
Analysis Comparison for Rapid Identification of Pathogenic Virus from Infected Tissue Samples


Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus and Cells
2.2. Virus Preparation and Titration
2.3. Virus Inoculation
2.4. Sample Preparation
2.5. RNA Extraction and cDNA Synthesis
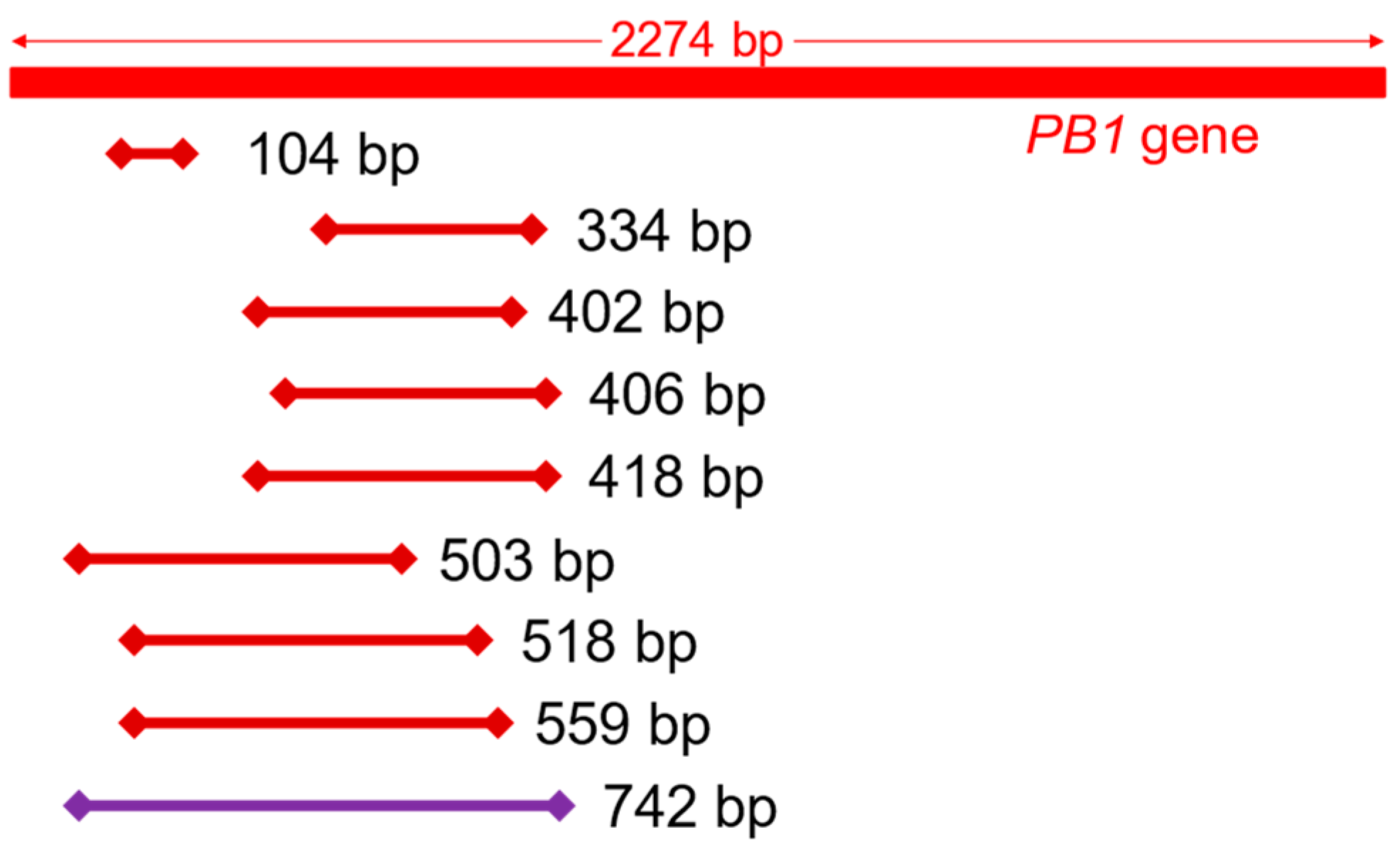
2.6. Amplification and Quantification of the PB1 Gene
2.7. Measurement of the DNA Concentration
2.8. RDV Method
2.9. NGS
2.10. Statistical Analysis
3. Results
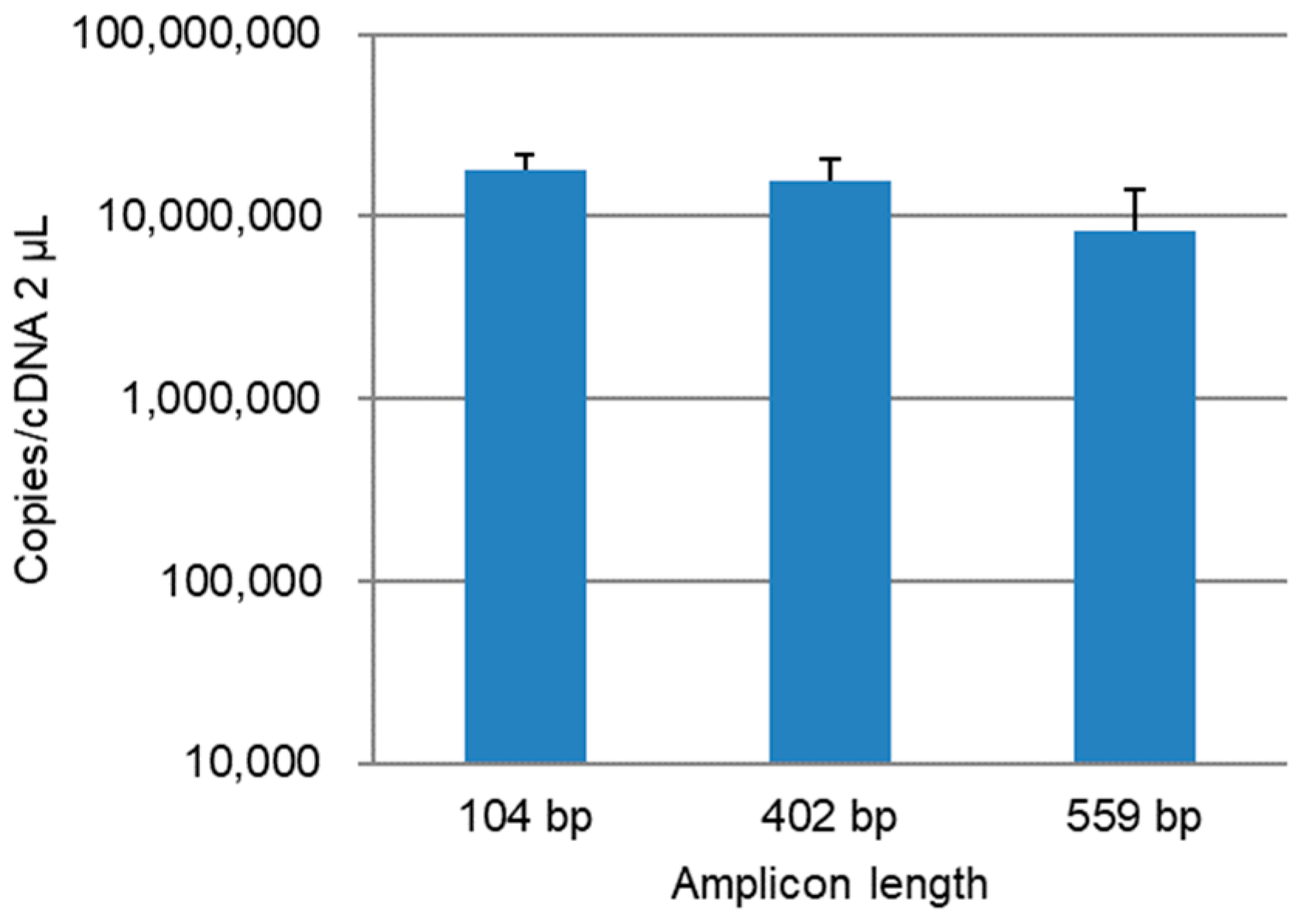
3.1. qPCR Assay Development
3.2. Quantitation of the PB1 Gene in Lung Homogenate Samples
3.3. Detection with RDV
3.4. Detection with NGS
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Landry, M.L.; Caliendo, A.M.; Ginocchio, C.C.; Tang, Y.-W.; Valsamakis, A. Algorithm for detection and identification of viruses. In Manual of Clinical Microbiology, 10th ed.; Versalovic, J., Carroll, K.C., Jorgensen, J.H., Funke, G., Landry, M.L., Warnock, D.W., Eds.; ASM Press: Washington, DC, USA, 2011; Volume 2, pp. 1297–1301. [Google Scholar]
- Mizutani, T.; Endoh, D.; Okamoto, M.; Shirato, K.; Shimizu, H.; Arita, M.; Fukushi, S.; Saijo, M.; Sakai, K.; Lim, C.K.; et al. Rapid genome sequencing of RNA viruses. Emerg. Infect. Dis. 2007, 13, 322–324. [Google Scholar] [CrossRef]
- Hosokawa-Muto, J.; Sakai, H.; Sassa, Y.; Fujinami, Y.; Kishimoto, M.; Nakahara, H. Rapid detection of pathogenic virus genome sequence from throat and nasal swab samples using an exhaustive gene amplification method. Forensic Sci. Med. Pathol. 2019, 15, 399–403. [Google Scholar] [CrossRef]
- Wollants, E.; Smolders, D.; Naesens, R.; Bruynseels, P.; Lagrou, K.; Matthijnssens, J.; Van Ranst, M. Use of Next-Generation Sequencing for Diagnosis of West Nile Virus Infection in Patient Returning to Belgium from Hungary. Emerg. Infect. Dis. 2018, 24, 2380–2382. [Google Scholar] [CrossRef]
- Perlejewski, K.; Popiel, M.; Laskus, T.; Nakamura, S.; Motooka, D.; Stokowy, T.; Lipowski, D.; Pollak, A.; Lechowicz, U.; Caraballo Cortes, K.; et al. Next-generation sequencing (NGS) in the identification of encephalitis-causing viruses: Unexpected detection of human herpesvirus 1 while searching for RNA pathogens. J. Virol. Methods 2015, 226, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Whitmer, S.L.M.; Yadav, P.D.; Sarkale, P.; Chaubal, G.Y.; Francis, A.; Klena, J.; Nichol, S.T.; Stroher, U.; Mourya, D.T. Characterization of Unknown Orthobunya-Like Viruses from India. Viruses 2018, 10, 451. [Google Scholar] [CrossRef] [Green Version]
- Sano, K.; Okazaki, S.; Taniguchi, S.; Masangkay, J.S.; Puentespina, R., Jr.; Eres, E.; Cosico, E.; Quibod, N.; Kondo, T.; Shimoda, H.; et al. Detection of a novel herpesvirus from bats in the Philippines. Virus Genes 2015, 51, 136–139. [Google Scholar] [CrossRef]
- Smith, J.A.L. Collection and Preservation of Microbial Forensic Samples. In Microbial Forensics, 2nd ed.; Budowle, B., Schutzer, S.E., Breeze, R.G., Keim, P.S., Morse, S.A., Eds.; Academic Press: Cambridge, MA, USA, 2011; pp. 379–392. [Google Scholar]
- Murray, C.L.; Oh, T.S.; Rice, C.M. Keeping Track of Viruses. In Microbial Forensics, 2nd ed.; Budowle, B., Schutzer, S.E., Breeze, R.G., Keim, P.S., Morse, S.A., Eds.; Academic Press: Cambridge, MA, USA, 2011; pp. 137–153. [Google Scholar]
- Gonzalez-Candelas, F.; Bracho, M.A.; Wrobel, B.; Moya, A. Molecular evolution in court: Analysis of a large hepatitis C virus outbreak from an evolving source. BMC Biol. 2013, 11, 76. [Google Scholar] [CrossRef] [Green Version]
- Scaduto, D.I.; Brown, J.M.; Haaland, W.C.; Zwickl, D.J.; Hillis, D.M.; Metzker, M.L. Source identification in two criminal cases using phylogenetic analysis of HIV-1 DNA sequences. Proc. Natl. Acad. Sci. USA 2010, 107, 21242–21247. [Google Scholar] [CrossRef] [Green Version]
- Ciesielski, C.; Marianos, D.; Ou, C.Y.; Dumbaugh, R.; Witte, J.; Berkelman, R.; Gooch, B.; Myers, G.; Luo, C.C.; Schochetman, G.; et al. Transmission of human immunodeficiency virus in a dental practice. Ann. Intern. Med. 1992, 116, 798–805. [Google Scholar] [CrossRef]
- Hall, C.L.; Zascavage, R.R.; Sedlazeck, F.J.; Planz, J.V. Potential applications of nanopore sequencing for forensic analysis. Forensic Sci. Rev. 2020, 32, 23–54. [Google Scholar]
- Zascavage, R.R.; Shewale, S.J.; Planz, J.V. Deep-Sequencing Technologies and Potential Applications in Forensic DNA Testing. Forensic Sci. Rev. 2013, 25, 79–105. [Google Scholar] [PubMed]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef]
- Deamer, D.; Akeson, M.; Branton, D. Three decades of nanopore sequencing. Nat. Biotechnol. 2016, 34, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Ip, C.L.C.; Loose, M.; Tyson, J.R.; de Cesare, M.; Brown, B.L.; Jain, M.; Leggett, R.M.; Eccles, D.A.; Zalunin, V.; Urban, J.M.; et al. MinION Analysis and Reference Consortium: Phase 1 data release and analysis. F1000Research 2015, 4, 1075. [Google Scholar] [CrossRef]
- Hosokawa-Muto, J.; Fujinami, Y.; Mizuno, N. Evaluation of the Universal Viral Transport system for long-term storage of virus specimens for microbial forensics. J. Forensic Leg. Med. 2015, 34, 29–33. [Google Scholar] [CrossRef]
- Kärber, G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Arch. Exp. Pathol. Pharmakol. 1931, 162, 480–483. [Google Scholar] [CrossRef]
- Hedlund, M.; Aschenbrenner, L.M.; Jensen, K.; Larson, J.L.; Fang, F. Sialidase-based anti-influenza virus therapy protects against secondary pneumococcal infection. J. Infect. Dis. 2010, 201, 1007–1015. [Google Scholar] [CrossRef]
- Urushisaki, T.; Takemura, T.; Tazawa, S.; Fukuoka, M.; Hosokawa-Muto, J.; Araki, Y.; Kuwata, K. Caffeoylquinic acids are major constituents with potent anti-influenza effects in brazilian green propolis water extract. Evid. Based Complement. Alternat. Med. 2011, 2011, 254914. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, M.; Yoshizumi, S.; Ishida, S.; Komagome, R.; Nagano, H.; Kudo, S.; Okano, M. Usefulness of the rapid determination system of viral genome sequences in human stool specimens. J. Virol. Methods 2012, 179, 256–260. [Google Scholar] [CrossRef]
- Sim, J.; Chapman, B. In-field whole genome sequencing using the MinION nanopore sequencer to detect the presence of high-prized military targets. Aust. J. Forensic Sci. 2019, 51, S86–S90. [Google Scholar] [CrossRef]
- Loman, N.J.; Quick, J.; Simpson, J.T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 2015, 12, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Phillippy, A.M. One chromosome, one contig: Complete microbial genomes from long-read sequencing and assembly. Curr. Opin. Microbiol. 2015, 23, 110–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal-McKinney, J.M.; Liu, K.C.; Lock, C.M.; Wu, W.H.; Hu, J. Comparison of MiSeq, MinION, and hybrid genome sequencing for analysis of Campylobacter jejuni. Sci. Rep. 2021, 11, 5676. [Google Scholar] [CrossRef] [PubMed]
- Oxford Nanopore Technologies. Accuracy. Available online: https://nanoporetech.com/accuracy (accessed on 15 October 2021).


{kind=link}
{kind=link}
{kind=link}
| Primer Name | Forward Primer Sequence (5′–3′) Reverse Primer Sequence (5′–3′) | Amplicon Length (bp) | Reference |
|---|---|---|---|
| - - | GATGGACAACAAACACCGAAACT TACACAATGTTTGGGCATAACC | 104 | [21] |
| PB1-334F PB1-334R | CAGATCAAATGGCCTCACGG ACTCCTTGCCAGTGTCTCAAC | 334 | This study |
| PB1-402F PB1-402R | GGAGGTTGTTCAGCAAACACG CCTGGGGTTGCAATTGCTCT | 402 | This study |
| PB1-406F PB1-406R | CCGACAGACCTATGACTGGAC CCTTGCCAGTGTCTCAACAA | 406 | This study |
| PB1-418F PB1-418R | GAGGTTGTTCAGCAAACACGA ACCCCCTTATTTGCATCCCTG | 418 | This study |
| PB1-503F PB1-503R | CTTACAGCCATGGGACAGGA AGTCATATTGTCTCTCACCCGTC | 503 | This study |
| - PB1-518R | GATGGACAACAAACACCGAAACT TGTGTTCAGGGTCAATGCTCT | 518 | [21] This study |
| - PB1-559R | GATGGACAACAAACACCGAAACT GCTCTCCGTTTTAGCTTCCC | 559 | [21] This study |
| PB1-742F PB1-742R | CCTCCTTACAGCCATGGGAC CTCCAACTGGCAACCCTGAT | 742 | This study |
| Read Detail | Number of Reads |
|---|---|
| 18S ribosomal RNA genes of mammals | 12 |
| 28S ribosomal RNA genes of mammals | 2 |
| 45S ribosomal RNA genes of mammals | 2 |
| Chromosomal sequences of mice | 2 |
| Chromosomal sequences of fish | 2 |
| Mouse tumor necrosis factor mRNA | 1 |
| Mouse ribosomal protein S9 mRNA | 1 |
| Mouse Dab2ip mRNA | 1 |
| 18S ribosomal RNA gene of nematodes | 1 |
| Total | 24 |
| MiSeq Reads | MinION Reads | |||
|---|---|---|---|---|
| Sample 1 | Sample 2 | Sample 1 | Sample 2 | |
| Mapped to influenza A virus | 2142 | 2678 | 30 | 147 |
| (0.12%) | (0.15%) | (0.07%) | (0.10%) | |
| Mapped to the mouse genome | 1,800,025 | 1,659,092 | 10,829 | 77,556 |
| (97.03%) | (95.25%) | (23.54%) | (54.43%) | |
| Un-mapped | 52,877 | 80,034 | 35,138 | 64,772 |
| (2.85%) | (4.60%) | (76.39%) | (45.47%) | |
| Total reads | 1,855,044 | 1,741,804 | 45,997 | 142,475 |
| MiSeq | MinION | |
|---|---|---|
| Input DNA for library preparation | 1 ng/sample | 86 ng/sample |
| Time for library preparation | approximately 3 h 1 | approximately 30 min |
| Time for sequence preparation | approximately 1.5 h | approximately 10 min |
| Total sequencing time | 24 h | 16 h |
| Average read length | 132 bases | 1603 bases |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosokawa-Muto, J.; Sassa-O’Brien, Y.; Fujinami, Y.; Nakahara, H. Analysis Comparison for Rapid Identification of Pathogenic Virus from Infected Tissue Samples. Diagnostics 2022, 12, 196. https://doi.org/10.3390/diagnostics12010196
Hosokawa-Muto J, Sassa-O’Brien Y, Fujinami Y, Nakahara H. Analysis Comparison for Rapid Identification of Pathogenic Virus from Infected Tissue Samples. Diagnostics. 2022; 12(1):196. https://doi.org/10.3390/diagnostics12010196
Chicago/Turabian StyleHosokawa-Muto, Junji, Yukiko Sassa-O’Brien, Yoshihito Fujinami, and Hiroaki Nakahara. 2022. "Analysis Comparison for Rapid Identification of Pathogenic Virus from Infected Tissue Samples" Diagnostics 12, no. 1: 196. https://doi.org/10.3390/diagnostics12010196