiographic Assessment
Initial radiographic assessment of the female proband (III.1.) included: anterior-posterior cephalometric and panoramic X-rays which showed multiple osteomas affecting the skull bones and mandible, slight facial asymmetry and several impacted teeth (Figure 2a–c). CBCT revealed diffuse sclerosis of the jaw bones (Figure 2d–g), expansion of the body of the mandible, multiple peripheral osteomas including a large osteoma at the left angle of the mandible associated with facial asymmetry (Figure 2d–j). Small osteomas were registered within the anterior ethmoidal cells, frontal sinus, several skull bones (frontal, temporal, parietal, occipital), zygomatic arch, nasal bones (Figure 2g–j) and the left external auditory canal with a discrete decrease of its diameter (Figure 2i).
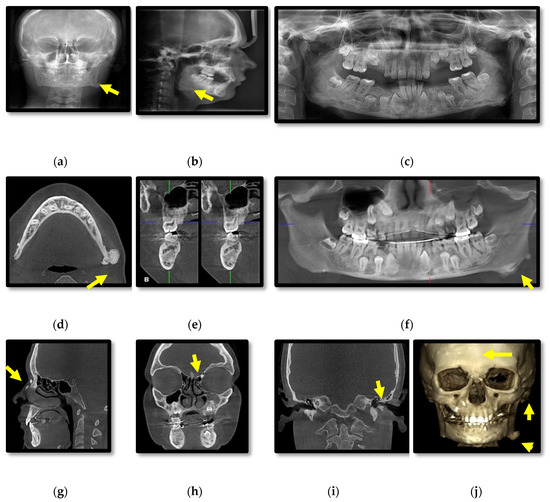
Figure 2.
Patient III.1. Cephalometric (a,b) and panoramic (c) radiographs; Cone-beam computed tomography (CBCT) with axial (d), cross-section (e), panoramic (f), sagittal (g), coronal (h,i) views and 3D reconstructions of bone tissue (j).
Cephalometric and panoramic radiographs of the male patient (III.2.) showing multiple radiopaque masses with relatively well-defined borders affecting the jaw bones, diffuse sclerosis of the body of the mandible and large masses near the gonions (Figure 3a,b). CBCT axial, cross-section, panoramic, sagittal, coronal, 3D lateral-oblique and 3D sagittal reconstructions showing multiple enostoses located in the medullary bone of the body of the maxilla and mandible, delayed dental eruptions and hypercementosis (Figure 3c–i). Bilateral mushroom-shaped osteomas affect the angle of the mandible, temporal and frontal bones (Figure 3h,i).
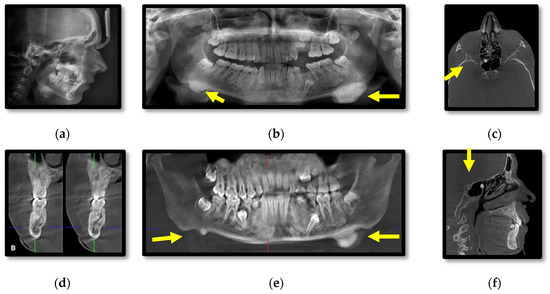
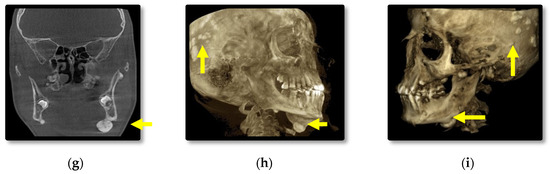
Figure 3.
Patient III.2. Cephalometric (a) and panoramic (b) radiographs; CBCT axial (c), cross-section (d), panoramic (e), sagittal (f), coronal (g), 3D lateral-oblique (h) and 3D sagittal (i).
In comparison, the male patient presented overall larger osteomas affecting the left angle of the mandible and both temporal bones, left coronoid process, impacted teeth, and increased bone sclerosis of the jaw bones. Additionally, a breast lipoma was observed in the male patient. Specific for this case is the presence of a pedunculated osteoma attached to the anterior wall of the right sphenoidal sinus and mucositis of the posterior ethmoidal cells.
3.2. Pathological Examination
The pathological examination showed compact bone tissue, consisting of mature bone lamellae with Haversian canals and paucicellular fibrous stroma (Figure 4). These morphological aspects confirm the presence of osteoma.
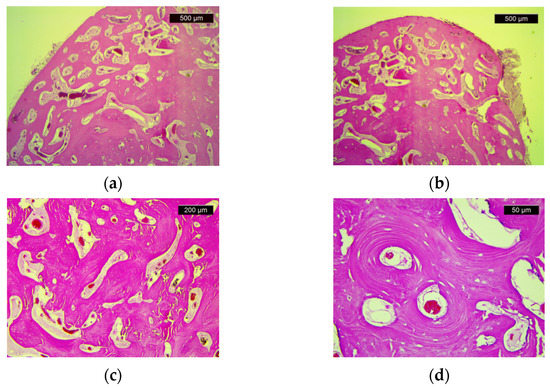
Figure 4.
Histologic features of osteoma: (a) Dense cortical lamellar bone tissue (HE, ×25); (b) Dense, compact bone tissue with marked paucicellular and congestive stromal fibrosis (HE, ×25); (c) Compact, dense bone with visible Haversian canals (HE, ×50); (d) Haversian canals (HE, ×200).
3.3. Digestive Endoscopic Evaluation
Upper digestive endoscopy in the proband showed a normal aspect of esophageal and gastric mucosa. A pseudopolipoid appearance (<2 mm) and hypertrophic papilla were observed in the duodenum. Lower digestive endoscopy revealed multiple polyps (approximately 50–70), the largest located in the sigmoid colon with a diameter of 5–6 mm. Polypoid lesions were observed in cecum (a 2 mm polyp) and at 5 cm from the ileocecal valve (also a 2 mm polyp).
Pathological examination of the biopsied fragments shows the structure of tubular adenomatous polyp with low-grade epithelial dysplasia. The colonic mucosa fragment has glandular architecture and preserved mucosecretion, with moderate lymphoplasmacytic inflammatory infiltrate and chorion edema.
Similar aspects of colon were discovered by endoscopy in the male patient.
3.4. Genetic Testing
Gene sequencing (by NGS) showed a novel pathogenic variant—c.4609dup (p.Thr1537Asnfs*7)—in heterozygous status in the APC gene in the siblings. The reference sequence is: APC: NM_000038.5. The novel sequence has a premature translational stop signal in the APC gene (p.Thr1537Asnfs*7). While this is not anticipated to result in nonsense mediated decay, it is expected to disrupt the last 1307 amino acids of the APC protein. This variant is not present in population databases (ExAC no frequency) [10]. It is expected to disrupt the domains of the protein, which mediate interactions with the cytoskeleton. A different truncation (p.Tyr2645Lysfs*14) that lies downstream of this variant has been determined to be pathogenic. This suggests that deletion of this region of the APC protein is causative of disease. For these reasons, this variant has been classified as pathogenic [10]. No pathogenic variants were identified in the other genes analyzed.
Ophthalmologic and endocrine examinations did not reveal particular aspects concordant with diagnosis of Gardner syndrome.
The diagnosis of Gardner syndrome was established based on the presence of multiple adenomatous polyps in the colon, the presence of extracolonic manifestations (multiple osteomas, dental abnormalities, soft tissue tumor), suggestive familial history for FAP, being confirmed by the discovery of a pathogenic variant in the APC gene.
So far, the female patient underwent surgical removal of the mandibular osteoma and orthodontic treatment. The male patient benefitted only from orthodontic treatment due to incomplete ossification. Both patients entered the surveillance oncological program with an annual endoscopic evaluation of the digestive mucosa and biopsy of the lesions.
4. Discussion
The APC protein is a complex protein, having several domains with different cellular roles. The APC protein contains an oligomerization domain; an armadillo repeat-domain (implied in cell migration and cell adhesion by link of Asef, KAP3 and IQGAP1); a 15–20 aa repeats that fix β-catenin and participates in cell adhesion, proliferation and differentiation; SAM repeats that allow connection with axin; a basic region for microtubule binding and C-terminal domains for EB1 binding. The last two domains are implied in chromosomal segregation and mitotic progression [11].
The majority of mutations are missense or frameshift, produce a truncated protein and modify the last exon that contains 75% of the coding sequence of the gene [12,13]. The region between codons 1250 and 1464 is considered a mutation cluster region (MCR) and mutations of this region have a higher frequency in the general population (15.49% of total) [14]. The significant consequences seem to be mutations in domains implied in binding β-catenin, EB1 and microtubules. These mutations disrupt cellular processes and generate the appearance of tumors by stimulation of cell migration, activation of proliferation and inhibition of differentiation. Additionally, chromosomal instability and cell immortalization are produced [11]. The most common mutations are in codon 1309 (10% of patients with FAP) and codon 1061 (5% of patients); 25–30% of mutations are de novo. In these cases, there is no evidence of FAP phenotype or mutations in the APC gene in family members. In some cases, the mutation exists only in the gametes (germinal mosaicism) [15].
The types of mutations in the APC gene included in the Human Gene Mutation Database (HGMD®) are: missense/nonsense (578 variants), splicing substitutions (125 variants), regulatory substitutions (13 variants), small deletions (20 pb or less; 796 variants), small insertions/duplications (20 pb or less; 343 variants), small indels (20 pb or less; 51 variants), gross deletions (143 variants), gross insertions/duplications (16 variants), complex rearrangements (13 variants) (HGMD® Professional 2021.1, accessed on 26 June 2021) [16]. The distribution of germline mutations among exons is presented in Figure 5.
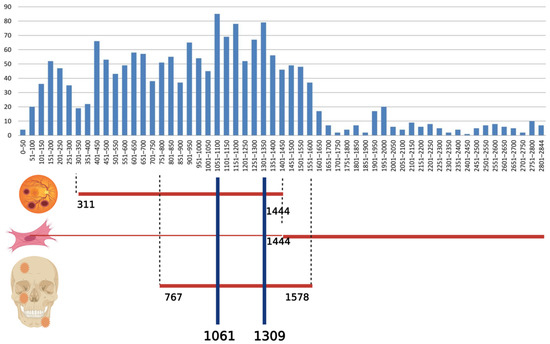
Figure 5.
Distribution of germline mutations in APC gene (data from the online database HGMD® Professional 2021.1, accessed on 26 June 2021) and correlation with congenital hypertrophy of the retinal pigment epithelium, desmoid tumors and osteomas. The abscissa represents the APC codon number and the ordinate represents the number of different mutations. Created with BioRender.com.
The gene panel used for our patients contains genes involved in hereditary polyposis syndromes, some of which are relatively new but also genes involved in other types of hereditary cancers. Using such a panel offers accurate differential diagnosis with the intestinal polyposis syndromes. Among those genes, we mention: NTHL1 (nth like DNA glycosylase 1), POLE (DNA polymerase epsilon, catalytic subunit), POLD1 (DNA polymerase delta 1, catalytic subunit), MSH3 (mutS homolog 3), AXIN2 (axin 2), GREM1 (gremlin 1, DAN family BMP antagonist), MUTYH (mutY DNA glycosylase), STK11 (serine/threonine kinase 11), SMAD4 (SMAD family member 4) and BMPR1A (bone morphogenetic protein receptor type 1A). All these genes are found in the panel of genes recommended for testing for inherited colorectal cancer and polyposis by the American College of Medical Genetics and Genomics [9]. The use of targeted gene panel-based next-generation sequencing technology is recommended for patients with FAP because it is cost effective, and less time consuming. This approach is more targeted but delivers less information than whole genome sequencing (WGS) as only a subset of genes is evaluated [17].
4.1. Extracolonic Manifestations in FAP
Penetrance is almost complete for colonic manifestations, but variable for extra colonic manifestations. The development of adenomatous polyps begins in childhood and adolescence. It is estimated that 50% of patients with FAP have colorectal adenomas by age 15 and the percentage increases to 95% by age 25 [18]. The age of onset of colonic symptoms correlates with the location of the mutation: age 20 (variants at 1309 codon), age 30 (variants between 168 and 1580 codons except for the 1309 codon), age 52 (variants in 5′ of codon 168 and 3′ of codon 1580) [19].
There are two categories of extracolonic manifestations in FAP: malignant and non-malignant. They are present in 70% of FAP cases [20]. Non-malignant extra intestinal manifestations are represented by: osteomas, dental abnormalities, congenital hypertrophy of the retinal pigment epithelium (CHRPE), benign cutaneous lesions, desmoid tumors and adrenal masses [21]. Malignant manifestations are located in the small bowel (duodenum, periampulla or distal to the duodenum), pancreas, thyroid, CNS, liver, bile ducts and stomach [21].
4.2. Osteomas
Osteomas are present in 60–80% of patients with FAP and 50% of all osteomas occur in FAP [22,23]. The prevalence of osteomas is 1–2% in the general population [24]. Osteomas are benign tumors characterized by compact lamellar cortical or cancellous bone [25]. There are three types of osteomas: central (originates in the endosteum), peripheral (originates in the periosteum) and extra skeletal soft tissue osteoma (developing in the muscles) [26]. They are most commonly located in the skull (jaw bones and paranasal sinuses), rarely in long bones or muscles. Osteomas are more common in the mandible than in the maxilla [27].
In descending order of the frequency of osteomas at the level of the sinuses, we mention: frontal sinus, ethmoid, maxillary and sphenoid sinuses. The prevalence in the paranasal sinuses is different depending on the imaging method used for their detection: conventional examination shows a prevalence of about 1% in the general population, while the use of computed tomography has a higher detection rate showing a prevalence of about 3% in the general population [28]. The presence of multiple osteomas (more than three osteomas) is suggestive for Gardner syndrome [28,29]. In a prospective study of paranasal osteomas, Erdogan et al. showed that in 7% of cases, osteomas were detected in more than one sinus [28].
Osteomas are important in the early management of FAP due to the fact that they occur before intestinal manifestations—the detection of osteomas precedes the diagnosis of FAP by 17 years [23]. The appearance of osteomas is associated with mutations between codons 767 and 1578 [24]. In our case the mutation is present in the same region.
Another particularity of our patients is the presence of multiple osteomas in cranial bones, jaw bones and paranasal sinuses. However, osteoma of sphenoid sinus (present in patient III.2) is a less common location.
4.3. Dental Abnormalities
Dental abnormalities are present in 30–75% of patients with FAP [30]. The meta-analysis performed by Almeida et al. in 2015 shows a frequency of dental anomalies of 30.48% [31]. Dental abnormalities in FAP are represented by: impacted teeth, congenitally missing teeth, supernumerary teeth, dentiferous cysts, compound odontomas, hypercementosis [21,22]. Supernumerary teeth were identified in 11–27% of FAP patients compared to 0–4% in the general population. The most common location is anterior and around canines, in the alveolar bone between the teeth or attached to the follicle of an impacted tooth. However, this location is not specific to FAP [32,33]. The literature is poor in studies about the correlation between the presence of dental abnormalities and the location of mutations in the APC gene. It was assumed that there was the same correlation as in the case of desmoid tumors and osteomas [34,35].
Due to the existence of dental abnormalities, a dentist may be involved in the early diagnosis of familial adenomatous polyposis.
Supernumerary teeth can be an important clue for early diagnosis. We found dental modifications in our cases. The two siblings have impacted teeth and hypercementosis, while their mother and their maternal grandfather showed supernumerary teeth.
4.4. Desmoid Tumors
Desmoid tumors (DT) appear in the musculoaponeurotic tissue in any region of the body. There are 3 common locations: extremities (proximal or at the girdle), abdominal wall and intra-abdominal (intestinal wall and mesentery) [36]. They represent 3% of soft tissue tumors and 0.03% of all neoplasms [36]. DT occurs in 12–15% of FAP patients [37]. They generate a high mortality in patients with FAP, with a rate between 10% and 50% [38]. The patient with FAP has an 800-fold higher risk of developing DT and the risk increases if there is a mutation after exon 1399 in the APC gene [37]. DT in FAP appear earlier than sporadic DT [39]. In 80% of patients with DT, they appear until the age of 40 with a peak in the second and third decade of life [37]; 65% of DT occur intra-abdominally [37]. There are four risk factors for DT: a positive family history for DT, mutation location, surgical trauma, female sex [37].
The correlation between the appearance of DT and the location of the mutation is not highly specific. Friedl et al. found a 60% frequency of DT in patients with FAP caused by mutations in codons 1445–1580, but a 20% frequency of DT in patients with mutations before codon 1444 in the FAP gene [19].
4.5. Congenital Hypertrophy of the Retinal Pigment Epithelium
Congenital hypertrophy of the retinal pigment epithelium (CHRPE) is a pigmented lesion with a depigmented halo in the retina and the most common extracolonic manifestation found in 74% of FAP patients compared to 1.2–4.4% in the general population [40,41]. It has no clinical significance and it is asymptomatic. It is the earliest extracolonic sign present at birth or in the neonatal period [41]. It is associated with mutations in the codon region 311–1444 [42]; 70.74% of the mutations described in HGMD are in this codon range.
4.6. Malignant Extracolonic Manifestations
Malignant extracolonic manifestations in FAP are represented by an additional cancer in addition to colon cancer in FAP. Lifetime risk for cancer in FAP differs among cancer types [21]. Some entities are rare, such as small bowel cancer distal to the duodenum, others are more common, such as thyroid cancer or small bowel cancer (duodenum or periampulla) (Table 1). Studies concerning this type of cancer are rare, in correlation with their small frequency.

Table 1.
Extracolonic cancer in FAP—lifetime risk (modified after Jasperson) [21].
Mutations in the APC gene 697–1224 codons are associated with a 3-fold higher risk of brain tumor and a 13-fold higher risk of medulloblastoma [43]. Duodenal adenomas have a 3–4 times higher risk of developing if the mutation in APC is between codons 976–1067 [44].
4.7. Thyroid Cancer
The incidence of thyroid cancer is 2–12% in cases with FAP and 10% in the Gardner variant with a cumulative risk of 2.8% by age 60 [45]. This neoplasia is multifocal and bilateral. In cases with FAP, the majority of cases are cribriform morular variant of papillary CT [46]. It is associated with germline mutations between the codons 1286 and 1513 in the APC gene [45,47]. In the respective codons interval, there are 251 variants which represent 14.40% of the total different mutations reported in HGMD up to date. It predominates in women and is diagnosed earlier than sporadic with an average diagnostic age of 29.2 years [47,48,49]. In a systematic review, Septer et al. concluded that there is an increased risk of thyroid cancer associated with a mutation in codon 1061 [49].
5. Conclusions
The genetic syndromes predisposed to colorectal cancer (including FAP) are characterized by pleiotropy and genetic heterogeneity. Mutations in the APC gene, which later lead to familial adenomatous polyposis manifest earlier as osteomas, dental abnormalities, congenital hypertrophy of the retinal pigment epithelium, benign cutaneous lesions, desmoid tumors and adrenal masses. An early diagnosis via these early manifestations allows a proper oncological surveillance. Furthermore, a detailed family history in patients with these extracolonic manifestations is very important in identifying the family members at risk.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/diagnostics11091560/s1, Table S1: Invitae Multi-Cancer panel (2019).
Author Contributions
Conceptualization, L.C. and E.V.G.; methodology, L.C. and E.V.G.; software, L.C.; validation, D.H., L.C. and E.V.G.; formal analysis, M.L.C.; investigation, C.A., M.L.C., V.-L.D., O.-B.B., B.I.D., M.-C.P., N.C.G., V.V.L. and D.D.; resources, L.C.; data curation, L.C. and M.L.C.; writing—original draft preparation, M.L.C. and L.C.; writing—review and editing, L.C., C.G. and E.V.G.; visualization, D.H.; supervision, D.H., L.C., C.G. and E.V.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Committee of “St. Spiridon” Hospital, Iasi, Romania (32/7 June 2021).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patients to publish this paper.
Data Availability Statement
Some data were obtained from Human Gene Mutation Database (HGMD® Professional 2021.1, accessed on 26 June 2021) available online: http://www.hgmd.cf.ac.uk/ac/all.php.
Acknowledgments
The authors thank Catalina Cucu (“St. Spiridon” Hospital, Iasi, Romania) for her support. Pedigree was constructed and drawn using (Progeny Clinical Version N/Progeny Lab Version N) (Progeny Genetics LLC, Delray Beach, FL, www.progenygenetics.com) (accessed on 18 June 2021).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, N.; Ugai, T.; Zhong, R.; Hamada, T.; Fujiyoshi, K.; Giannakis, M.; Wu, K.; Cao, Y.; Ng, K.; Ogino, S. Rising incidence of early-onset colorectal cancer—A call to action. Nat. Rev. Clin. Oncol. 2020, 1–14. [Google Scholar] [CrossRef]
- Silla, I.O.; Rueda, D.; Rodríguez, Y.; García, J.L.; de la Cruz Vigo, F.; Perea, J. Early-onset colorectal cancer: A separate subset of colorectal cancer. World J. Gastroenterol. 2014, 20, 17288. [Google Scholar] [CrossRef]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef]
- Patel, S.G.; Ahnen, D.J. Colorectal cancer in the young. Curr. Hepatol. Rep. 2018, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, E.M.; Koeppe, E.; Everett, J.; Ulintz, P.; Kiel, M.; Osborne, J.; Williams, L.; Hanson, K.; Gruber, S.B.; Rozek, L.S. Germline genetic features of young individuals with colorectal cancer. Gastroenterology 2018, 154, 897–905. [Google Scholar] [CrossRef]
- Mork, M.E.; You, Y.N.; Ying, J.; Bannon, S.A.; Lynch, P.M.; Rodriguez-Bigas, M.A.; Vilar, E. High prevalence of hereditary cancer syndromes in adolescents and young adults with colorectal cancer. J. Clin. Oncol. 2015, 33, 3544. [Google Scholar] [CrossRef]
- Yalcin, S.; Philip, P.A. (Eds.) Textbook of Gastrointestinal Oncology; Springer Nature: Cham, Switzerland, 2019. [Google Scholar]
- Mao, R.; Krautscheid, P.; Graham, R.P.; Ganguly, A.; Shankar, S.; Ferber, M.; Hegde, M. Genetic testing for inherited colorectal cancer and polyposis, 2021 revision: A technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 1–11. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. ClinVar; [VCV000858116.2]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000858116.2 (accessed on 8 August 2021).
- Zhang, L.; Shay, J.W. Multiple roles of APC and its therapeutic implications in colorectal cancer. JNCI J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Morin, P.J.; Weeraratna, A.T. The APC tumor suppressor pathway. In Tumor Suppressor Genes; Humana Press: Totowa, NJ, USA, 2003; pp. 21–40. [Google Scholar]
- Aitchison, A.; Hakkaart, C.; Day, R.C.; Morrin, H.R.; Frizelle, F.A.; Keenan, J.I. APC Mutations Are Not Confined to Hotspot Regions in Early-Onset Colorectal Cancer. Cancers 2020, 12, 3829. [Google Scholar] [CrossRef]
- Cetta, F.; Dhamo, A. Inherited multitumoral syndromes including colorectal carcinoma. Surg. Oncol. 2007, 16, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Half, E.; Bercovich, D.; Rozen, P. Familial adenomatous polyposis. Orphanet J. Rare Dis. 2009, 4, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Qual. Life Res. 2017, 136, 665–677. Available online: http://www.hgmd.cf.ac.uk/ac/all.php (accessed on 26 June 2021). [CrossRef] [PubMed]
- Wang, D.; Liang, S.; Zhang, X.; Dey, S.K.; Li, Y.; Xu, C.; Yu, Y.; Li, M.; Zhao, G.; Zhang, Z. Targeted next-generation sequencing approach for molecular genetic diagnosis of hereditary colorectal cancer: Identification of a novel single nucleotide germline insertion in adenomatous polyposis coli gene causes familial adenomatous polyposis. Mol. Genet. Genom. Med. 2019, 7, e00505. [Google Scholar] [CrossRef] [PubMed]
- Brosens, L.A.; van Hattem, W.A.; Jansen, M.; de Leng, W.W.; Giardiello, F.M.; Offerhaus, G.J.A. Gastrointestinal polyposis syndromes. Curr. Mol. Med. 2007, 7, 29–46. [Google Scholar] [CrossRef]
- Friedl, W.; Caspari, R.; Sengteller, M.; Uhlhaas, S.; Lamberti, C.; Jungck, M.; Kadmon, M.; Wolf, M.; Fahnenstich, J.; Gebert, J.; et al. Can APC mutation analysis contribute to therapeutic decisions in familial adenomatous polyposis? Experience from 680 FAP families. Gut 2001, 48, 515–521. [Google Scholar] [CrossRef]
- Ficari, F.; Cama, A.; Valanzano, R.; Curia, M.C.; Palmirotta, R.; Aceto, G.; Esposito, D.L.; Crognale, S.; Lombardi, A.; Messerini, L.; et al. APC gene mutations and colorectal adenomatosis in familial adenomatous polyposis. Br. J. Cancer 2000, 82, 348–353. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jasperson, K.W.; Patel, S.G.; Ahnen, D.J. APC-Associated Polyposis Conditions. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; 18 December 1998 [Updated 2 February 2017]; University of Washington: Seattle, WA, USA, 1993–2021; Available online: https://www.ncbi.nlm.nih.gov/books/NBK1345/ (accessed on 6 June 2021).
- Dinarvand, P.; Davaro, E.P.; Doan, J.V.; Ising, M.E.; Evans, N.R.; Phillips, N.J.; Lai, J.; Guzman, M.A. Familial adenomatous polyposis syndrome: An update and review of extraintestinal manifestations. Arch. Pathol. Lab. Med. 2019, 143, 1382–1398. [Google Scholar] [CrossRef] [PubMed]
- Bülow, S. Incidence of associated diseases in familial polyposis coli. In Seminars in Surgical Oncology; John Wiley & Sons, Inc.: New York, NY, USA; Volume 3, pp. 84–87. [CrossRef]
- Groen, E.J.; Roos, A.; Muntinghe, F.L.; Enting, R.H.; de Vries, J.; Kleibeuker, J.H.; Witjes, M.J.; Links, T.P.; van Beek, A.P. Extra-intestinal manifestations of familial adenomatous polyposis. Ann. Surg. Oncol. 2008, 15, 2439–2450. [Google Scholar] [CrossRef]
- Gundewar, S.; Kothari, D.S.; Mokal, N.J.; Ghalme, A. Osteomas of the craniofacial region: A case series and review of literature. Indian J. Plast. Surg. 2013, 46, 479. [Google Scholar] [CrossRef]
- Orabona, G.D.A.; Salzano, G.; Iaconetta, G.; Piombino, P.; Ponzo, L.; Santella, A.; Astarita, F.; Solari, D.; Salzano, F.A.; Califano, L. Facial osteomas: Fourteen cases and a review of literature. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 1796–1802. [Google Scholar]
- Debta, P.; Debta, F.M.; Bussari, S.; Acharya, S.S.; Jeergal, V.A. Cancellous osteoma of maxilla: A rare case report. J. Int. Soc. Prev. Community Dent. 2016, 6, 261. [Google Scholar] [CrossRef]
- Erdogan, N.; Demir, U.; Songu, M.; Ozenler, N.K.; Uluç, E.; Dirim, B. A prospective study of paranasal sinus osteomas in 1889 cases: Changing patterns of localization. Laryngoscope 2009, 119, 2355–2359. [Google Scholar] [CrossRef]
- Payne, M.; Anderson, J.A.; Cook, J. Gardner’s syndrome—A case report. Br. Dent. J. 2002, 193, 383–384. [Google Scholar] [CrossRef]
- Cankaya, A.B.; Erdem, M.A.; Isler, S.C.; Cifter, M.; Olgac, V.; Kasapoglu, C.; Oral, C.K. Oral and maxillofacial considerations in Gardner’s syndrome. Int. J. Med. Sci. 2012, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Almeida, F.T.; Pachêco-Pereira, C.; Porporatti, A.L.; Flores-Mir, C.; Leite, A.F.; De Luca Canto, G.; Guerra, E.N.S. Oral manifestations in patients with familial adenomatous polyposis: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Wijn, M.A.; Keller, J.J.; Giardiello, F.M.; Brand, H.S. Oral and maxillofacial manifestations of familial adenomatous polyposis. Oral Dis. 2007, 13, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Ida, M.; Nakamura, T.; Utsunomiya, J. Osteomatous changes and tooth abnormalities found in the jaws of patients with adenomatosis coli. Oral Surg. Oral Med. Oral Pathol. 1981, 52, 2–11. [Google Scholar] [CrossRef]
- Oku, T.; Takayama, T.; Sato, Y.; Sato, Y.; Takada, K.; Hayashi, T.; Takahashi, M.; Kuroda, M.; Kato, J.; Niitsu, Y. A case of Gardner syndrome with a mutation at codon 1556 of APC: A suggested case of genotype–phenotype correlation in dental abnormality. Eur. J. Gastroenterol. Hepatol. 2004, 16, 101–105. [Google Scholar] [CrossRef]
- Järvinen, H.J.; Peltomäki, P. The complex genotype–phenotype relationship in familial adenomatous polyposis. Eur. J. Gastroenterol. Hepatol. 2004, 16, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Papagelopoulos, P.J.; Mavrogenis, A.F.; Mitsiokapa, E.A.; Papaparaskeva, K.T.; Galanis, E.C.; Soucacos, P.N. Current trends in the management of extra-abdominal desmoid tumours. World J. Surg. Oncol. 2006, 4, 1–8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sinha, A.; Tekkis, P.P.; Gibbons, D.C.; Phillips, R.K.; Clark, S.K. Risk factors predicting desmoid occurrence in patients with familial adenomatous polyposis: A meta-analysis. Colorectal Dis. 2011, 13, 1222–1229. [Google Scholar] [CrossRef]
- Clark, S.K.; Neale, K.F.; Landgrebe, J.C.; Phillips, R.K.S. Desmoid tumours complicating familial adenomatous polyposis. Br. J. Surg. 1999, 86, 1185–1189. [Google Scholar] [CrossRef] [PubMed]
- Koskenvuo, L.; Ristimäki, A.; Lepistö, A. Comparison of sporadic and FAP-associated desmoid-type fibromatoses. J. Surg. Oncol. 2017, 116, 716–721. [Google Scholar] [CrossRef]
- Tiret, A.; Parc, C. Fundus lesions of adenomatous polyposis. Curr. Opin. Ophthalmol. 1999, 10, 168–172. [Google Scholar] [CrossRef]
- Nusliha, A.; Dalpatadu, U.; Amarasinghe, B.; Chandrasinghe, P.C.; Deen, K.I. Congenital hypertrophy of retinal pigment epithelium (CHRPE) in patients with familial adenomatous polyposis (FAP); a polyposis registry experience. BMC Res. Notes 2014, 7, 1–4. [Google Scholar] [CrossRef]
- Nieuwenhuis, M.H.; Vasen, H.F.A. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): A review of the literature. Crit. Rev. Oncol. Hematol. 2007, 61, 153–161. [Google Scholar] [CrossRef]
- Attard, T.M.; Giglio, P.; Koppula, S.; Snyder, C.; Lynch, H.T. Brain tumors in individuals with familial adenomatous polyposis: A cancer registry experience and pooled case report analysis. Cancer 2007, 109, 761–766. [Google Scholar] [CrossRef]
- Wiik, M.U.; Talseth-Palmer, B.A. Familial Adenomatous Polyposis. In Handbook of Tumor Syndromes; Liu, D., Ed.; CRC Press—Taylor & Francis Group: Boca Raton, FL, USA, 2020. [Google Scholar]
- Pichert, G.; Jacobs, C. (Eds.) Rare Hereditary Cancers: Diagnosis and Management; Springer International Publishing: Cham, Switzerland, 2016; Volume 205. [Google Scholar]
- Steinhagen, E.; Guillem, J.G.; Chang, G.; Salo-Mullen, E.E.; Shia, J.; Fish, S.; Stadler, Z.K.; Markowitz, A.J. The prevalence of thyroid cancer and benign thyroid disease in patients with familial adenomatous polyposis may be higher than previously recognized. Clin. Colorectal Cancer 2012, 11, 304–308. [Google Scholar] [CrossRef]
- Truta, B.; Allen, B.A.; Conrad, P.G.; Kim, Y.S.; Berk, T.; Gallinger, S.; Bapat, B.; Terdiman, J.P.; Sleisenger, M.H. Genotype and phenotype of patients with both familial adenomatous polyposis and thyroid carcinoma. Fam. Cancer 2003, 2, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Cetta, F.; Montalto, G.; Gori, M.; Curia, M.C.; Cama, A.; Olschwang, S. Germline mutations of the APC gene in patients with familial adenomatous polyposis-associated thyroid carcinoma: Results from a European cooperative study. J. Clin. Endocrinol. Metab. 2000, 85, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Septer, S.; Slowik, V.; Morgan, R.; Dai, H.; Attard, T. Thyroid cancer complicating familial adenomatous polyposis: Mutation spectrum of at-risk individuals. Hered. Cancer Clin. Pract. 2013, 11, 1–6. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).