
Rare X-Linked Hypohidrotic Ectodermal Dysplasia in Females Associated with Ectodysplasin-A Variants and the X-Chromosome Inactivation Pattern
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Variants Analysis
2.3. Conservation and Protein Conformational Analyses
2.4. Analysis of XCI
3. Results
3.1. Clinical Findings
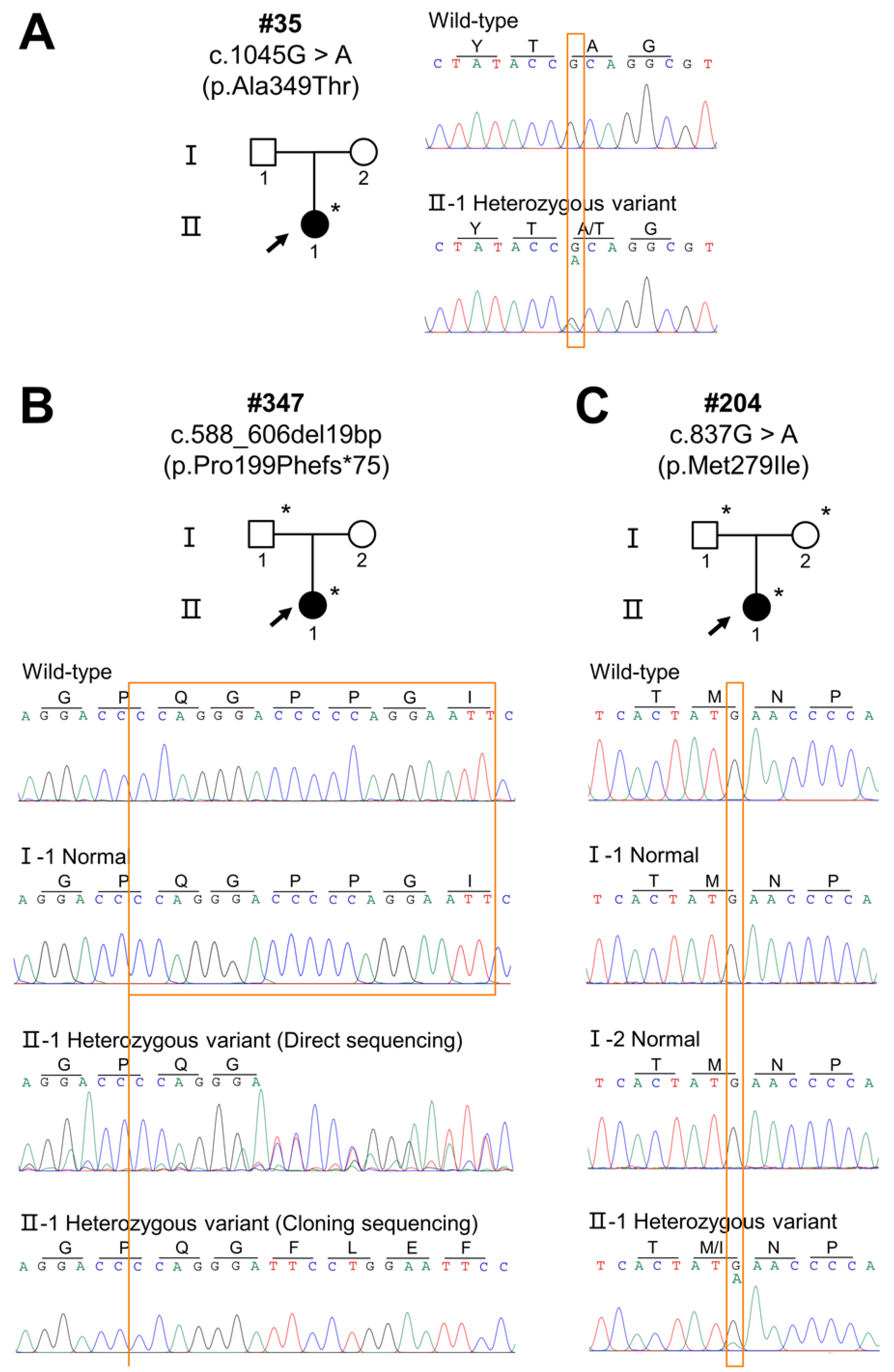
3.2. EDA Variants
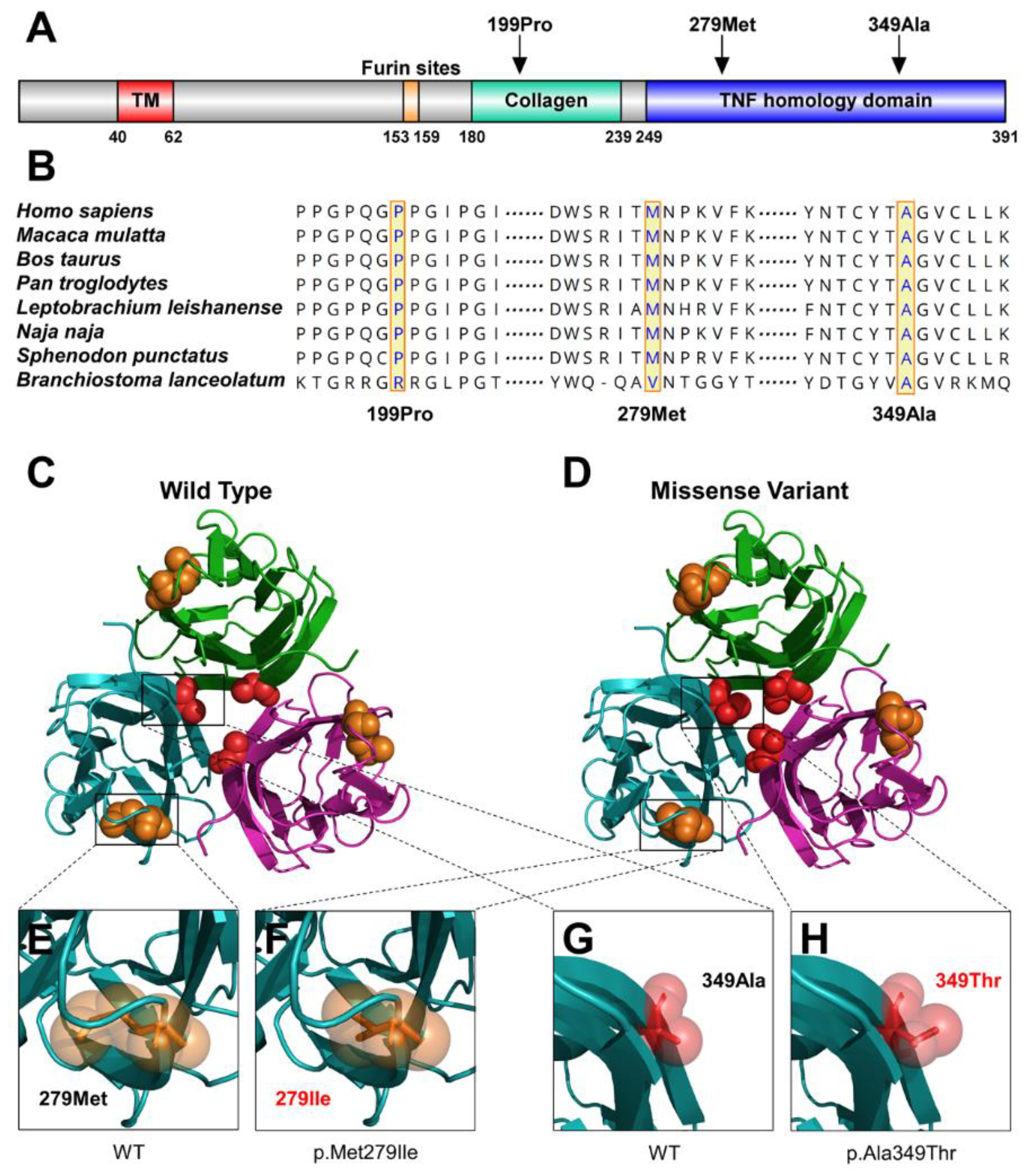
3.3. Bioinformatics Findings
3.4. Skewed XCI
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Visinoni, Á.F.; Lisboa-Costa, T.; Pagnan, N.A.B.; Chautard-Freire-Maia, E.A. Ectodermal dysplasias: Clinical and molecular review. Am. J. Med. Genet. Part A 2009, 149A, 1980–2002. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Reali, J.; Mendoza-Ramos, M.I.; Garrido-Guerrero, E.; Méndez-Catalá, C.F.; Méndez-Cruz, A.R.; Pozo-Molina, G. Hypohidrotic ectodermal dysplasia: Clinical and molecular review. Int. J. Dermatol. 2018, 57, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Mou, C.; Thomason, H.A.; Willan, P.M.; Clowes, C.; Harris, W.E.; Drew, C.F.; Dixon, J.; Dixon, M.J.; Headon, D.J. Enhanced ectodysplasin-A receptor (EDAR) signaling alters multiple fiber characteristics to produce the East Asian hair form. Hum. Mutat. 2008, 29, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Monreal, A.W.; Zonana, J.; Ferguson, B. Identification of a new splice form of the EDA1 gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations. Am. J. Hum. Genet. 1998, 63, 380–389. [Google Scholar] [CrossRef]
- Han, D.; Gong, Y.; Wu, H.; Zhang, X.; Yan, M.; Wang, X.; Qu, H.; Feng, H.; Song, S. Novel EDA mutation resulting in X-linked non-syndromic hypodontia and the pattern of EDA-associated isolated tooth agenesis. Eur. J. Med. Genet. 2008, 51, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, D.; Song, S.; Wang, Y.; Zhao, H.; Pan, S.; Bai, B.; Feng, H. Correlation between the phenotypes and genotypes of X-linked hypohidrotic ectodermal dysplasia and non-syndromic hypodontia caused by ectodysplasin-A mutations. Eur. J. Med. Genet. 2011, 54, e377–e382. [Google Scholar] [CrossRef]
- Burger, K.; Schneider, A.-T.; Wohlfart, S.; Kiesewetter, F.; Huttner, K.; Johnson, R.; Schneider, H. Genotype-phenotype correlation in boys with X-linked hypohidrotic ectodermal dysplasia. Am. J. Med. Genet. Part A 2014, 164A, 2424–2432. [Google Scholar] [CrossRef]
- Wright, J.T.; Grange, D.K.; Fete, M. Hypohidrotic Ectodermal Dysplasia. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Cambiaghi, S.; Restano, L.; Pääkkönen, K.; Caputo, R.; Kere, J. Clinical Findings in Mosaic Carriers of Hypohidrotic Ectodermal Dysplasia. Arch. Dermtol. 2000, 136, 217–224. [Google Scholar] [CrossRef]
- Ørstavik, K.H.; Knudsen, G.P.S.; Nordgarden, H.; Ormerod, E.; Strømme, P.; Lazarou, L.P.; Rosser, L.G.; Prescott, T.; Houge, G. Severe hypohidrotic ectodermal dysplasia in a girl caused by a de novo 9;X insertion that includes XIST and disrupts the EDA gene. Am. J. Med. Genet. Part A 2007, 143A, 1510–1513. [Google Scholar] [CrossRef]
- Pavlovsky, M.; Fuchs-Telem, D.; Nousbeck, J.; Sarig, O.; Sprecher, E. Molecular evidence for the role of X-chromosome inactivation in linear presentation of X-linked hypohidrotic ectodermal dysplasia: Correspondence. Clin. Exp. Dermatol. 2012, 37, 186–188. [Google Scholar] [CrossRef]
- Prasad, M.K.; Geoffroy, V.; Vicaire, S.; Jost, B.; Dumas, M.; Le Gras, S.; Switala, M.; Gasse, B.; Laugel-Haushalter, V.; Paschaki, M.; et al. A targeted next-generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvement. J. Med. Genet. 2016, 53, 98–110. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.C.; Zoghbi, H.Y.; Moseley, A.B.; Rosenblatt, H.M.; Belmont, J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992, 51, 1229–1239. [Google Scholar] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Pääkkönen, K.; Cambiaghi, S.; Novelli, G.; Ouzts, L.V.; Penttinen, M.; Kere, J.; Srivastava, A.K. The mutation spectrum of the EDA gene in X-linked anhidrotic ectodermal dysplasia: Mutations in Brief. Hum. Mutat. 2001, 17, 349. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Ko, J.M.; Chae, J.-H. Novel and Private EDA Mutations and Clinical Phenotypes of Korean Patients with X-Linked Hypohidrotic Ectodermal Dysplasia. Cytogenet. Genome Res. 2019, 158, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huang, Y.; Hua, R.; Zhao, X.; Yang, W.; Liu, Y.; Zhang, X. Mutation Screening of the EDA Gene in Seven Chinese Families with X-Linked Hypohidrotic Ectodermal Dysplasia. Genet. Test. Mol. Biomark. 2018, 22, 487–491. [Google Scholar] [CrossRef]
- Na, G.Y.; Kim, D.W.; Lee, S.J.; Chung, S.L.; Park, D.J.; Kim, J.C.; Kim, M.K. Mutation in the ED1 Gene, Ala349Thr, in a Korean Patient with X-Linked Hypohidrotic Ectodermal Dysplasia Developing de novo. Pediatr. Dermatol. 2004, 21, 568–572. [Google Scholar] [CrossRef]
- Conte, C.; Gambardella, S.; Bulli, C.; Rinaldi, F.; Di Marino, D.; Falconi, M.; Bramanti, P.; Desideri, A.; Novelli, G. Screening of EDA1 gene in X-linked anhidrotic ectodermal dysplasia using DHPLC: Identification of 14 novel mutations in Italian patients. Genet. Test. 2008, 12, 437–442. [Google Scholar] [CrossRef]
- Kobielak, K.; Kobielak, A.; Roszkiewicz, J.; Wierzba, J.; Limon, J.; Trzeciak, W.H. Mutations in the EDA gene in three unrelated families reveal no apparent correlation between phenotype and genotype in the patients with an X-linked anhidrotic ectodermal dysplasia. Am. J. Med. Genet. 2001, 100, 191–197. [Google Scholar] [CrossRef]
- RamaDevi, A.R.; Reddy, E.C.; Ranjan, S.; Bashyam, M.D. Molecular genetic analysis of patients from India with hypohidrotic ectodermal dysplasia reveals novel mutations in the EDA and EDAR genes. Br. J. Dermatol. 2008, 158, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Präger, T.M.; Finke, C.; Miethke, R.-R. Dental findings in patients with ectodermal dysplasia. J. Orofac. Orthop. 2006, 67, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.; Dória, S. X-chromosome inactivation: Implications in human disease. J. Genet. 2021, 100, 63. [Google Scholar] [CrossRef] [PubMed]
- Belmont, J.W. Genetic control of X inactivation and processes leading to X-inactivation skewing. Am. J. Hum. Genet. 1996, 58, 1101–1108. [Google Scholar]
- Cantone, I.; Fisher, A.G. Human X chromosome inactivation and reactivation: Implications for cell reprogramming and disease. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160358. [Google Scholar] [CrossRef]
- Lei, K.; Zhang, Y.; Dong, Z.; Sun, Y.; Yi, Z.; Chen, Z. A novel 1-bp deletion mutation and extremely skewed X-chromosome inactivation causing severe X-linked hypohidrotic ectodermal dysplasia in a Chinese girl. Clin. Exp. Dermatol. 2018, 43, 60–62. [Google Scholar] [CrossRef]
- GIEDE (Spanish Multidisciplinary Research Group for Ectodermal Dysplasia); Martínez-Romero, M.C.; Ballesta-Martínez, M.J.; López-González, V.; Sánchez-Soler, M.J.; Serrano-Antón, A.T.; Barreda-Sánchez, M.; Rodriguez-Peña, L.; Martínez-Menchon, M.T.; Frías-Iniesta, J.; et al. EDA, EDAR, EDARADD and WNT10A allelic variants in patients with ectodermal derivative impairment in the Spanish population. Orphanet J. Rare Dis. 2019, 14, 281. [Google Scholar] [CrossRef]
- Körber, L.; Schneider, H.; Fleischer, N.; Maier-Wohlfart, S. No evidence for preferential X-chromosome inactivation as the main cause of divergent phenotypes in sisters with X-linked hypohidrotic ectodermal dysplasia. Orphanet J. Rare Dis. 2021, 16, 98. [Google Scholar] [CrossRef]
- Sharp, A.; Robinson, D.; Jacobs, P. Age- and tissue-specific variation of X chromosome inactivation ratios in normal women. Hum. Genet. 2000, 107, 343–349. [Google Scholar] [CrossRef]
- Phung, T.N.; Olney, K.C.; Pinto, B.J.; Silasi, M.; Perley, L.; O’Bryan, J.; Kliman, H.J.; Wilson, M.A. X chromosome inactivation in the human placenta is patchy and distinct from adult tissues. HGG Adv. 2022, 3, 100121. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Features | #35 | #347 | #204 |
|---|---|---|---|
| Gender | Female | Female | Female |
| Age (years) | 8 | 21 | 14 |
| Permanent dentition | |||
| Missing number (excluding third molar, n = 28) | 19 | 24 | 20 |
| Peg-shaped teeth | − | + | − |
| Hair | |||
| Sparse scalp hair | − | +, brown | +, thin |
| Sparse eyebrows | + | + | + |
| Skin | |||
| Erythematous patches | − | + | + |
| Hyperpigmentation | + | + | − |
| Perioral and periocular wrinkles | + | + | − |
| Dry skin | + | + | + |
| Gland function | |||
| Hypohidrosis | + | − | + |
| Anhidrosis | − | + | − |
| Dry eyes | − | + | + |
| Xerostomia | − | + | − |
| Variant | Patient | Domain | Mutation Taster | Fathmm | PolyPhen-2 | gnomAD, dbSNP, 1000G | ACMG Classification (Evidence of Pathogenicity) |
|---|---|---|---|---|---|---|---|
| c.1045G>A (p.Ala349Thr) | #35 | TNF | Disease causing | DAMAGING (−5.92) | Probably damaging (0.996) | rs132630317 | Pathogenic PS1 + PM1 + PM2 + PP2 + PP3 + PP4 |
| c.588_606del19bp (p.Pro199Phefs*75) | #347 | Collagen | Disease causing | - | - | Not found | Pathogenic PVS1 + PM1 + PM2 + PM4 + PP3 + PP4 |
| c.837G>A (p.Met279Ile) | #204 | TNF | Disease causing | DAMAGING (−5.80) | Benign (0.038) | Not found | Pathogenic PS2 + PM1 + PM2 + PP2 + PP3 + PP4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Su, L.; Liu, H.; Zheng, J.; Feng, H.; Liu, Y.; Yu, M.; Han, D. Rare X-Linked Hypohidrotic Ectodermal Dysplasia in Females Associated with Ectodysplasin-A Variants and the X-Chromosome Inactivation Pattern. Diagnostics 2022, 12, 2300. https://doi.org/10.3390/diagnostics12102300
Liu H, Su L, Liu H, Zheng J, Feng H, Liu Y, Yu M, Han D. Rare X-Linked Hypohidrotic Ectodermal Dysplasia in Females Associated with Ectodysplasin-A Variants and the X-Chromosome Inactivation Pattern. Diagnostics. 2022; 12(10):2300. https://doi.org/10.3390/diagnostics12102300
Chicago/Turabian StyleLiu, Haochen, Lanxin Su, Hangbo Liu, Jinglei Zheng, Hailan Feng, Yang Liu, Miao Yu, and Dong Han. 2022. "Rare X-Linked Hypohidrotic Ectodermal Dysplasia in Females Associated with Ectodysplasin-A Variants and the X-Chromosome Inactivation Pattern" Diagnostics 12, no. 10: 2300. https://doi.org/10.3390/diagnostics12102300