
Novel Mutation in APC Gene Associated with Multiple Osteomas in a Family and Review of Genotype-Phenotype Correlations of Extracolonic Manifestations in Gardner Syndrome

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
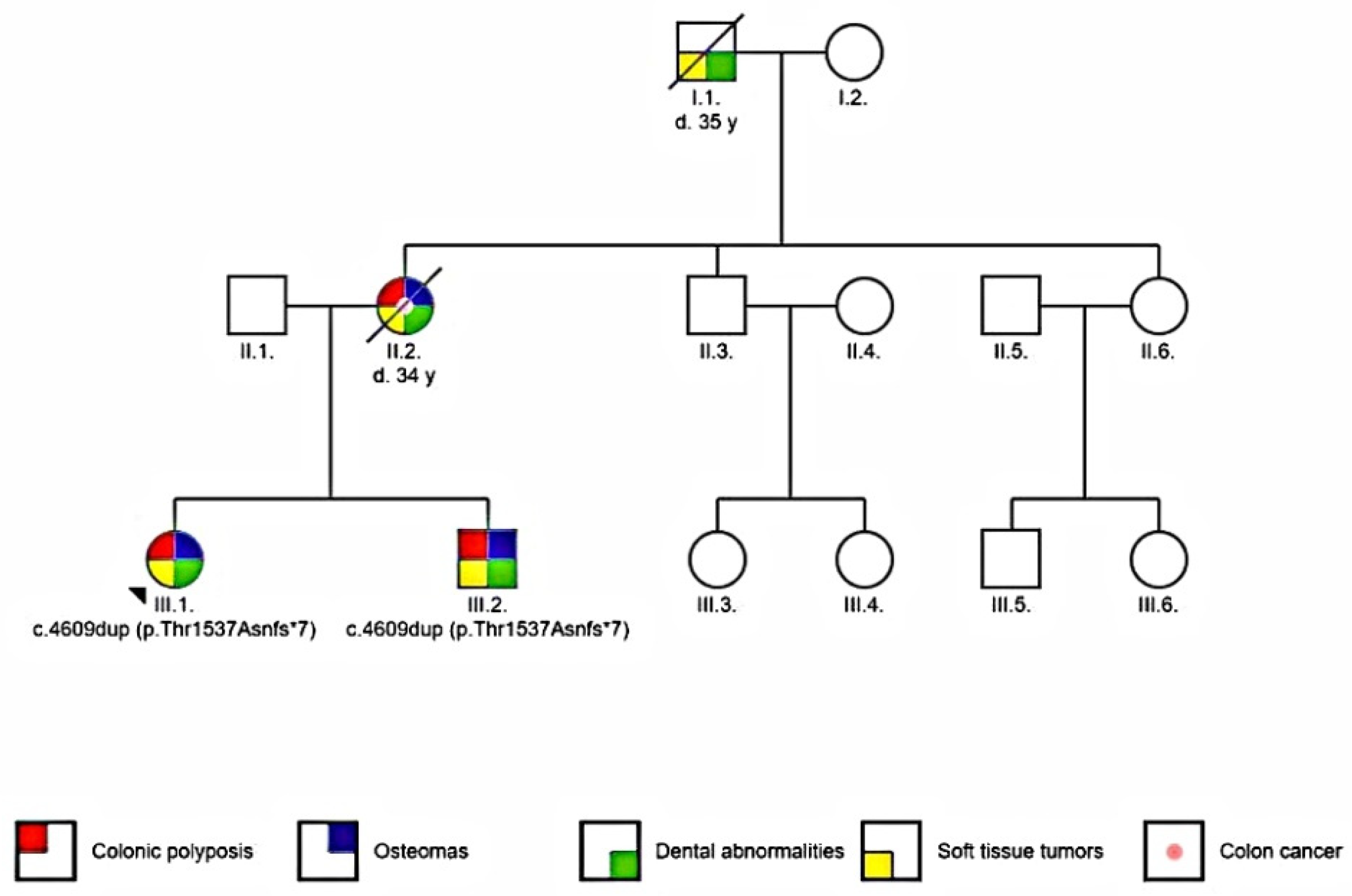
2.1. Patients
2.2. Methods
3. Results
3.1. Radiographic Assessment
3.2. Pathological Examination
3.3. Digestive Endoscopic Evaluation
3.4. Genetic Testing
4. Discussion
4.1. Extracolonic Manifestations in FAP
4.2. Osteomas
4.3. Dental Abnormalities
4.4. Desmoid Tumors
4.5. Congenital Hypertrophy of the Retinal Pigment Epithelium
4.6. Malignant Extracolonic Manifestations
4.7. Thyroid Cancer
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akimoto, N.; Ugai, T.; Zhong, R.; Hamada, T.; Fujiyoshi, K.; Giannakis, M.; Wu, K.; Cao, Y.; Ng, K.; Ogino, S. Rising incidence of early-onset colorectal cancer—A call to action. Nat. Rev. Clin. Oncol. 2020, 1–14. [Google Scholar] [CrossRef]
- Silla, I.O.; Rueda, D.; Rodríguez, Y.; García, J.L.; de la Cruz Vigo, F.; Perea, J. Early-onset colorectal cancer: A separate subset of colorectal cancer. World J. Gastroenterol. 2014, 20, 17288. [Google Scholar] [CrossRef]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef]
- Patel, S.G.; Ahnen, D.J. Colorectal cancer in the young. Curr. Hepatol. Rep. 2018, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, E.M.; Koeppe, E.; Everett, J.; Ulintz, P.; Kiel, M.; Osborne, J.; Williams, L.; Hanson, K.; Gruber, S.B.; Rozek, L.S. Germline genetic features of young individuals with colorectal cancer. Gastroenterology 2018, 154, 897–905. [Google Scholar] [CrossRef]
- Mork, M.E.; You, Y.N.; Ying, J.; Bannon, S.A.; Lynch, P.M.; Rodriguez-Bigas, M.A.; Vilar, E. High prevalence of hereditary cancer syndromes in adolescents and young adults with colorectal cancer. J. Clin. Oncol. 2015, 33, 3544. [Google Scholar] [CrossRef] [Green Version]
- Yalcin, S.; Philip, P.A. (Eds.) Textbook of Gastrointestinal Oncology; Springer Nature: Cham, Switzerland, 2019. [Google Scholar]
- Mao, R.; Krautscheid, P.; Graham, R.P.; Ganguly, A.; Shankar, S.; Ferber, M.; Hegde, M. Genetic testing for inherited colorectal cancer and polyposis, 2021 revision: A technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 1–11. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. ClinVar; [VCV000858116.2]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000858116.2 (accessed on 8 August 2021).
- Zhang, L.; Shay, J.W. Multiple roles of APC and its therapeutic implications in colorectal cancer. JNCI J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [Green Version]
- Morin, P.J.; Weeraratna, A.T. The APC tumor suppressor pathway. In Tumor Suppressor Genes; Humana Press: Totowa, NJ, USA, 2003; pp. 21–40. [Google Scholar]
- Aitchison, A.; Hakkaart, C.; Day, R.C.; Morrin, H.R.; Frizelle, F.A.; Keenan, J.I. APC Mutations Are Not Confined to Hotspot Regions in Early-Onset Colorectal Cancer. Cancers 2020, 12, 3829. [Google Scholar] [CrossRef]
- Cetta, F.; Dhamo, A. Inherited multitumoral syndromes including colorectal carcinoma. Surg. Oncol. 2007, 16, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Half, E.; Bercovich, D.; Rozen, P. Familial adenomatous polyposis. Orphanet J. Rare Dis. 2009, 4, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Qual. Life Res. 2017, 136, 665–677. Available online: http://www.hgmd.cf.ac.uk/ac/all.php (accessed on 26 June 2021). [CrossRef] [PubMed] [Green Version]
- Wang, D.; Liang, S.; Zhang, X.; Dey, S.K.; Li, Y.; Xu, C.; Yu, Y.; Li, M.; Zhao, G.; Zhang, Z. Targeted next-generation sequencing approach for molecular genetic diagnosis of hereditary colorectal cancer: Identification of a novel single nucleotide germline insertion in adenomatous polyposis coli gene causes familial adenomatous polyposis. Mol. Genet. Genom. Med. 2019, 7, e00505. [Google Scholar] [CrossRef] [PubMed]
- Brosens, L.A.; van Hattem, W.A.; Jansen, M.; de Leng, W.W.; Giardiello, F.M.; Offerhaus, G.J.A. Gastrointestinal polyposis syndromes. Curr. Mol. Med. 2007, 7, 29–46. [Google Scholar] [CrossRef]
- Friedl, W.; Caspari, R.; Sengteller, M.; Uhlhaas, S.; Lamberti, C.; Jungck, M.; Kadmon, M.; Wolf, M.; Fahnenstich, J.; Gebert, J.; et al. Can APC mutation analysis contribute to therapeutic decisions in familial adenomatous polyposis? Experience from 680 FAP families. Gut 2001, 48, 515–521. [Google Scholar] [CrossRef] [Green Version]
- Ficari, F.; Cama, A.; Valanzano, R.; Curia, M.C.; Palmirotta, R.; Aceto, G.; Esposito, D.L.; Crognale, S.; Lombardi, A.; Messerini, L.; et al. APC gene mutations and colorectal adenomatosis in familial adenomatous polyposis. Br. J. Cancer 2000, 82, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Jasperson, K.W.; Patel, S.G.; Ahnen, D.J. APC-Associated Polyposis Conditions. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; 18 December 1998 [Updated 2 February 2017]; University of Washington: Seattle, WA, USA, 1993–2021; Available online: https://www.ncbi.nlm.nih.gov/books/NBK1345/ (accessed on 6 June 2021).
- Dinarvand, P.; Davaro, E.P.; Doan, J.V.; Ising, M.E.; Evans, N.R.; Phillips, N.J.; Lai, J.; Guzman, M.A. Familial adenomatous polyposis syndrome: An update and review of extraintestinal manifestations. Arch. Pathol. Lab. Med. 2019, 143, 1382–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bülow, S. Incidence of associated diseases in familial polyposis coli. In Seminars in Surgical Oncology; John Wiley & Sons, Inc.: New York, NY, USA; Volume 3, pp. 84–87. [CrossRef]
- Groen, E.J.; Roos, A.; Muntinghe, F.L.; Enting, R.H.; de Vries, J.; Kleibeuker, J.H.; Witjes, M.J.; Links, T.P.; van Beek, A.P. Extra-intestinal manifestations of familial adenomatous polyposis. Ann. Surg. Oncol. 2008, 15, 2439–2450. [Google Scholar] [CrossRef] [Green Version]
- Gundewar, S.; Kothari, D.S.; Mokal, N.J.; Ghalme, A. Osteomas of the craniofacial region: A case series and review of literature. Indian J. Plast. Surg. 2013, 46, 479. [Google Scholar] [CrossRef]
- Orabona, G.D.A.; Salzano, G.; Iaconetta, G.; Piombino, P.; Ponzo, L.; Santella, A.; Astarita, F.; Solari, D.; Salzano, F.A.; Califano, L. Facial osteomas: Fourteen cases and a review of literature. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 1796–1802. [Google Scholar]
- Debta, P.; Debta, F.M.; Bussari, S.; Acharya, S.S.; Jeergal, V.A. Cancellous osteoma of maxilla: A rare case report. J. Int. Soc. Prev. Community Dent. 2016, 6, 261. [Google Scholar] [CrossRef] [Green Version]
- Erdogan, N.; Demir, U.; Songu, M.; Ozenler, N.K.; Uluç, E.; Dirim, B. A prospective study of paranasal sinus osteomas in 1889 cases: Changing patterns of localization. Laryngoscope 2009, 119, 2355–2359. [Google Scholar] [CrossRef]
- Payne, M.; Anderson, J.A.; Cook, J. Gardner’s syndrome—A case report. Br. Dent. J. 2002, 193, 383–384. [Google Scholar] [CrossRef] [Green Version]
- Cankaya, A.B.; Erdem, M.A.; Isler, S.C.; Cifter, M.; Olgac, V.; Kasapoglu, C.; Oral, C.K. Oral and maxillofacial considerations in Gardner’s syndrome. Int. J. Med. Sci. 2012, 9, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, F.T.; Pachêco-Pereira, C.; Porporatti, A.L.; Flores-Mir, C.; Leite, A.F.; De Luca Canto, G.; Guerra, E.N.S. Oral manifestations in patients with familial adenomatous polyposis: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Wijn, M.A.; Keller, J.J.; Giardiello, F.M.; Brand, H.S. Oral and maxillofacial manifestations of familial adenomatous polyposis. Oral Dis. 2007, 13, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Ida, M.; Nakamura, T.; Utsunomiya, J. Osteomatous changes and tooth abnormalities found in the jaws of patients with adenomatosis coli. Oral Surg. Oral Med. Oral Pathol. 1981, 52, 2–11. [Google Scholar] [CrossRef]
- Oku, T.; Takayama, T.; Sato, Y.; Sato, Y.; Takada, K.; Hayashi, T.; Takahashi, M.; Kuroda, M.; Kato, J.; Niitsu, Y. A case of Gardner syndrome with a mutation at codon 1556 of APC: A suggested case of genotype–phenotype correlation in dental abnormality. Eur. J. Gastroenterol. Hepatol. 2004, 16, 101–105. [Google Scholar] [CrossRef]
- Järvinen, H.J.; Peltomäki, P. The complex genotype–phenotype relationship in familial adenomatous polyposis. Eur. J. Gastroenterol. Hepatol. 2004, 16, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Papagelopoulos, P.J.; Mavrogenis, A.F.; Mitsiokapa, E.A.; Papaparaskeva, K.T.; Galanis, E.C.; Soucacos, P.N. Current trends in the management of extra-abdominal desmoid tumours. World J. Surg. Oncol. 2006, 4, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, A.; Tekkis, P.P.; Gibbons, D.C.; Phillips, R.K.; Clark, S.K. Risk factors predicting desmoid occurrence in patients with familial adenomatous polyposis: A meta-analysis. Colorectal Dis. 2011, 13, 1222–1229. [Google Scholar] [CrossRef]
- Clark, S.K.; Neale, K.F.; Landgrebe, J.C.; Phillips, R.K.S. Desmoid tumours complicating familial adenomatous polyposis. Br. J. Surg. 1999, 86, 1185–1189. [Google Scholar] [CrossRef] [PubMed]
- Koskenvuo, L.; Ristimäki, A.; Lepistö, A. Comparison of sporadic and FAP-associated desmoid-type fibromatoses. J. Surg. Oncol. 2017, 116, 716–721. [Google Scholar] [CrossRef]
- Tiret, A.; Parc, C. Fundus lesions of adenomatous polyposis. Curr. Opin. Ophthalmol. 1999, 10, 168–172. [Google Scholar] [CrossRef]
- Nusliha, A.; Dalpatadu, U.; Amarasinghe, B.; Chandrasinghe, P.C.; Deen, K.I. Congenital hypertrophy of retinal pigment epithelium (CHRPE) in patients with familial adenomatous polyposis (FAP); a polyposis registry experience. BMC Res. Notes 2014, 7, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Nieuwenhuis, M.H.; Vasen, H.F.A. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): A review of the literature. Crit. Rev. Oncol. Hematol. 2007, 61, 153–161. [Google Scholar] [CrossRef]
- Attard, T.M.; Giglio, P.; Koppula, S.; Snyder, C.; Lynch, H.T. Brain tumors in individuals with familial adenomatous polyposis: A cancer registry experience and pooled case report analysis. Cancer 2007, 109, 761–766. [Google Scholar] [CrossRef]
- Wiik, M.U.; Talseth-Palmer, B.A. Familial Adenomatous Polyposis. In Handbook of Tumor Syndromes; Liu, D., Ed.; CRC Press—Taylor & Francis Group: Boca Raton, FL, USA, 2020. [Google Scholar]
- Pichert, G.; Jacobs, C. (Eds.) Rare Hereditary Cancers: Diagnosis and Management; Springer International Publishing: Cham, Switzerland, 2016; Volume 205. [Google Scholar]
- Steinhagen, E.; Guillem, J.G.; Chang, G.; Salo-Mullen, E.E.; Shia, J.; Fish, S.; Stadler, Z.K.; Markowitz, A.J. The prevalence of thyroid cancer and benign thyroid disease in patients with familial adenomatous polyposis may be higher than previously recognized. Clin. Colorectal Cancer 2012, 11, 304–308. [Google Scholar] [CrossRef]
- Truta, B.; Allen, B.A.; Conrad, P.G.; Kim, Y.S.; Berk, T.; Gallinger, S.; Bapat, B.; Terdiman, J.P.; Sleisenger, M.H. Genotype and phenotype of patients with both familial adenomatous polyposis and thyroid carcinoma. Fam. Cancer 2003, 2, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Cetta, F.; Montalto, G.; Gori, M.; Curia, M.C.; Cama, A.; Olschwang, S. Germline mutations of the APC gene in patients with familial adenomatous polyposis-associated thyroid carcinoma: Results from a European cooperative study. J. Clin. Endocrinol. Metab. 2000, 85, 286–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Septer, S.; Slowik, V.; Morgan, R.; Dai, H.; Attard, T. Thyroid cancer complicating familial adenomatous polyposis: Mutation spectrum of at-risk individuals. Hered. Cancer Clin. Pract. 2013, 11, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extracolonic Cancer | Lifetime Risk for Cancer Observations |
|---|---|
| small bowel (duodenum or periampulla) cancer | 4–12% |
| small bowel (distal to the duodenum) cancer | rare |
| pancreas cancer | 1% |
| thyroid cancer | 1–12% |
| CNS cancer | <1% |
| liver cancer | 1.6% |
| bile ducts cancer | Low, but increased |
| stomach cancer | <1% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antohi, C.; Haba, D.; Caba, L.; Ciofu, M.L.; Drug, V.-L.; Bărboi, O.-B.; Dobrovăț, B.I.; Pânzaru, M.-C.; Gorduza, N.C.; Lupu, V.V.; et al. Novel Mutation in APC Gene Associated with Multiple Osteomas in a Family and Review of Genotype-Phenotype Correlations of Extracolonic Manifestations in Gardner Syndrome. Diagnostics 2021, 11, 1560. https://doi.org/10.3390/diagnostics11091560
Antohi C, Haba D, Caba L, Ciofu ML, Drug V-L, Bărboi O-B, Dobrovăț BI, Pânzaru M-C, Gorduza NC, Lupu VV, et al. Novel Mutation in APC Gene Associated with Multiple Osteomas in a Family and Review of Genotype-Phenotype Correlations of Extracolonic Manifestations in Gardner Syndrome. Diagnostics. 2021; 11(9):1560. https://doi.org/10.3390/diagnostics11091560
Chicago/Turabian StyleAntohi, Cristina, Danisia Haba, Lavinia Caba, Mihai Liviu Ciofu, Vasile-Liviu Drug, Oana-Bogdana Bărboi, Bogdan Ionuț Dobrovăț, Monica-Cristina Pânzaru, Nicoleta Carmen Gorduza, Vasile Valeriu Lupu, and et al. 2021. "Novel Mutation in APC Gene Associated with Multiple Osteomas in a Family and Review of Genotype-Phenotype Correlations of Extracolonic Manifestations in Gardner Syndrome" Diagnostics 11, no. 9: 1560. https://doi.org/10.3390/diagnostics11091560
APA StyleAntohi, C., Haba, D., Caba, L., Ciofu, M. L., Drug, V.-L., Bărboi, O.-B., Dobrovăț, B. I., Pânzaru, M.-C., Gorduza, N. C., Lupu, V. V., Dimofte, D., Gug, C., & Gorduza, E. V. (2021). Novel Mutation in APC Gene Associated with Multiple Osteomas in a Family and Review of Genotype-Phenotype Correlations of Extracolonic Manifestations in Gardner Syndrome. Diagnostics, 11(9), 1560. https://doi.org/10.3390/diagnostics11091560