
Recognized and Emerging Features of Erythropoietic and X-Linked Protoporphyria

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Cutaneous Manifestations
3. Liver Involvement
4. Anemia
5. Vitamin D Deficiency and Osteoporosis
6. Systemic Inflammation
7. Management
7.1. Photoprotection
7.2. Photosensitivity
7.3. Liver Disease
7.4. Microcytic Anemia
7.5. Vitamin D Deficiency
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Magnus, I.A.; Jarrett, A.; Prankerd, T.A.; Rimington, C. Erythropoietic protoporphyria. A new porphyria syndrome with solar urticaria due to protoporphyrinaemia. Lancet 1961, 2, 448–451. [Google Scholar] [CrossRef]
- Anderson, K.; Sassa, A.; Bishop, D.; Desnick, R. Disorders of Heme Biosynthesis: X-Linked Sideroblastic Anemia and the Porphyrias; Scriver, C., Beaudet, A., Sly, W., Valle, D., Eds.; The metabolic and molecular basis of inherited diseases; McGraw-Hill: New York, NY, USA, 2001; pp. 2961–3062. [Google Scholar]
- Whatley, S.D.; Ducamp, S.; Gouya, L.; Grandchamp, B.; Beaumont, C.; Badminton, M.N.; Elder, G.H.; Holme, S.A.; Anstey, A.V.; Parker, M.; et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am. J. Hum. Genet. 2008, 83, 408–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balwani, M.; Bloomer, J.; Desnick, R. Erythropoietic Protoporphyria, Autosomal Recessive; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Gouya, L.; Puy, H.; Robreau, A.M.; Bourgeois, M.; Lamoril, J.; Da Silva, V.; Grandchamp, B.; Deybach, J.C. The penetrance of dominant erythropoietic protoporphyria is modulated by expression of wildtype FECH. Nat. Genet. 2002, 30, 27–28. [Google Scholar] [CrossRef] [PubMed]
- Chiara, M.; Primon, I.; Tarantini, L.; Agnelli, L.; Brancaleoni, V.; Granata, F.; Bollati, V.; Di Pierro, E. Targeted resequencing of FECH locus reveals that a novel deep intronic pathogenic variant and eQTLs may cause erythropoietic protoporphyria (EPP) through a methylation-dependent mechanism. Genet. Med. 2020, 22, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Whatley, S.D.; Mason, N.G.; Holme, S.A.; Anstey, A.V.; Elder, G.H.; Badminton, M.N. Molecular epidemiology of erythropoietic protoporphyria in the U.K. Br. J. Dermatol. 2010, 162, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M.; Doheny, D.; Bishop, D.F.; Nazarenko, I.; Yasuda, M.; Dailey, H.A.; Anderson, K.E.; Bissell, D.M.; Bloomer, J.; Bonkovsky, H.L.; et al. Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and x-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria. Mol. Med. 2013, 19, 26–35. [Google Scholar]
- Kadirvel, S.; Furuyama, K.; Harigae, H.; Kaneko, K.; Tamai, Y.; Ishida, Y.; Shibahara, S. The carboxyl-terminal region of erythroid-specific 5-aminolevulinate synthase acts as an intrinsic modifier for its catalytic activity and protein stability. Exp. Hematol. 2012, 40, 477–486. [Google Scholar] [CrossRef]
- Brancaleoni, V.; Balwani, M.; Granata, F.; Graziadei, G.; Missineo, P.; Fiorentino, V.; Fustinoni, S.; Cappellini, M.D.; Naik, H.; Desnick, R.J.; et al. X-chromosomal inactivation directly influences the phenotypic manifestation of X-linked protoporphyria. Clin. Genet. 2016, 89, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Yien, Y.Y.; Ducamp, S.; van der Vorm, L.N.; Kardon, J.R.; Manceau, H.; Kannengiesser, C.; Bergonia, H.A.; Kafina, M.D.; Karim, Z.; Gouya, L.; et al. Mutation in human CLPX elevates levels of delta-aminolevulinate synthase and protoporphyrin IX to promote erythropoietic protoporphyria. Proc. Nat.l Acad. Sci. USA 2017, 114, E8045–E8052. [Google Scholar] [CrossRef] [Green Version]
- Lecha, M.; Puy, H.; Deybach, J.C. Erythropoietic protoporphyria. Orphanet J. Rare Dis. 2009, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Balwani, M. Erythropoietic Protoporphyria and X-Linked Protoporphyria: Pathophysiology, genetics, clinical manifestations, and management. Mol. Genet. Metab. 2019, 128, 298–303. [Google Scholar] [CrossRef]
- Poh-Fitzpatrick, M.B. Erythropoietic protoporphyria. Int. J. Dermatol. 1978, 17, 359–369. [Google Scholar] [CrossRef]
- Poh-Fitzpatrick, M.B. Porphyrias: Photosensitivity and phototherapy. Methods Enzym. 2000, 319, 485–493. [Google Scholar]
- Gou, E.W.; Balwani, M.; Bissell, D.M.; Bloomer, J.R.; Bonkovsky, H.L.; Desnick, R.J.; Naik, H.; Phillips, J.D.; Singal, A.K.; Wang, B.; et al. Pitfalls in Erythrocyte Protoporphyrin Measurement for Diagnosis and Monitoring of Protoporphyrias. Clin. Chem. 2015, 61, 1453–1456. [Google Scholar] [CrossRef] [Green Version]
- Di Pierro, E.; De Canio, M.; Mercadante, R.; Savino, M.; Granata, F.; Tavazzi, D.; Nicolli, A.M.; Trevisan, A.; Marchini, S.; Fustinoni, S. Laboratory Diagnosis of Porphyria. Diagnostics 2021, 11, 1343. [Google Scholar] [CrossRef]
- Elder, G.; Harper, P.; Badminton, M.; Sandberg, S.; Deybach, J.C. The incidence of inherited porphyrias in Europe. J. Inherit. Metab. Dis. 2013, 36, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Manceau, H.; Gouya, L.; Puy, H. Acute hepatic and erythropoietic porphyrias: From ALA synthases 1 and 2 to new molecular bases and treatments. Curr. Opin. Hematol. 2017, 24, 198–207. [Google Scholar] [CrossRef]
- Ventura, P.; Brancaleoni, V.; Di Pierro, E.; Graziadei, G.; Macri, A.; Carmine, G.C.; Nicolli, A.; Rossi, M.T.; Granata, F.; Fiorentino, V.; et al. Clinical and molecular epidemiology of erythropoietic protoporphyria in Italy. Eur. J. Dermatol. 2020, 30, 532–540. [Google Scholar] [CrossRef]
- Rutter, K.J.; Ashraf, I.; Cordingley, L.; Rhodes, L.E. Quality of life and psychological impact in the photodermatoses: A systematic review. Br. J. Dermatol. 2020, 182, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.; Poblete-Gutierrez, P. Delayed diagnosis and diminished quality of life in erythropoietic protoporphyria: Results of a cross-sectional study in Sweden. J. Intern Med. 2011, 269, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Bharati, A.; Badminton, M.N.; Whatley, S.D.; O’Brien, D.V.; Bell, H.K. Late-onset erythropoietic protoporphyria in association with haematological malignancy. Clin. Exp. Dermatol. 2006, 31, 668–670. [Google Scholar] [CrossRef]
- Poh-Fitzpatrick, M.B. The “priming phenomenon” in the acute phototoxicity of erythropoietic protoporphyria. J. Am. Acad. Dermatol. 1989, 21, 311. [Google Scholar] [CrossRef]
- Lecha, M. Erythropoietic protoporphyria. Photodermatol. Photoimmunol. Photomed. 2003, 19, 142–146. [Google Scholar] [CrossRef] [Green Version]
- Balwani, M.; Naik, H.; Anderson, K.E.; Bissell, D.M.; Bloomer, J.; Bonkovsky, H.L.; Phillips, J.D.; Overbey, J.R.; Wang, B.; Singal, A.K.; et al. Clinical, Biochemical, and Genetic Characterization of North American Patients With Erythropoietic Protoporphyria and X-linked Protoporphyria. JAMA Dermatol. 2017, 153, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M.; Desnick, R.J. The porphyrias: Advances in diagnosis and treatment. Blood 2012, 120, 4496–4504. [Google Scholar] [CrossRef]
- Minder, E.I.; Schneider-Yin, X.; Mamet, R.; Horev, L.; Neuenschwander, S.; Baumer, A.; Austerlitz, F.; Puy, H.; Schoenfeld, N. A homoallelic FECH mutation in a patient with both erythropoietic protoporphyria and palmar keratoderma. J. Eur. Acad. Dermatol. Venereol. 2010, 24, 1349–1353. [Google Scholar] [CrossRef]
- Taniguchi, M.; Lindsey, J.S. Database of Absorption and Fluorescence Spectra of >300 Common Compounds for use in PhotochemCAD. Photochem. Photobiol. 2018, 94, 290–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Maronese, C.A.; Moltrasio, C.; Piccinno, R.; Marletta, D.A.; De Luca, G.; Graziadei, G.; Granata, F.; Di Pierro, E.; Cappellini, M.D.; et al. Ultraviolet A phototest positivity is associated with higher free erythrocyte protoporphyrin IX concentration and lower transferrin saturation values in erythropoietic protoporphyria. Photodermatol. Photoimmunol. Photomed. 2021. [Google Scholar] [CrossRef] [PubMed]
- Holme, S.A.; Anstey, A.V.; Finlay, A.Y.; Elder, G.H.; Badminton, M.N. Erythropoietic protoporphyria in the U.K. Clinical features and effect on quality of life. Br. J. Dermatol. 2006, 155, 574–581. [Google Scholar] [CrossRef]
- Ibrahim, G.W.; Watson, C.J. Enterohepatic circulation and conversion of protoporphyrin to bile pigment in man. Proc. Soc. Exp. Biol. Med. 1968, 127, 890–895. [Google Scholar] [CrossRef]
- Poh-Fitzpatrick, M.B.; Whitlock, R.T.; Leftkowitch, J.H. Changes in protoporphyrin distribution dynamics during liver failure and recovery in a patient with protoporphyria and Epstein-Barr viral hepatitis. Am. J. Med. 1986, 80, 943–950. [Google Scholar] [CrossRef]
- Singal, A.K.; Parker, C.; Bowden, C.; Thapar, M.; Liu, L.; McGuire, B.M. Liver transplantation in the management of porphyria. Hepatology 2014, 60, 1082–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlin, S.; Stal, P.; Adam, R.; Karam, V.; Porte, R.; Seehofer, D.; Gunson, B.K.; Hillingso, J.; Klempnauer, J.L.; Schmidt, J.; et al. Liver transplantation for erythropoietic protoporphyria in Europe. Liver Transpl. 2011, 17, 1021–1026. [Google Scholar] [CrossRef]
- Anstey, A.V.; Hift, R.J. Liver disease in erythropoietic protoporphyria: Insights and implications for management. Gut 2007, 56, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Casanova-Gonzalez, M.J.; Trapero-Marugan, M.; Jones, E.A.; Moreno-Otero, R. Liver disease and erythropoietic protoporphyria: A concise review. World J. Gastroenterol. 2010, 16, 4526–4531. [Google Scholar] [CrossRef]
- Wensink, D.; Coenen, S.; Wilson, J.H.P.; Wagenmakers, M.A.E.M.; Langendonk, J.G. Liver involvement in patients with erythropoietic protoporphyria. Dig. Liver Dis. 2021. [Google Scholar] [CrossRef]
- Rademakers, L.H.; Cleton, M.I.; Kooijman, C.; Baart de la Faille, H.; van Hattum, J. Early involvement of hepatic parenchymal cells in erythrohepatic protoporphyria? An ultrastructural study of patients with and without overt liver disease and the effect of chenodeoxycholic acid treatment. Hepatology 1990, 11, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.G.; Avner, D.L.; Berenson, M.M. Structure-function relationships of protoporphyrin-induced liver injury. Arch. Pathol. Lab. Med. 1984, 108, 744–746. [Google Scholar] [PubMed]
- Cox, T.M.; Alexander, G.J.; Sarkany, R.P. Protoporphyria. Semin. Liver Dis. 1998, 18, 85–93. [Google Scholar] [CrossRef]
- Coffey, A.; Leung, D.H.; Quintanilla, N.M. Erythropoietic Protoporphyria: Initial Diagnosis With Cholestatic Liver Disease. Pediatrics 2018, 141, S445–S450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maitra, D.; Carter, E.L.; Richardson, R.; Rittie, L.; Basrur, V.; Zhang, H.; Nesvizhskii, A.I.; Osawa, Y.; Wolf, M.W.; Ragsdale, S.W.; et al. Oxygen and Conformation Dependent Protein Oxidation and Aggregation by Porphyrins in Hepatocytes and Light-Exposed Cells. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 659–682. [Google Scholar] [CrossRef] [Green Version]
- Maitra, D.; Bragazzi, C.J.; Elenbaas, J.S.; Bonkovsky, H.L.; Shavit, J.A.; Omary, M.B. Porphyrin-Induced Protein Oxidation and Aggregation as a Mechanism of Porphyria-Associated Cell Injury. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 535–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libbrecht, L.; Meerman, L.; Kuipers, F.; Roskams, T.; Desmet, V.; Jansen, P. Liver pathology and hepatocarcinogenesis in a long-term mouse model of erythropoietic protoporphyria. J. Pathol. 2003, 199, 191–200. [Google Scholar] [CrossRef]
- Meerman, L.; Koopen, N.R.; Bloks, V.; Van Goor, H.; Havinga, R.; Wolthers, B.G.; Kramer, W.; Stengelin, S.; Muller, M.; Kuipers, F.; et al. Biliary fibrosis associated with altered bile composition in a mouse model of erythropoietic protoporphyria. Gastroenterology 1999, 117, 696–705. [Google Scholar] [CrossRef]
- Lyoumi, S.; Abitbol, M.; Rainteau, D.; Karim, Z.; Bernex, F.; Oustric, V.; Millot, S.; Letteron, P.; Heming, N.; Guillmot, L.; et al. Protoporphyrin retention in hepatocytes and Kupffer cells prevents sclerosing cholangitis in erythropoietic protoporphyria mouse model. Gastroenterology 2011, 141, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Sachar, M.; Lu, J.; Shehu, A.I.; Zhu, J.; Chen, J.; Liu, K.; Anderson, K.E.; Xie, W.; Gonzalez, F.J.; et al. The essential role of the transporter ABCG2 in the pathophysiology of erythropoietic protoporphyria. Sci. Adv. 2019, 5, eaaw6127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abitbol, M.; Bernex, F.; Puy, H.; Jouault, H.; Deybach, J.C.; Guenet, J.L.; Montagutelli, X. A mouse model provides evidence that genetic background modulates anemia and liver injury in erythropoietic protoporphyria. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1208–G1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, Y.; Cheong, P.L.; Lynch, J.; Brighton, C.; Frase, S.; Kargas, V.; Rampersaud, E.; Wang, Y.; Sankaran, V.G.; Yu, B.; et al. The severity of hereditary porphyria is modulated by the porphyrin exporter and Lan antigen ABCB6. Nat. Commun. 2016, 7, 12353. [Google Scholar] [CrossRef] [PubMed]
- Holme, S.A.; Worwood, M.; Anstey, A.V.; Elder, G.H.; Badminton, M.N. Erythropoiesis and iron metabolism in dominant erythropoietic protoporphyria. Blood 2007, 110, 4108–4110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaby, C.; Lyoumi, S.; Ducamp, S.; Martin-Schmitt, C.; Gouya, L.; Deybach, J.C.; Beaumont, C.; Puy, H. Excessive erythrocyte PPIX influences the hematologic status and iron metabolism in patients with dominant erythropoietic protoporphyria. Cell Mol. Biol. 2009, 55, 45–52. [Google Scholar]
- Bossi, K.; Lee, J.; Schmeltzer, P.; Holburton, E.; Groseclose, G.; Besur, S.; Hwang, S.; Bonkovsky, H.L. Homeostasis of iron and hepcidin in erythropoietic protoporphyria. Eur. J. Clin. Investig. 2015, 45, 1032–1041. [Google Scholar] [CrossRef]
- Barman-Aksoezen, J.; Girelli, D.; Aurizi, C.; Schneider-Yin, X.; Campostrini, N.; Barbieri, L.; Minder, E.I.; Biolcati, G. Disturbed iron metabolism in erythropoietic protoporphyria and association of GDF15 and gender with disease severity. J. Inherit. Metab. Dis. 2017, 40, 433–441. [Google Scholar] [CrossRef]
- Lyoumi, S.; Abitbol, M.; Andrieu, V.; Henin, D.; Robert, E.; Schmitt, C.; Gouya, L.; de Verneuil, H.; Deybach, J.C.; Montagutelli, X.; et al. Increased plasma transferrin, altered body iron distribution, and microcytic hypochromic anemia in ferrochelatase-deficient mice. Blood 2007, 109, 811–818. [Google Scholar] [CrossRef]
- Barman-Aksozen, J.; Halloy, F.; Iyer, P.S.; Schumperli, D.; Minder, A.E.; Hall, J.; Minder, E.I.; Schneider-Yin, X. Delta-aminolevulinic acid synthase 2 expression in combination with iron as modifiers of disease severity in erythropoietic protoporphyria. Mol. Genet. Metab. 2019, 128, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Shea, A.L. Vitamin D, the natural way. Clin. Nutr. ESPEN 2021, 41, 10–12. [Google Scholar] [CrossRef]
- Touvier, M.; Deschasaux, M.; Montourcy, M.; Sutton, A.; Charnaux, N.; Kesse-Guyot, E.; Assmann, K.E.; Fezeu, L.; Latino-Martel, P.; Druesne-Pecollo, N.; et al. Determinants of vitamin D status in Caucasian adults: Influence of sun exposure, dietary intake, sociodemographic, lifestyle, anthropometric, and genetic factors. J. Investig. Dermatol. 2015, 135, 378–388. [Google Scholar] [CrossRef] [Green Version]
- Major, J.M.; Graubard, B.I.; Dodd, K.W.; Iwan, A.; Alexander, B.H.; Linet, M.S.; Freedman, D.M. Variability and reproducibility of circulating vitamin D in a nationwide U.S. population. J. Clin. Endocrinol. Metab. 2013, 98, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Holick, M.F. The vitamin D deficiency pandemic and consequences for nonskeletal health: Mechanisms of action. Mol. Asp. Med. 2008, 29, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Holme, S.A.; Anstey, A.V.; Badminton, M.N.; Elder, G.H. Serum 25-hydroxyvitamin D in erythropoietic protoporphyria. Br. J. Dermatol. 2008, 159, 211–213. [Google Scholar] [CrossRef]
- Spelt, J.M.; de Rooij, F.W.; Wilson, J.H.; Zandbergen, A.A. Vitamin D deficiency in patients with erythropoietic protoporphyria. J. Inherit. Metab. Dis. 2010, 33 (Suppl. 3), S1–S4. [Google Scholar] [CrossRef]
- Rhodes, L.E.; Webb, A.R.; Berry, J.L.; Felton, S.J.; Marjanovic, E.J.; Wilkinson, J.D.; Vail, A.; Kift, R. Sunlight exposure behaviour and vitamin D status in photosensitive patients: Longitudinal comparative study with healthy individuals at U.K. Latitude. Br. J. Dermatol. 2014, 171, 1478–1486. [Google Scholar] [CrossRef] [PubMed]
- Allo, G.; del Carmen Garrido-Astray, M.; Mendez, M.; De Salamanca, R.E.; Martinez, G.; Hawkins, F. Bone mineral density and vitamin D levels in erythropoietic protoporphyria. Endocrine 2013, 44, 803–807. [Google Scholar] [CrossRef]
- Biewenga, M.; Matawlie, R.H.S.; Friesema, E.C.H.; Koole-Lesuis, H.; Langeveld, M.; Wilson, J.H.P.; Langendonk, J.G. Osteoporosis in patients with erythropoietic protoporphyria. Br. J. Dermatol. 2017, 177, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Scholz, M.; Croizat, G.; Psencik, J.; Dedic, R.; Nonell, S.; Wagnieres, G. Understanding delayed fluorescence and triplet decays of Protoporphyrin IX under hypoxic conditions. Photochem. Photobiol. Sci. 2021, 20, 843–857. [Google Scholar] [CrossRef]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, H.W.; Perez, H.D.; Goldstein, I.M.; Gigli, I. Complement-derived chemotactic activity is generated in human serum containing uroporphyrin after irradiation with 405 nm light. J. Clin. Investig. 1981, 67, 1072–1077. [Google Scholar] [CrossRef]
- Lim, H.W.; Gigli, I. Role of complement in porphyrin-induced photosensitivity. J. Investig. Dermatol. 1981, 76, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.W.; Poh-Fitzpatrick, M.B.; Gigli, I. Activation of the complement system in patients with porphyrias after irradiation in vivo. J. Clin. Investig. 1984, 74, 1961–1965. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.W.; Perez, H.D.; Poh-Fitzpatrick, M.; Goldstein, I.M.; Gigli, I. Generation of chemotactic activity in serum from patients with erythropoietic protoporphyria and porphyria cutanea tarda. N. Engl. J. Med. 1981, 304, 212–216. [Google Scholar] [CrossRef]
- Gigli, I.; Schothorst, A.A.; Soter, N.A.; Pathak, M.A. Erythropoietic protoporphyria. Photoactivation of the complement system. J. Clin. Investig. 1980, 66, 517–522. [Google Scholar] [CrossRef] [Green Version]
- Smith-Jackson, K.; Marchbank, K.J. Targeting properdin in the treatment of atypical haemolytic uraemic syndrome: Better than eculizumab? Ann. Transl. Med. 2018, 6, S62. [Google Scholar] [CrossRef] [PubMed]
- Granata, F.; Duca, L.; Graziadei, G.; Brancaleoni, V.; Missineo, P.; De Luca, G.; Fustinoni, S.; Di Pierro, E. Inflammatory involvement into phototoxic reaction in erythropoietic protoporphyria (EPP) patients. Immunol. Res. 2019, 67, 382–389. [Google Scholar] [CrossRef]
- Granata, F.; Duca, L.; Brancaleoni, V.; Fustinoni, S.; De Luca, G.; Motta, I.; Graziadei, G.; Di Pierro, E. Alternative Pathway Involvement in Protoporphyria Patients Related to Sun Exposure. Front. Immunol. 2021, 12, 615620. [Google Scholar] [CrossRef]
- Schiekofer, C.; Vogt, T.; Reichrath, J. Erythropoietic protoporphyria. A rare differential diagnosis among photosensitive diseases. Hautarzt 2012, 63, 961–964. [Google Scholar] [CrossRef]
- Kaye, E.T.; Levin, J.A.; Blank, I.H.; Arndt, K.A.; Anderson, R.R. Efficiency of opaque photoprotective agents in the visible light range. Arch. Dermatol. 1991, 127, 351–355. [Google Scholar] [CrossRef]
- Wahlin, S.; Srikanthan, N.; Hamre, B.; Harper, P.; Brun, A. Protection from phototoxic injury during surgery and endoscopy in erythropoietic protoporphyria. Liver Transpl. 2008, 14, 1340–1346. [Google Scholar] [CrossRef]
- Hanaki, T.; Noda, T.; Eguchi, H.; Iwagami, Y.; Akita, H.; Asaoka, T.; Gotoh, K.; Kobayashi, S.; Umeshita, K.; Mori, M.; et al. Successful Liver Transplantation for Liver Failure With Erythropoietic Protoporphyria by Covering the Operating Theater Lights With Polyimide Film: A Case Report. Transpl. Proc. 2020, 52, 625–629. [Google Scholar] [CrossRef]
- Minder, E.I.; Schneider-Yin, X.; Steurer, J.; Bachmann, L.M. A systematic review of treatment options for dermal photosensitivity in erythropoietic protoporphyria. Cell Mol. Biol. 2009, 55, 84–97. [Google Scholar] [PubMed]
- Corbett, M.F.; Herxheimer, A.; Magnus, I.A.; Ramsay, C.A.; Kobza-Black, A. The long term treatment with beta-carotene in erythropoietic protoporphyria: A controlled trial. Br. J. Dermatol. 1977, 97, 655–662. [Google Scholar] [CrossRef]
- Di Pierro, E.; Granata, F. Nutrients and Porphyria: An Intriguing Crosstalk. Int. J. Mol. Sci. 2020, 21, 3462. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.; Ferguson, J. Narrow-band UVB (TL-01) phototherapy: An effective preventative treatment for the photodermatoses. Br. J. Dermatol. 1995, 132, 956–963. [Google Scholar] [CrossRef]
- Warren, L.J.; George, S. Erythropoietic protoporphyria treated with narrow-band (TL-01) UVB phototherapy. Australas. J. Dermatol. 1998, 39, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Roelandts, R. Photo(chemo)therapy and general management of erythropoietic protoporphyria. Dermatology 1995, 190, 330–331. [Google Scholar] [CrossRef]
- Bijlmer-Iest, J.C.; van Asbeck, B.S.; van Asbeck, B.S.; van Hattum, J.; van Weelden, H.; Marx, J.J.; Koningsberger, J.C. Protoporphyrin photosensitivity cannot be attenuated by oral N-acetylcysteine. Photodermatol. Photoimmunol. Photomed. 1992, 9, 245–249. [Google Scholar]
- Norris, P.G.; Baker, C.S.; Roberts, J.E.; Hawk, J.L. Treatment of erythropoietic protoporphyria with N-acetylcysteine. Arch. Dermatol. 1995, 131, 354–355. [Google Scholar] [CrossRef] [PubMed]
- Mathews-Roth, M.M.; Rosner, B.; Benfell, K.; Roberts, J.E. A double-blind study of cysteine photoprotection in erythropoietic protoporphyria. Photodermatol. Photoimmunol. Photomed. 1994, 10, 244–248. [Google Scholar] [PubMed]
- Mathews-Roth, M.M.; Rosner, B. Long-term treatment of erythropoietic protoporphyria with cysteine. Photodermatol. Photoimmunol. Photomed. 2002, 18, 307–309. [Google Scholar] [CrossRef]
- Boffa, M.J.; Ead, R.D.; Reed, P.; Weinkove, C. A double-blind, placebo-controlled, crossover trial of oral vitamin C in erythropoietic protoporphyria. Photodermatol. Photoimmunol. Photomed. 1996, 12, 27–30. [Google Scholar] [CrossRef]
- Parker, C.J.; Desnick, R.J.; Bissel, M.D.; Bloomer, J.R.; Singal, A.; Gouya, L.; Puy, H.; Anderson, K.E.; Balwani, M.; Phillips, J.D. Results of a pilot study of isoniazid in patients with erythropoietic protoporphyria. Mol. Genet. Metab. 2019, 128, 309–313. [Google Scholar] [CrossRef]
- Langendonk, J.G.; Balwani, M.; Anderson, K.E.; Bonkovsky, H.L.; Anstey, A.V.; Bissell, D.M.; Bloomer, J.; Edwards, C.; Neumann, N.J.; Parker, C.; et al. Afamelanotide for Erythropoietic Protoporphyria. N. Engl. J. Med. 2015, 373, 48–59. [Google Scholar] [CrossRef]
- Wensink, D.; Wagenmakers, M.A.E.M.; Langendonk, J.G. Afamelanotide for prevention of phototoxicity in erythropoietic protoporphyria. Expert. Rev. Clin. Pharm. 2021, 14, 151–160. [Google Scholar] [CrossRef]
- Biolcati, G.; Marchesini, E.; Sorge, F.; Barbieri, L.; Schneider-Yin, X.; Minder, E.I. Long-term observational study of afamelanotide in 115 patients with erythropoietic protoporphyria. Br. J. Dermatol. 2015, 172, 1601–1612. [Google Scholar] [CrossRef]
- Wensink, D.; Wagenmakers, M.A.E.M.; Barman-Aksozen, J.; Friesema, E.C.H.; Wilson, J.H.P.; van Rosmalen, J.; Langendonk, J.G. Association of Afamelanotide With Improved Outcomes in Patients With Erythropoietic Protoporphyria in Clinical Practice. JAMA Dermatol. 2020, 156, 570–575. [Google Scholar] [CrossRef]
- Barman-Aksozen, J.; Nydegger, M.; Schneider-Yin, X.; Minder, A.E. Increased phototoxic burn tolerance time and quality of life in patients with erythropoietic protoporphyria treated with afamelanotide—A three years observational study. Orphanet. J. Rare Dis. 2020, 15, 213. [Google Scholar] [CrossRef]
- Wensink, D.; Langendonk, J.G.; Overbey, J.R.; Balwani, M.; Van Broekhoven, E.J.E.; Wagenmakers, M.A.E.M.; Wilson, J.H.P.; Wheeden, K.; Naik, H.; Desnick, R.J. Erythropoietic protoporphyria: Time to prodrome, the warning signal to exit sun exposure without pain-a patient-reported outcome efficacy measure. Genet. Med. 2021, 23, 1616–1623. [Google Scholar] [CrossRef]
- Minder, E.I. Afamelanotide, an agonistic analog of alpha-melanocyte-stimulating hormone, in dermal phototoxicity of erythropoietic protoporphyria. Expert. Opin. Investig. Drugs. 2010, 19, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Pathak, M.A. Photoprotective Role of Melanin (Eumelanin) in Human Skin. In Light in Biology and Medicine; Springer: Berlin/Heidelberg, Germany; Boston, MA, USA, 1988; pp. 337–344. [Google Scholar]
- Minder, E.I.; Barman-Aksoezen, J.; Schneider-Yin, X. Pharmacokinetics and Pharmacodynamics of Afamelanotide and its Clinical Use in Treating Dermatologic Disorders. Clin. Pharm. 2017, 56, 815–823. [Google Scholar] [CrossRef]
- Haylett, A.K.; Nie, Z.; Brownrigg, M.; Taylor, R.; Rhodes, L.E. Systemic photoprotection in solar urticaria with alpha-melanocyte-stimulating hormone analogue [Nle4-D-Phe7]-alpha-MSH. Br. J. Dermatol. 2011, 164, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Biolcati, G.; Aurizi, C.; Barbieri, L.; Cialfi, S.; Screpanti, I.; Talora, C. Efficacy of the melanocortin analogue Nle4-D-Phe7-alpha-melanocyte-stimulating hormone in the treatment of patients with Hailey-Hailey disease. Clin. Exp. Dermatol. 2014, 39, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.W.; Grimes, P.E.; Agbai, O.; Hamzavi, I.; Henderson, M.; Haddican, M.; Linkner, R.V.; Lebwohl, M. Afamelanotide and narrowband UV-B phototherapy for the treatment of vitiligo: A randomized multicenter trial. JAMA Dermatol. 2015, 151, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Erwin, A.L.; Balwani, M. Porphyrias in the Age of Targeted Therapies. Diagnostics 2021, 11, 1795. [Google Scholar] [CrossRef]
- Ardalan, Z.S.; Chandran, S.; Vasudevan, A.; Angus, P.W.; Grigg, A.; He, S.; Macdonald, G.A.; Strasser, S.I.; Tate, C.J.; Kennedy, G.A.; et al. Management of Patients With Erythropoietic Protoporphyria-Related Progressive Liver Disease. Liver Transpl. 2019, 25, 1620–1633. [Google Scholar] [CrossRef] [PubMed]
- Bonkovsky, H.L.; Schned, A.R. Fatal liver failure in protoporphyria. Synergism between ethanol excess and the genetic defect. Gastroenterology 1986, 90, 191–201. [Google Scholar] [CrossRef]
- Komatsu, H.; Ishii, K.; Imamura, K.; Maruyama, K.; Yonei, Y.; Masuda, H.; Tsuchihashi, T.; Sajima, Y. A case of erythropoietic protoporphyria with liver cirrhosis suggesting a therapeutic value of supplementation with alpha-tocopherol. Hepatol. Res. 2000, 18, 298–309. [Google Scholar] [CrossRef]
- Pirlich, M.; Lochs, H.; Schmidt, H.H. Liver cirrhosis in erythropoietic protoporphyria: Improvement of liver function with ursodeoxycholic acid. Am. J. Gastroenterol. 2001, 96, 3468–3469. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.; Doss, M.O. Liver cirrhosis in protoporphyria: Bile acid therapy and liver transplantation. Z. Gastroenterol. 1995, 33, 399–403. [Google Scholar] [PubMed]
- Abitbol, M.; Puy, H.; Sabate, J.M.; Guenet, J.L.; Deybach, J.C.; Montagutelli, X. Ursodesoxycholic acid and heme-arginate are unable to improve hematopoiesis and liver injury in an erythropoietic protoporphyria mouse model. Physiol. Res. 2006, 55 (Suppl. 2), S93–S101. [Google Scholar] [PubMed]
- Beukeveld, G.J.; Wolthers, B.G. Cholestyramine orally administered to patients with erythropoietic protoporphyria results in urinary excretion of protoporphyrin: To be explained by the Herbst-Volkheimer effect? Clin. Chim. Acta 1995, 233, 119–126. [Google Scholar] [CrossRef]
- McCullough, A.J.; Barron, D.; Mullen, K.D.; Petrelli, M.; Park, M.C.; Mukhtar, H.; Bickers, D.R. Fecal protoporphyrin excretion in erythropoietic protoporphyria: Effect of cholestyramine and bile acid feeding. Gastroenterology 1988, 94, 177–181. [Google Scholar] [CrossRef]
- Tewari, A.; Marsden, J.; Naik, H.; Benton, E.C.; Sarkany, R. Oral cholestyramine is not an effective treatment for uncomplicated erythropoietic protoporphyria. J. Am. Acad. Dermatol. 2012, 67, 1383–1384. [Google Scholar] [CrossRef]
- Fujimori, N.; Komatsu, M.; Tanaka, N.; Iwaya, M.; Nakano, H.; Sugiura, A.; Yamazaki, T.; Shibata, S.; Iwaya, Y.; Muraki, T.; et al. Cimetidine/lactulose therapy ameliorates erythropoietic protoporphyria-related liver injury. Clin. J. Gastroenterol. 2017, 10, 452–458. [Google Scholar] [CrossRef]
- Bloomer, J.R.; Pierach, C.A. Effect of hematin administration to patients with protoporphyria and liver disease. Hepatology 1982, 2, 817–821. [Google Scholar] [CrossRef]
- Dellon, E.S.; Szczepiorkowski, Z.M.; Dzik, W.H.; Graeme-Cook, F.; Ades, A.; Bloomer, J.R.; Cosimi, A.B.; Chung, R.T. Treatment of recurrent allograft dysfunction with intravenous hematin after liver transplantation for erythropoietic protoporphyria. Transplantation 2002, 73, 911–915. [Google Scholar] [CrossRef]
- Lamon, J.M.; Poh-Fitzpatrick, M.B.; Lamola, A.A. Hepatic protoporphyrin production in human protoporphyria. Effects of intravenous hematin and analysis of erythrocyte protoporphyrin distribution. Gastroenterology 1980, 79, 115–125. [Google Scholar] [CrossRef]
- Wahlin, S.; Harper, P. Pretransplant albumin dialysis in erythropoietic protoporphyria: A costly detour. Liver Transpl. 2007, 13, 1614–1615. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Kawakami, Y.; Kan, H.; Fujino, H.; Fukuhara, T.; Naeshiro, N.; Miyaki, D.; Kawaoka, T.; Hiramatsu, A.; Tsuge, M.; et al. A second attack of cholestasis associated with erythropoietic protoporphyria was successfully treated by plasma exchange and blood transfusion. Clin. J. Gastroenterol. 2014, 7, 333–337. [Google Scholar] [CrossRef] [PubMed]
- van Wijk, H.J.; van Hattum, J.; De La Faille, H.B.; van den Berg, J.W.; Edixhoven-Bosdijk, A.; Wilson, J.H. Blood exchange and transfusion therapy for acute cholestasis in protoporphyria. Dig. Dis. Sci. 1988, 33, 1621–1625. [Google Scholar] [CrossRef]
- Wensink, D.; Wagenmakers, M.A.; Wilson, J.P.; Langendonk, J.G. Letter to the editor: Diagnosis of erythropoietic protoporphyria with severe liver injury—A case report. World J. Gastroenterol. 2019, 25, 4292–4293. [Google Scholar] [CrossRef] [PubMed]
- Wahlin, S.; Aschan, J.; Bjornstedt, M.; Broome, U.; Harper, P. Curative bone marrow transplantation in erythropoietic protoporphyria after reversal of severe cholestasis. J. Hepatol. 2007, 46, 174–179. [Google Scholar] [CrossRef]
- Rand, E.B.; Bunin, N.; Cochran, W.; Ruchelli, E.; Olthoff, K.M.; Bloomer, J.R. Sequential liver and bone marrow transplantation for treatment of erythropoietic protoporphyria. Pediatrics 2006, 118, e1896–e1899. [Google Scholar] [CrossRef] [PubMed]
- Dowman, J.K.; Gunson, B.K.; Mirza, D.F.; Badminton, M.N.; Newsome, P.N. UK experience of liver transplantation for erythropoietic protoporphyria. J. Inherit. Metab. Dis. 2011, 34, 539–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlin, S.; Harper, P. The role for BMT in erythropoietic protoporphyria. Bone Marrow Transpl. 2010, 45, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Landefeld, C.; Kentouche, K.; Gruhn, B.; Stauch, T.; Rossler, S.; Schuppan, D.; Whatley, S.D.; Beck, J.F.; Stolzel, U. X-linked protoporphyria: Iron supplementation improves protoporphyrin overload, liver damage and anaemia. Br. J. Haematol. 2016, 173, 482–484. [Google Scholar] [CrossRef] [PubMed]
- Thunell, S.; Harper, P.; Brun, A. Porphyrins, porphyrin metabolism and porphyrias. IV. Pathophysiology of erythyropoietic protoporphyria--diagnosis, care and monitoring of the patient. Scand. J. Clin. Lab. Investig. 2000, 60, 581–604. [Google Scholar]
- Holick, M.F.; Binkley, N.C.; Bischoff-Ferrari, H.A.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Murad, M.H.; Weaver, C.M. Evaluation, treatment, and prevention of vitamin D deficiency: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1911–1930. [Google Scholar] [CrossRef] [Green Version]
- Orekoya, O.; Rhodes, L.E.; Osman, J.E.; Webb, A.R.; Farrar, M.D. A qualitative study of knowledge, behaviour and attitudes regarding vitamin D acquisition among patients with photosensitivity disorders. Photodermatol. Photoimmunol. Photomed. 2020, 36, 378–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heerfordt, I.M.; Lerche, C.M.; Philipsen, P.A.; Wulf, H.C. The effect of vitamin D recommendations on serum 25-hydroxyvitamin D level in patients with erythropoietic protoporphyria. Nutrition 2021, 93, 111477. [Google Scholar] [CrossRef] [PubMed]
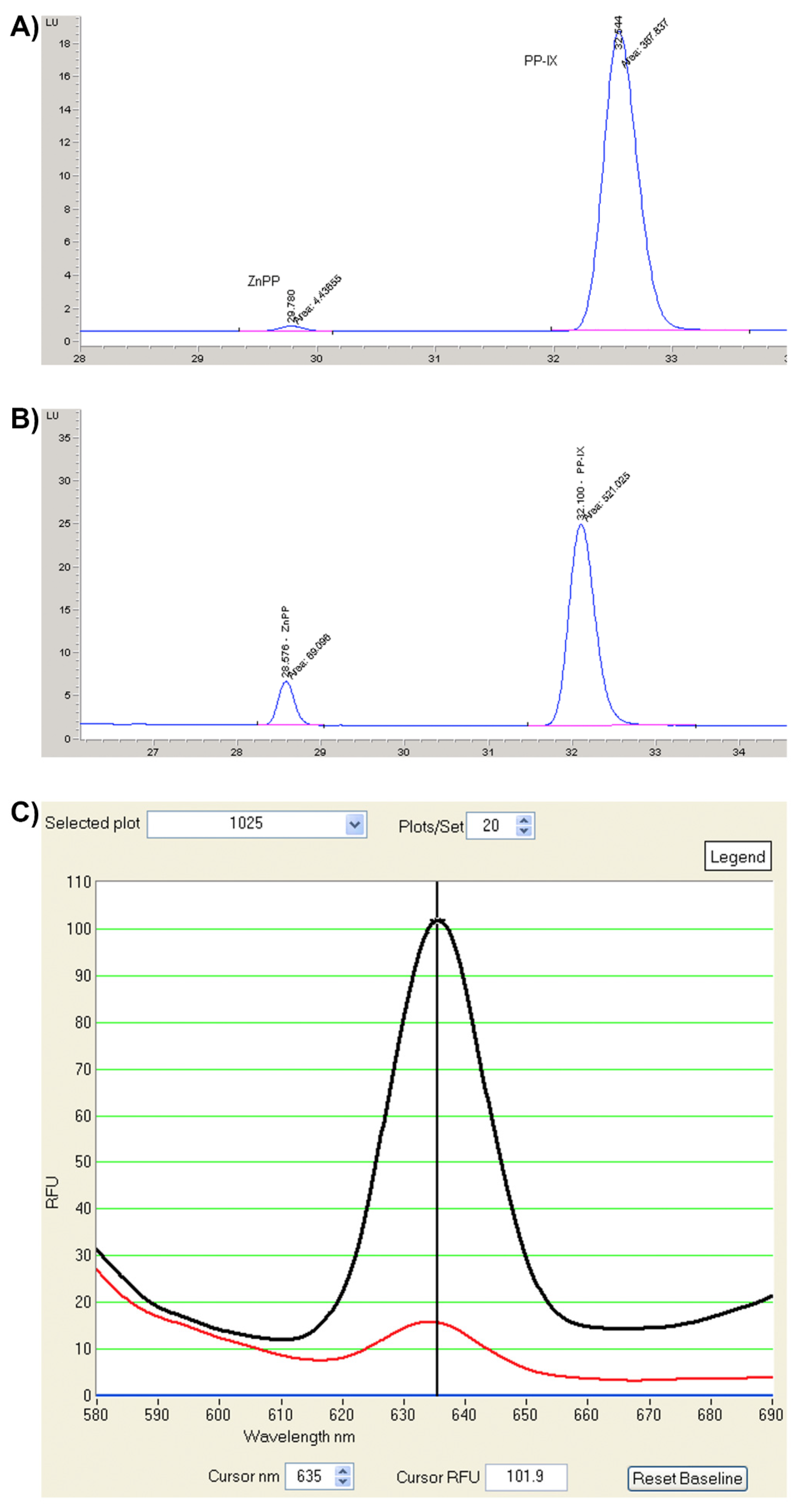
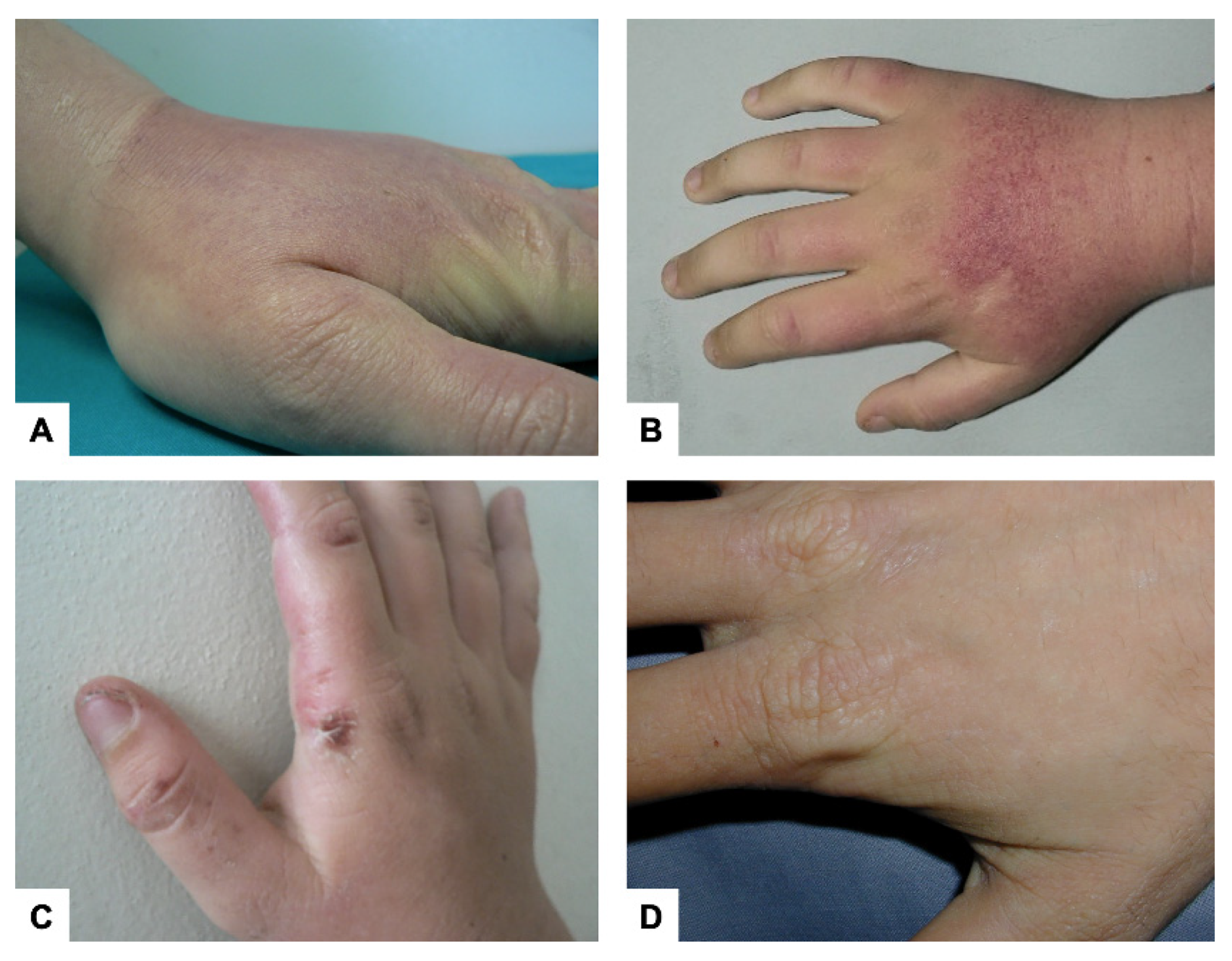
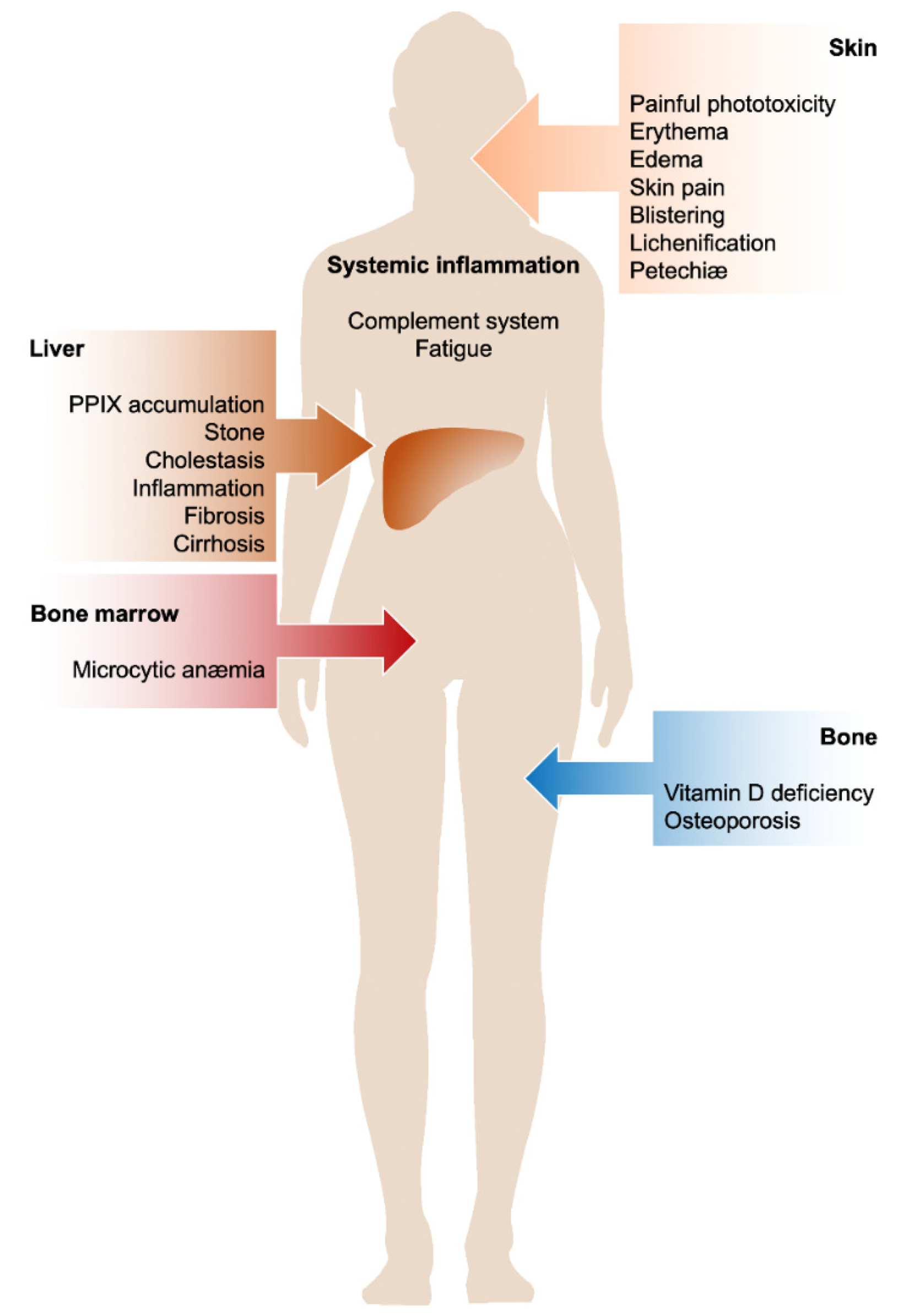

{kind=link}
{kind=link}
{kind=link}
| Symptoms | Treatment Measurements | Recommendations |
|---|---|---|
| Cutaneous Manifestations | ||
| Photoprotection | Protective clothing such as hats, long sleeves, gloves and trousers are beneficial. | Sunscreens mainly protect against UV radiation and do not protect against light that causes EPP symptoms. Windows in houses and cars offer no protection since harmful visible light passes. |
| Application of yellow filters to lamps in the operating room | Only in PP patients who are undergoing long-lasting abdominal surgery such as liver transplantation. | |
| Photosensitivity | Afamelanotide (Scenesse®) | 16 mg subcutaneous implant every 60 days with a maximum of four implants per year |
| Liver involvement | ||
| Gallstones | Ursodeoxycholic acid | Monitoring of erythrocyte protoporphyrins, liver function tests, and necrosis and stasis indices. Abdominal imaging (ultrasound or computed tomography) every 6 to 12 months depending on the patient |
| Cholestasis | Plasma exchange and erythrocyte transfusion | |
| Acute liver failure | Liver or bone marrow transplantation | Bone marrow transplantation can restore liver function without the need for a liver transplantation if the cholestatic liver disease can be stabilised by medical treatment and the fibrosis grade is not advanced |
| Iron deficiency anemia | Oral iron supplementation | In EPP patients, oral iron supplementation should be considered only for severe iron deficiency and not just routinely when the patient is slightly anemic. Monitoring of complete blood count, ferritin, transferrin saturation and erythrocyte protoporphyrins every 6 months. |
| Osteopenia, osteoporosis | Adequate diet and vitamin D supplementation | Monitoring of serum vitamin D and bone indices every year. Bone mineral density every year if in treatment; every 3–5 y if normal scans are detected. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Pierro, E.; Granata, F.; De Canio, M.; Rossi, M.; Ricci, A.; Marcacci, M.; De Luca, G.; Sarno, L.; Barbieri, L.; Ventura, P.; et al. Recognized and Emerging Features of Erythropoietic and X-Linked Protoporphyria. Diagnostics 2022, 12, 151. https://doi.org/10.3390/diagnostics12010151
Di Pierro E, Granata F, De Canio M, Rossi M, Ricci A, Marcacci M, De Luca G, Sarno L, Barbieri L, Ventura P, et al. Recognized and Emerging Features of Erythropoietic and X-Linked Protoporphyria. Diagnostics. 2022; 12(1):151. https://doi.org/10.3390/diagnostics12010151
Chicago/Turabian StyleDi Pierro, Elena, Francesca Granata, Michele De Canio, Mariateresa Rossi, Andrea Ricci, Matteo Marcacci, Giacomo De Luca, Luisa Sarno, Luca Barbieri, Paolo Ventura, and et al. 2022. "Recognized and Emerging Features of Erythropoietic and X-Linked Protoporphyria" Diagnostics 12, no. 1: 151. https://doi.org/10.3390/diagnostics12010151
APA StyleDi Pierro, E., Granata, F., De Canio, M., Rossi, M., Ricci, A., Marcacci, M., De Luca, G., Sarno, L., Barbieri, L., Ventura, P., & Graziadei, G. (2022). Recognized and Emerging Features of Erythropoietic and X-Linked Protoporphyria. Diagnostics, 12(1), 151. https://doi.org/10.3390/diagnostics12010151