
Coexistence of Primary Myelofibrosis and Paroxysmal Nocturnal Hemoglobinuria Clone with JAK2 V617F, U2AF1 and SETBP1 Mutations: A Case Report and Brief Review of Literature


Abstract
:1. Introduction
2. Case Presentation
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Hussaini, M.; Zhang, H.; Shao, H.; Qin, D.; Zhang, X.; Ma, Z.; Hussnain Naqvi, S.M.; Zhang, L.; Moscinski, L.C. Comparison of the Mutational Profiles of Primary Myelofibrosis, Polycythemia Vera, and Essential Thrombocytosis. Am. J. Clin. Pathol. 2017, 147, 444–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients with Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.; Kelly, R.J.; Hillmen, P. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood 2013, 121, 4985–4996. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Clemente, M.J.; Hosono, N.; Yoshida, K.; Przychodzen, B.; Yoshizato, T.; Shiraishi, Y.; Miyano, S.; Ogawa, S.; Maciejewski, J.P.; et al. Deep sequencing reveals stepwise mutation acquisition in paroxysmal nocturnal hemoglobinuria. J. Clin. Investig. 2014, 124, 4529–4538. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lin, Y.; Chen, L.; Qin, L.; Tan, H.; Zou, J.; Zhang, D.; Nie, Y.; Wang, G.; Zhang, H.; et al. Identification of acquired PIGA mutations and additional variants by next-generation sequencing in paroxysmal nocturnal hemoglobinuria. Int. J. Lab. Hematol. 2020, 42, 473–481. [Google Scholar] [CrossRef]
- Chen, F.; Hu, S.; Ruan, J.; Chen, M.; Han, B. Mutational landscape and its clinical significance in paroxysmal nocturnal hemoglobinuria. Blood Cancer J. 2021, 11, 58. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, S.P., 2nd; Talwalkar, S.S.; Simons, R.; Yam, L. Acute lymphoblastic leukemic transformation in a patient with chronic idiopathic myelofibrosis and paroxysmal nocturnal hemoglobinuria: A case report and review of the literature. Arch. Pathol. Lab. Med. 2005, 129, 96–99. [Google Scholar] [CrossRef]
- Sugimori, C.; Padron, E.; Caceres, G.; Shain, K.; Sokol, L.; Zhang, L.; Tiu, R.; O’Keefe, C.L.; Afable, M.; Clemente, M.; et al. Paroxysmal nocturnal hemoglobinuria and concurrent JAK2(V617F) mutation. Blood Cancer J. 2012, 2, e63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraiman, Y.S.; Cuka, N.; Batista, D.; Vuica-Ross, M.; Moliterno, A.R. Development of paroxysmal nocturnal hemoglobinuria in CALR-positive myeloproliferative neoplasm. J. Blood Med. 2016, 7, 107–110. [Google Scholar]
- Gaidano, V.; Geuna, M.; Cignetti, A.; Carnuccio, F.; Bernabei, P.; Santoro, N.; Saglio, G.; Sivera, P. Myeloproliferative neoplasms, thrombosis and paroxysmal nocturnal hemoglobinuria: Is this triad more frequent than we thought? Hematol. Med. Oncol. 2017, 2, 4–5. [Google Scholar] [CrossRef] [Green Version]
- Gutwein, O.; Englander, Y.; Herzog-Tzarfati, K.; Filipovich-Rimon, T.; Apel, A.; Marcus, R.; Rahimi-Levene, N.; Koren-Michowitz, M. Prevalence of Paroxysmal Nocturnal Hemoglobinuria Clones in Myeloproliferative Neoplasm Patients: A Cross-Sectional Study. Clin. Lymphoma Myeloma Leuk. 2019, 19, 812–814. [Google Scholar] [CrossRef] [PubMed]
- Kirito, K. Expansion of paroxysmal nocturnal hemoglobinuria clones in MPLW515L mutation harboring primary myelofibrosis. Ann. Hematol. 2020, 99, 2707–2709. [Google Scholar] [CrossRef]
- Chatzidavid, S.; Giannakopoulou, N.; Diamantopoulos, P.T.; Gavriilaki, E.; Katsiampoura, P.; Lakiotaki, E.; Sakellariou, S.; Viniou, N.A.; Dryllis, G. JAK2V617F positive polycythemia vera with paroxysmal nocturnal hemoglobinuria and visceral thromboses: A case report and review of the literature. Thromb. J. 2021, 19, 16. [Google Scholar] [CrossRef]
- Fattizzo, B.; Ireland, R.; Dunlop, A.; Yallop, D.; Kassam, S.; Large, J.; Gandhi, S.; Muus, P.; Manogaran, C.; Sanchez, K.; et al. Clinical and prognostic significance of small paroxysmal nocturnal hemoglobinuria clones in myelodysplastic syndrome and aplastic anemia. Leukemia 2021. [Google Scholar] [CrossRef]
- Gangat, N.; Caramazza, D.; Vaidya, R.; George, G.; Begna, K.; Schwager, S.; Van Dyke, D.; Hanson, C.; Wu, W.; Pardanani, A.; et al. DIPSS plus: A refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J. Clin. Oncol. 2011, 29, 392–397. [Google Scholar] [CrossRef]
- Emanuel, R.M.; Dueck, A.C.; Geyer, H.L.; Kiladjian, J.J.; Slot, S.; Zweegman, S.; te Boekhorst, P.A.; Commandeur, S.; Schouten, H.C.; Sackmann, F.; et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: Prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J. Clin. Oncol. 2012, 30, 4098–4103. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. U2AF1 mutation types in primary myelofibrosis: Phenotypic and prognostic distinctions. Leukemia 2018, 32, 2274–2278. [Google Scholar] [CrossRef]
- Engle, E.K.; Fisher, D.A.; Miller, C.A.; McLellan, M.D.; Fulton, R.S.; Moore, D.M.; Wilson, R.K.; Ley, T.J.; Oh, S.T. Clonal evolution revealed by whole genome sequencing in a case of primary myelofibrosis transformed to secondary acute myeloid leukemia. Leukemia 2015, 29, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Meggendorfer, M.; Bacher, U.; Alpermann, T.; Haferlach, C.; Kern, W.; Gambacorti-Passerini, C.; Haferlach, T.; Schnittger, S. SETBP1 mutations occur in 9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i(17)(q10), ASXL1 and CBL mutations. Leukemia 2013, 27, 1852–1860. [Google Scholar] [CrossRef]
- Eder-Azanza, L.; Hurtado, C.; Navarro-Herrera, D.; Calavia, D.; Novo, F.J.; Vizmanos, J.L. Analysis of genes encoding epigenetic regulators in myeloproliferative neoplasms: Coexistence of a novel SETBP1 mutation in a patient with a p.V617F JAK2 positive myelofibrosis. Mol. Clin. Oncol. 2019, 10, 639–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albano, F.; Anelli, L.; Zagaria, A.; Coccaro, N.; Casieri, P.; Minervini, A.; Specchia, G. SETBP1 and miR_4319 dysregulation in primary myelofibrosis progression to acute myeloid leukemia. J. Hematol. Oncol. 2012, 5, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, H.; Uoshima, N.; Kamitsuji, Y.; Kawata, E.; Komori, Y.; Sasaki, N.; Tsutsumi, Y.; Tsukamoto, T.; Mizutani, S.; Nannya, Y.; et al. Paroxysmal nocturnal hemoglobinuria complicated with essential thrombocythemia harboring concomitant PIGA, CALR, and ASXL1 mutations. Ann. Hematol. 2021, 100, 2113–2115. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Finazzi, G.; Falanga, A. Myeloproliferative neoplasms and thrombosis. Blood 2013, 122, 2176–2184. [Google Scholar] [CrossRef]
- Rungjirajittranon, T.; Owattanapanich, W.; Ungprasert, P.; Siritanaratkul, N.; Ruchutrakool, T. A systematic review and meta-analysis of the prevalence of thrombosis and bleeding at diagnosis of Philadelphia-negative myeloproliferative neoplasms. BMC Cancer 2019, 19, 184. [Google Scholar] [CrossRef]
- Smalberg, J.H.; Arends, L.R.; Valla, D.C.; Kiladjian, J.J.; Janssen, H.L.; Leebeek, F.W. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: A meta-analysis. Blood 2012, 120, 4921–4928. [Google Scholar] [CrossRef]
- Hall, C.; Richards, S.; Hillmen, P. Primary prophylaxis with warfarin prevents thrombosis in paroxysmal nocturnal hemoglobinuria (PNH). Blood 2003, 102, 3587–3591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaman, M.A.; Ursuleac, I.; Coriu, D. Paroxysmal nocturnal hemoglobinuria: Pandora’s box? J. Mind Med. Sci. 2020, 7, 19. [Google Scholar] [CrossRef]
- Gaman, M.A.; Cozma, M.A.; Dobrica, E.C.; Cretoiu, S.M.; Gaman, A.M.; Diaconu, C.C. Liquid Biopsy and Potential Liquid Biopsy-Based Biomarkers in Philadelphia-Negative Classical Myeloproliferative Neoplasms: A Systematic Review. Life 2021, 11, 677. [Google Scholar] [CrossRef] [PubMed]
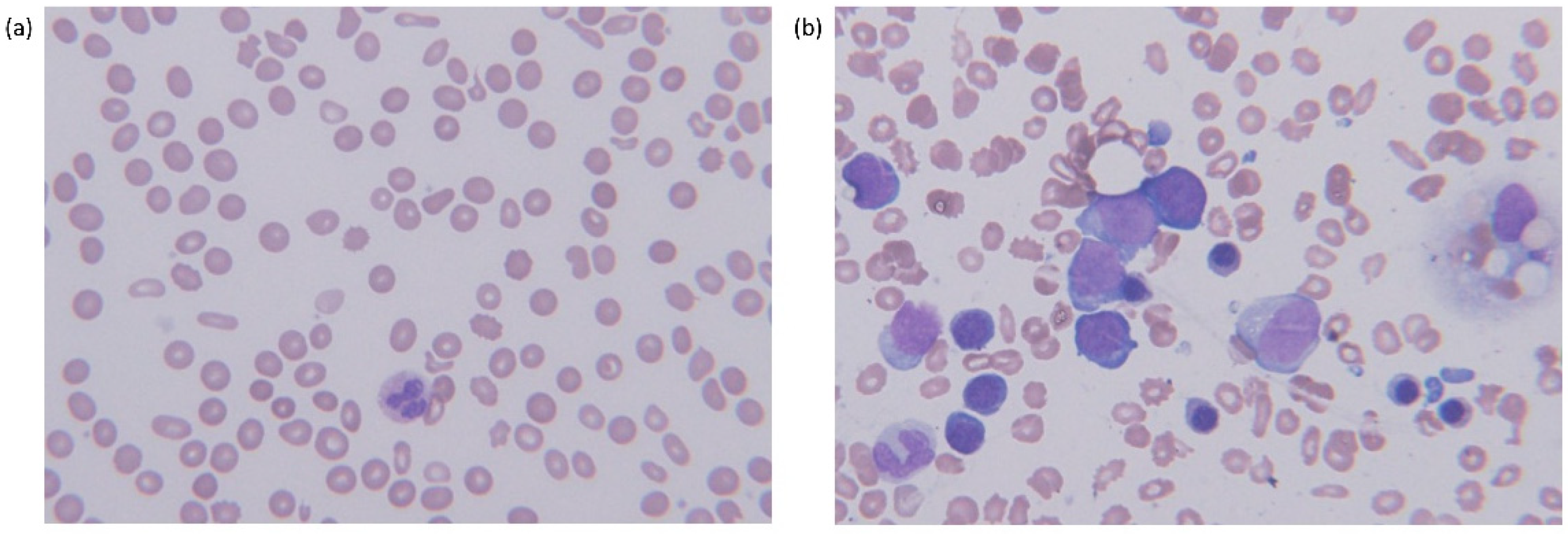
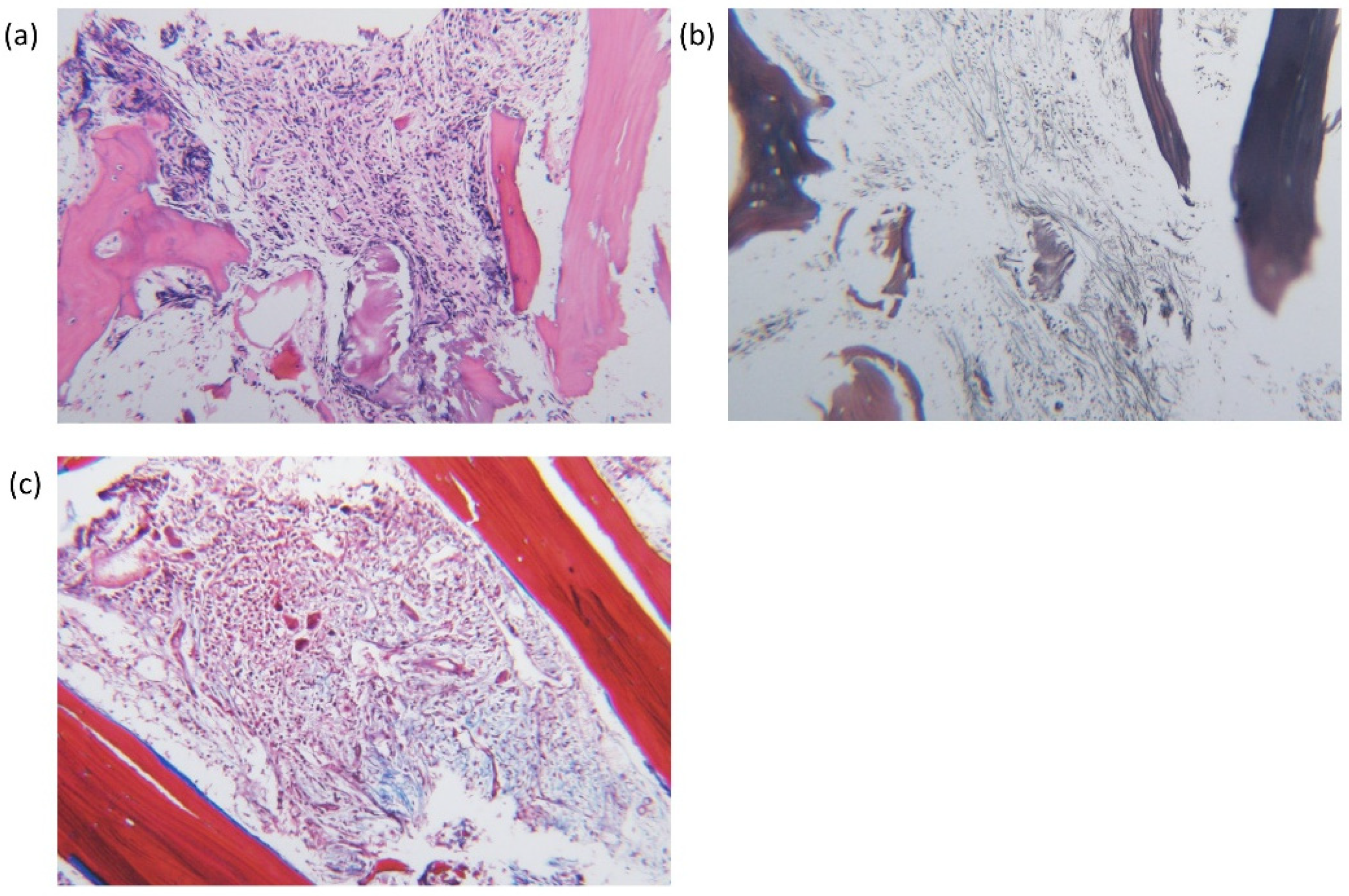

{kind=link}
{kind=link}
| Laboratory Parameter (Unit) | At Diagnosis | 2 Weeks under Ruxolitinib | 4 Weeks under Ruxolitinib | 4 Weeks Off Ruxolitinib | Reference Range |
|---|---|---|---|---|---|
| Hgb (g/dL) | 6.0 | 8.3 | 6.1 | 6.6 | 14.0–16.5 |
| Hct (%) | 19.0 | 26.4 | 19.3 | 20.3 | 42–50 |
| MCV (fL) | 84.4 | 85.2 | 84.6 | 87.5 | 80–96 |
| MCH (pg) | 26.7 | 26.8 | 26.8 | 28.6 | 27–33 |
| MCHC (g/dL) | 31.6 | 31.4 | 31.6 | 32.7 | 33–36 |
| Reticulocyte (%) | 2.58 | ND | ND | 0.81 | 0.6–2.4 |
| WBC (×109/L) | 5.86 | 5.88 | 7.44 | 7.36 | 4.0–10.0 |
| PLT (×109/L) | 128 | 74 | 53 | 53 | 150–450 |
| LDH (IU/L) | 640 | 537 | 559 | 401 | 106–211 |
| Total bilirubin (mg/dL) | 0.88 | 0.78 | 0.90 | 1.22 | 0.2–1.2 |
| Direct bilirubin (mg/dL) | 0.17 | 0.16 | 0.18 | 0.33 | 0–0.4 |
| AST (IU/L) | 20 | 33 | 24 | 16 | 0–50 |
| ALT (IU/L) | 9 | 27 | 16 | 16 | 0–50 |
| Uric acid (mg/dL) | 9.1 | 9.2 | 8.8 | 7.8 | 2.6–7.6 |
| Haptoglobin (mg/dL) | <8 | ND | ND | ND | 30–200 |
| EPO (mIU/mL) | 189.0 | ND | ND | ND | 4.3–29.0 |
| Plasma Hgb (g/dL) | 4.85 | ND | ND | ND | 0–5.0 |
| Blood, urine | Negative | ND | ND | ND | Negative |
| Hemosiderin | Negative | ND | ND | ND | Negative |
| Direct antiglobulin test (Direct Coombs’ test) | Negative | ND | ND | ND | Negative |
| Indirect antiglobulin test (Indirect Coombs’ test) | Negative | ND | ND | ND | Negative |
| References | Age (yr)/Gender at Dignosis | Presenting Signs and Symptoms | Diagnosis | Genetic Mutation | PNH Clones (% of RBC Granulocytes/Monocytes) | Treatment | Complication | Prognosis | |
|---|---|---|---|---|---|---|---|---|---|
| Init. | F/U (Time) | ||||||||
| Shaheen S.P. 2nd, et al., 2005 [11] | 53/M | Splenomegaly, leukocytosis | MPN | PMF PNH (2 yrs) | ND | ND | Hydroxyurea, interferon | Myocardial infarction, precursor B lymphoblastic leukemia | Died for intracranial hemorrhagesepsis |
| Sugimori C., et al., 2012 [12] | #1: 51/M | Stroke, Budd Chiari syndrome | PMF | PNH (ND) | JAK2 V617F | 13%/99%/ND | Eculizumab | Multiple complications of Budd Chiari syndrome, including esophageal variceal bleeding | Alive |
| #2: 65/M | Dark urine, transfusion dependent anemia | PNH | PMF (2 yrs) | JAK2 V617F | 40%/76.7%/ND | Hydroxyurea, anticoagulant, eculizumab | Splenic infarction, portal vein thrombosis | Died for liver failure | |
| #3: 78/M | Progressive anemia, dark urine | PNH | PMF (1 yr) | JAK2 V617F | 53%/73%/ND | Eculizamab, anticoagulant | Pneumonia | Died for Clostridium difficile infection | |
| Gaidano V., et al., 2017 [14] | 72/F | Portal vein thrombosis | Post ET-MF? PNH | JAK2 V617F | 71%/88.6%/86.9% | Hydroxyurea, anticoagulant | None | Alive | |
| Kirito K. 2020 [16] | 49/M | Anemia, thrombocythe-mia, dark urine | PMF PNH | MPLW515L | ND/99%/99% | ND | ND | ND | |
| Our case | 87/M | Progressive anemia, splenomegaly, lymphadenopathy | PNH PMF | JAK2 V617F U2AF1Q157 SETBP1 | 3.8%/48.6%/77.2% | Ruxolitinib | Pneumonia | Alive | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; So, M.-K.; Cho, M.-S.; Kim, D.-Y.; Huh, J. Coexistence of Primary Myelofibrosis and Paroxysmal Nocturnal Hemoglobinuria Clone with JAK2 V617F, U2AF1 and SETBP1 Mutations: A Case Report and Brief Review of Literature. Diagnostics 2021, 11, 1644. https://doi.org/10.3390/diagnostics11091644
Park S, So M-K, Cho M-S, Kim D-Y, Huh J. Coexistence of Primary Myelofibrosis and Paroxysmal Nocturnal Hemoglobinuria Clone with JAK2 V617F, U2AF1 and SETBP1 Mutations: A Case Report and Brief Review of Literature. Diagnostics. 2021; 11(9):1644. https://doi.org/10.3390/diagnostics11091644
Chicago/Turabian StylePark, Sholhui, Min-Kyung So, Min-Sun Cho, Dae-Young Kim, and Jungwon Huh. 2021. "Coexistence of Primary Myelofibrosis and Paroxysmal Nocturnal Hemoglobinuria Clone with JAK2 V617F, U2AF1 and SETBP1 Mutations: A Case Report and Brief Review of Literature" Diagnostics 11, no. 9: 1644. https://doi.org/10.3390/diagnostics11091644