
De Novo Mosaic 6p23-p25.3 Tetrasomy Caused by a Small Supernumerary Marker Chromosome Presenting Trisomy Distal 6p Phenotype: A Case Report and Literature Review

 ,
,  ,
,  ,
,
Abstract
1. Introduction
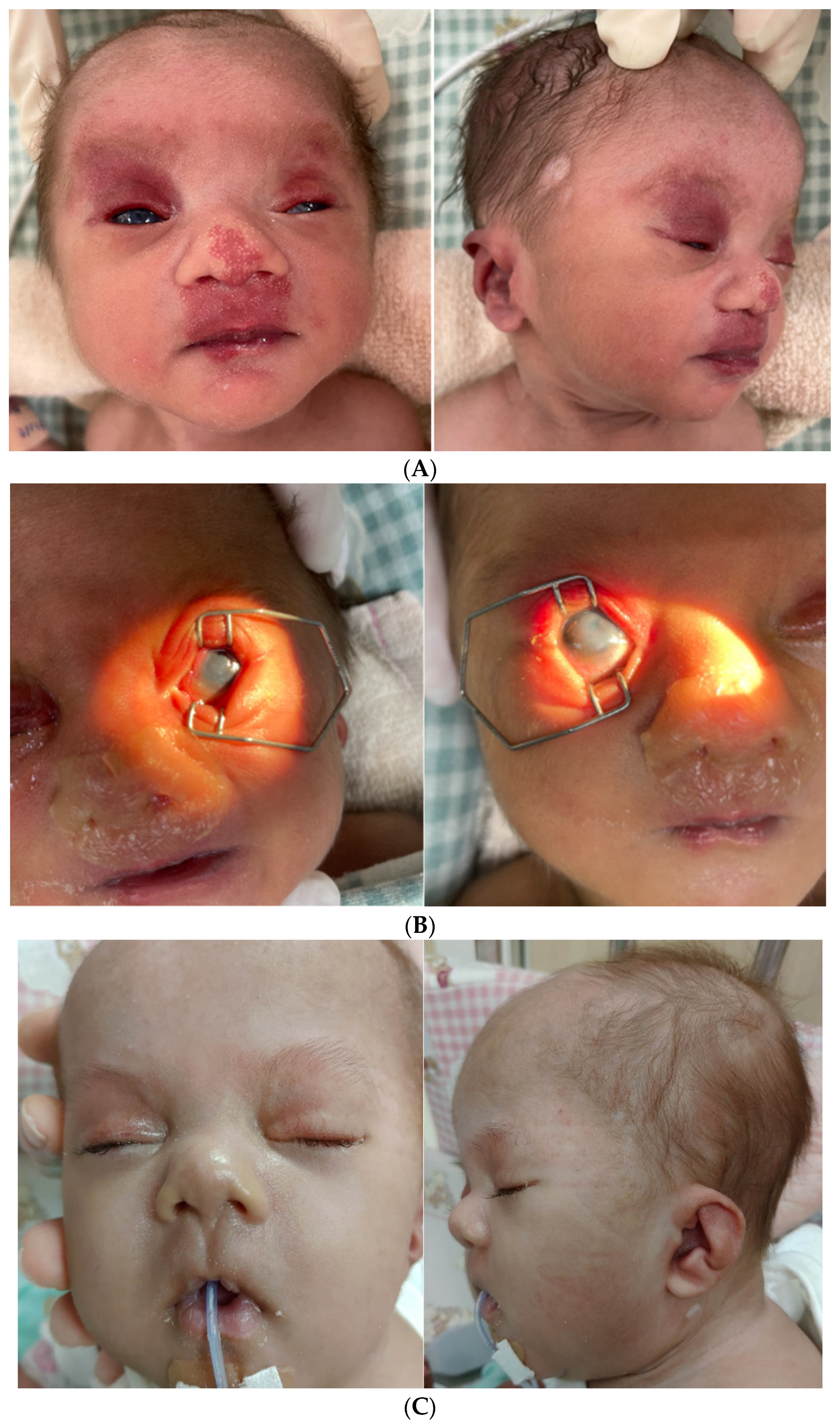
2. Case Report
3. Results
4. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Phelan et al. 1986 [29] | Breuning et al. 1977 [5] | Engelen et al. 2001 [6] | Petkovic et al. 2003 [7] | Scott et al. 2007 [8] | Kerrigan et al. 2007 [9] | Castiglioni et al. 2013 [10] | Chen et al. 2014 [11] | Jankauskienė et al. 2016 [12] | Sivasankaran et al. 2017 [4] | Fontana et al. 2017 [13] | Türkyılmaz et al. 2022 [14] | Stohler et al. 2007 [3] | Present Case |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Region | 6p21-pter | 6p21-p25 | 6p22.1-pter | 6p11.1-p25 | 6p22.2- p25.2 | 6p25.1-p25.3 | 6p23-p25.3 | 6p22.3- p25.3 | 6p22.1-p25.3 | 6p22.3-pter | 6p25.2-p25.3 | 6p22.1-p25.3 | Tetrasomy 6p25.1-6p25.3. | Mosaic Tetrasomy 6p23-6p25.3. |
| Age/Sex | NA | 2y8m/NA | NA | 3y/F | NA/F | 8.6y/F | 2y/F, 0y/F, 32y/M | Prenatal/F | 13y/F | 2y/M | 32y/F | 4m/F | 3y/F | 0y/M |
| Low birth weight | + | + | + | + | + | + | −, +, − | + | + | + | NA | + | NA | + |
| Growth retardation | + | + | - | + | + | + | −, +, − | + | + | + | + | + | + | + |
| Developmental delay | + | + | + | + | + | + | −, −, − | NA | + | + | - | + | + | + |
| Microcephaly | + | + | + | + | + | + | - | + | + | + | - | + | + | + |
| Craniosinostosis | NA | - | - | - | NA | - | - | + | - | - | - | - | - | - |
| Seizures | NA | - | - | NA | NA | + | - | NA | - | - | - | - | - | |
| Hemangioma | NA | - | + | NA | NA | + | +, −, − | NA | NA | NA | - | + | NA | + |
| Craniofacial abnormalities | + | + | + | + | + | + | +, +, − | + | + | + | - | + | + | + |
| Prominent/ high forehead | + | + | + | + | NA | + | −, −, − | NA | + | - | - | + | + | + |
| Low-set/ malformed ears | - | + | + | + | NA | NA | +, −, − | + | + | + | - | + | + | + |
| Bulbous nose | + | + | + | - | NA | + | +, −, − | + | NA | + | - | + | + | + |
| Choanal atresia | NA | - | - | - | NA | - | +, +, − | NA | - | NA | - | - | - | + |
| Blepharophimosis/ ptosis | + | NA | + | - | + | + | +, −, − | NA | + | + | - | + | + | - |
| ACED/Cataracts | NA | - | - | - | - | - | −, −, − | NA | - | NA | - | - | - | + |
| Hearing loss | NA | NA | - | - | - | - | −, −, + | NA | + | - | - | + | + | + |
| CHD | - | + | + | - | - | NA | +, −, − | NA | - | - | + | + | + | + |
| CAKUT | NA | + | + | NA | NA | NA | −, −, − | NA | + | - | - | + | + | + |
| Small kidney | NA | + | - | NA | NA | NA | −, −, − | NA | + | - | - | - | - | + |
| VUR | NA | NA | + | NA | NA | NA | −, −, − | NA | - | - | - | - | - | + |
| Hydronephrosis | NA | NA | + | NA | NA | NA | −, −, − | NA | - | - | - | + | + | + |
| Proteinuria | NA | + | NA | NA | NA | NA | −, −, − | NA | + | - | - | - | NA | + |
| Glomerulopathy | NA | + | NA | NA | NA | NA | −, −, − | NA | + | - | - | - | NA | + |
| Sacral dimple | NA | + | NA | NA | - | NA | −, −, − | NA | NA | - | - | + | + | + |
| Clinodactyly | NA | NA | NA | NA | - | NA | −, −, − | + | - | + | - | - | + | - |
| Additions features | Two-lobed right lung; mobile colon ascendens | Mild immunodeficiency (IgG 2/ IgG 4) | Left-sided posterolateral CDH | Hypothalamic hamartoma | Low maternal serum PAPP-A | Corpus callosum hypoplasia, internal hydrocephalus | Macrocephaly | Large AF, bilateral eyelid colobomas, astigmatism hyperopia |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| sSMC | small supernumerary marker chromosome |
| NG tube | nasogastric tube |
| EEG | electroencephalogram |
| aCGH | array comparative genomic hybridization |
References
- Jafari-Ghahfarokhi, H.; Moradi-Chaleshtori, M.; Liehr, T.; Hashemzadeh-Chaleshtori, M.; Teimori, H.; Ghasemi-Dehkordi, P. Small supernumerary marker chromosomes and their correlation with specific syndromes. Adv. Biomed. Res. 2015, 4, 140. [Google Scholar]
- Liehr, T.; Cirkovic, S.; Lalic, T.; Guc-Scekic, M.; De Almeida, C.; Weimer, J.; Iourov, I.; Melaragno, M.I.; Guilherme, R.S.; Stefanou, E.-G.G.; et al. Complex small supernumerary marker chromosomes—An update. Mol. Cytogenet. 2013, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Stohler, R.; Kucharski, E.; Farrow, E.; Delk, P.; Thurston, V.C.; Vance, G.H.; Torres-Martinez, W. A case of de novo partial tetrasomy of distal 6p and review of the literature. Am. J. Med. Genet. A 2007, 143A, 1978–1983. [Google Scholar] [CrossRef] [PubMed]
- Sivasankaran, A.; Murthy, K.; Oruganti, V.P.; Deenadayalu, A.; Samuel, C.R.; Kandukuri, L.R. De-novo ‘pure’ partial trisomy (6)(p22.3 → pter): A case report and review of the literature. Clin. Dysmorphol. 2017, 26, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Breuning, M.H.; Bijlsma, J.B.; de France, H.F. Partial trisomy 6p due to familial translocation t (6; 20) (p21; p13). A new syndrome? Hum. Genet. 1977, 38, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Engelen, J.J.M.; Marcelis, C.L.M.; Alofs, M.G.P.; Loneus, W.H.; Pulles-Heintzberger, C.F.M.; Hamers, A.J.H. De novo “pure” partial trisomy (6)(p22.1 → pter) in a chromosome 15 with an enlarged satellite, identified by microdissection. Am. J. Med. Genet. 2001, 99, 48–53. [Google Scholar] [CrossRef]
- Petković, I.; Barišić, I.; Bastić, M.; Hećimović, S.; Bago, R. Paternal origin of der (X) t (X; 6) in a girl with trisomy 6p and unbalanced t (6; 10) mosaicism in her mother. Am. J. Med. Genet. 2003, 120, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.A.; Klaassens, M.; Holder, A.M.; Lally, K.P.; Fernandes, C.; Galjaard, R.-J.; Tibboel, D.; De Klein, A.; Lee, B. Genome-wide oligonucleotide-based array comparative genome hybridization analysis of non-isolated congenital diaphragmatic hernia. Hum. Mol. Genet. 2007, 16, 424–430. [Google Scholar] [CrossRef]
- Kerrigan, J.F.; Kruer, M.C.; Corneveaux, J.; Panganiban, C.B.; Itty, A.; Reiman, D.; Ng, Y.-T.; Stephan, D.A.; Craig, D.W. Chromosomal abnormality at 6p25.1–25.3 identifies a susceptibility locus for hypothalamic hamartoma associated with epilepsy. Epilepsy Res. 2007, 75, 70–73. [Google Scholar] [CrossRef]
- Castiglione, A.; Guaran, V.; Astolfi, L.; Orioli, E.; Zeri, G.; Gemmati, D.; Bovo, R.; Montaldi, A.; Alghisi, A.; Martini, A. Karyotype-Phenotype Correlation in Partial Trisomies of the Short Arm of Chromosome 6: A Family Case Report and Review of the Literature. Cytogenet. Genome Res. 2013, 141, 243–259. [Google Scholar] [CrossRef]
- Chen, C.P.; Chen, M.; Chen, C.Y.; Chern, S.R.; Wu, P.S.; Chang, S.P.; Kuo, Y.L.; Chen, W.L.; Pan, C.W.; Wang, W. Prenatal diagnosis and molecular cytogenetic characterization of de novo pure partial trisomy 6p associated with microcephaly, craniosynostosis and abnormal maternal serum biochemistry. Gene 2014, 536, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Jankauskienė, A.; Koczkowska, M.; Bjerre, A.; Bernaciak, J.; Schaefer, F.; Lipska-Ziętkiewicz, B.S. Glomerulopathy in patients with distal duplication of chromosome 6p. BMC Nephrol. 2016, 17, 32. [Google Scholar]
- Fontana, P.; Tortora, C.; Petillo, R.; Malacarne, M.; Cavani, S.; Miniero, M.; D’Ambrosio, P.; De Brasi, D.; Pisanti, M.A. Brachydactyly type E in an Italian family with 6p25 trisomy. Eur. J. Med. Genet. 2017, 60, 195–199. [Google Scholar] [CrossRef]
- Türkyılmaz, A.; Cimbek, E.A.; Çebi, A.H.; Acar Arslan, E.; Karagüzel, G. De novo Pure Partial Trisomy 6p Associated with Facial Dysmorphism, Developmental Delay, Brain Anomalies, and Primary Congenital Hypothyroidism. Mol. Syndromol. 2022, 1–9. [Google Scholar] [CrossRef]
- Jang, W.; Chae, H.; Kim, J.; Son, J.; Kim, S.C.; Koo, B.K.; Kim, M.; Kim, Y.; Park, I.Y.; Sung, I.K. Identification of small marker chromosomes using microarray comparative genomic hybridization and multicolor fluorescent in situ hybridization. Mol. Cytogenet. 2016, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Liehr, T. Small Supernumerary Marker Chromosomes. 2022. Available online: http://cs-tl.de/DB/CA/sSMC/0-Start.html (accessed on 22 August 2022).
- De Blois, M.S.D.; Bachelot, A.; Ozilou, C.; Chevallier, S.; Waill-Perrier, M.C.; Romana, S.; Vekemans, M.; Turleau, C. Stable ring chromosome 6 due to a centromeric fission. In Proceedings of the 54th Annual Meeting of the American Society of Human Genetics, Toronto, ON, Canada, 26–30 October 2004; p. 175. [Google Scholar]
- Voullaire, L.E.; Slater, H.R.; Petrovic, V.; Choo, K.H. A functional marker centromere with no detectable alpha-satellite, satellite III, or CENP-B protein: Activation of a latent centromere? Am. J. Hum. Genet. 1993, 52, 1153–1163. [Google Scholar] [PubMed]
- Amor, D.J.; Choo, K.H. Neocentromeres: Role in human disease, evolution, and centromere study. Am. J. Hum. Genet. 2002, 71, 695–714. [Google Scholar] [CrossRef] [PubMed]
- Marshall, O.J.; Chueh, A.C.; Wong, L.H.; Choo, K.H. Neocentromeres: New insights into centromere structure, disease development, and karyotype evolution. Am. J. Hum. Genet. 2008, 82, 261–282. [Google Scholar] [CrossRef]
- Wang, X.; Liu, X.; Li, Y.; Yang, B.; Sun, X.; Yang, P.; Zhong, Z.; Chen, J. Identification and functional study of FOXC1 variants in Chinese families with glaucoma. Am. J. Med. Genet. A 2022, 188, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, O.J.; Ebenezer, N.D.; Jordan, T.; Fox, M.; Ocaka, L.; Payne, A.; Leroy, B.P.; Clark, B.J.; Hitchings, R.A.; Povey, S.; et al. Chromosomal duplication involving the forkhead transcription factor gene FOXC1 causes iris hypoplasia and glaucoma. Am. J. Hum. Genet. 2000, 67, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, D.Y.; Searby, C.C.; Alward, W.L.; Walton, D.; Craig, J.E.; Mackey, D.A.; Kawase, K.; Kanis, A.B.; Patil, S.R.; Stone, E.M.; et al. A spectrum of FOXC1 mutations suggests gene dosage as a mechanism for developmental defects of the anterior chamber of the eye. Am. J. Hum. Genet. 2001, 68, 364–372. [Google Scholar] [CrossRef]
- Lehmann, O.J.; Ebenezer, N.D.; Ekong, R.; Ocaka, L.; Mungall, A.J.; Fraser, S.; McGill, J.I.; Hitchings, R.A.; Khaw, P.T.; Sowden, J.C.; et al. Ocular developmental abnormalities and glaucoma associated with interstitial 6p25 duplications and deletions. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1843–1849. [Google Scholar]
- Chanda, B.; Asai-Coakwell, M.; Ye, M.; Mungall, A.; Barrow, M.; Dobyns, W.; Behesti, H.; Sowden, J.; Carter, N.P.; Walter, M.A.; et al. A novel mechanistic spectrum underlies glaucoma-associated chromosome 6p25 copy number variation. Hum. Mol. Genet. 2008, 17, 3446–3458. [Google Scholar] [CrossRef]
- Weh, E.; Reis, L.M.; Happ, H.C.; Levin, A.V.; Wheeler, P.G.; David, K.L.; Carney, E.; Angle, B.; Hauser, N.; Semina, E.V. Whole exome sequence analysis of Peters anomaly. Hum. Genet. 2014, 133, 1497–1511. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Jiang, W.; Phillips, F.M.; Haydon, R.C.; Peng, Y.; Zhou, L.; Luu, H.H.; An, N.; Breyer, B.; Vanichakarn, P.; et al. Osteogenic Activity of the Fourteen Types of Human Bone Morphogenetic Proteins (BMPs). J. Bone Jt. Surg. 2003, 85, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Watanabe, T.; Kyozuka, H.; Yamaguchi, A.; Sato, K.; Sato, M.; Go, H.; Fujimori, K. Perinatal diagnosis of a fetus with an unbalanced translocation 46, XY, der (10) t (6; 10) (p22; q26.1) with multiple malformations:a case report and literature review. Fukushima J. Med. Sci. 2021, 67, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Phelan, M.C.; Albiez, K.; Stevenson, R.E. Trisomy 6p due to a tandem duplication. Proc. Greenwood Genet. Ctr. 1986, 5, 39–43. [Google Scholar] [CrossRef]




| No. | Sex/ Age | Inheritance | Karyotype and Grade of Mosaicism | Result of SMC | Type of Rearrangement | Reference on sSMC Database [17] |
|---|---|---|---|---|---|---|
| 1 | F/ 1m | dn | 47,XX, + r [37]/ 46,XX [13] | r(6)(::p21.2→q10) | Interstitial deletion + r formation | 06-W-p21.2/1-1 |
| 2 | F/ prenatal | dn | 47,XY, + mar [12]/ 46,XY [3] | r(6)(::p21.2→q11.2::) | Interstitial deletion + r formation | 06-W-p21.2/2-1 |
| 3 | M/ 14y | NA | 47,XY, + mar [12]/ 46,XY [3] | r(6)(p21.2q12) | Interstitial deletion + r formation | 06-W-p21.2/2-2 |
| 4 | M/ 1m | dn | 47,XY, + mar [13]/ 46,XY [7] | min(6)(:p21.1→q11:) | Interstitial deletion | 06-W-p21.1/1-1 |
| 5 | M/ 16y | NA | 48,XY, + mar, + mar [8]/ 47,XY, + mar [10]/ 46,XY [3] | See note 1 below | Interstitial deletion/Interstitial deletion + r formation | 06-W-p12.3/1-1 |
| 6 | M/ 2y | NA | 47,XY, + r [17]/ 48,XY, + rx2[3] | r(6)(p12.3q12) | Interstitial deletion + r formation | 06-W-p12.3/2-1 |
| 7 | M/ 16y | NA | 47,XY, + mar [60%]/ 46,XY [40%] | See note 2 below | Interstitial deletion/Interstitial deletion + Inv dup | 06-W-p11.2/1-1 |
| 8 | M/ 2y | dn | 48,XY, + marx2[59]/47,XY, + mar [25]/ 46,XY [40] | min(6)(:p11.1→q12:) | Interstitial deletion | 06-W-p11.1/1-1 |
| 9 | F/ 12y | NA | 47,XX, + mar [17]/ 46,XX [3] | See note 3 below | Interstitial deletion/Interstitial deletion + r formation/Interstitial deletion + Inv dup | 06-W-p11.1/2-1 |
| No | Sex/ Age | Inheritance | Karyotype and Grade of Mosaicism | Result of SMC | Type of Rearrangement | Reference on sSMC Database [17] |
|---|---|---|---|---|---|---|
| 1 | M/ adult | NA | 47,XY,t(4;15), del(6)(q16.2q22.2), + r(6)(::q16.2→q22.2::)[100%] | r(6)(::q16.2→q22.2::) | r formation | 06-N-q16.2/1-1 |
| 2 | M/ 1y | NA | 47,XY, + mar [100%] | r(6)(::q24→q25::) | r formation | 06-N-q24/1-1 |
| 3 | NA./ prenatal | dn | 47, + mar [60%]/ 46[40%] mar not in fetal blood but in placenta | inv dup(6)(qter→q26: :q26→qter) | Terminal deletion + Inv dup | 06-N-qt26/1-1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Syu, Y.-M.; Ma, J.-Y.; Ou, T.-H.; Lee, C.-L.; Lin, H.-Y.; Lin, S.-P.; Lee, C.-J.; Chen, C.-P. De Novo Mosaic 6p23-p25.3 Tetrasomy Caused by a Small Supernumerary Marker Chromosome Presenting Trisomy Distal 6p Phenotype: A Case Report and Literature Review. Diagnostics 2022, 12, 2306. https://doi.org/10.3390/diagnostics12102306
Syu Y-M, Ma J-Y, Ou T-H, Lee C-L, Lin H-Y, Lin S-P, Lee C-J, Chen C-P. De Novo Mosaic 6p23-p25.3 Tetrasomy Caused by a Small Supernumerary Marker Chromosome Presenting Trisomy Distal 6p Phenotype: A Case Report and Literature Review. Diagnostics. 2022; 12(10):2306. https://doi.org/10.3390/diagnostics12102306
Chicago/Turabian StyleSyu, Yu-Min, Juine-Yih Ma, Tzu-Hsuen Ou, Chung-Lin Lee, Hsiang-Yu Lin, Shuan-Pei Lin, Chia-Jung Lee, and Chih-Ping Chen. 2022. "De Novo Mosaic 6p23-p25.3 Tetrasomy Caused by a Small Supernumerary Marker Chromosome Presenting Trisomy Distal 6p Phenotype: A Case Report and Literature Review" Diagnostics 12, no. 10: 2306. https://doi.org/10.3390/diagnostics12102306
APA StyleSyu, Y.-M., Ma, J.-Y., Ou, T.-H., Lee, C.-L., Lin, H.-Y., Lin, S.-P., Lee, C.-J., & Chen, C.-P. (2022). De Novo Mosaic 6p23-p25.3 Tetrasomy Caused by a Small Supernumerary Marker Chromosome Presenting Trisomy Distal 6p Phenotype: A Case Report and Literature Review. Diagnostics, 12(10), 2306. https://doi.org/10.3390/diagnostics12102306