

Proteomic Analysis of Pleural Effusions from COVID-19 Deceased Patients: Enhanced Inflammatory Markers

 , and
, and
Abstract
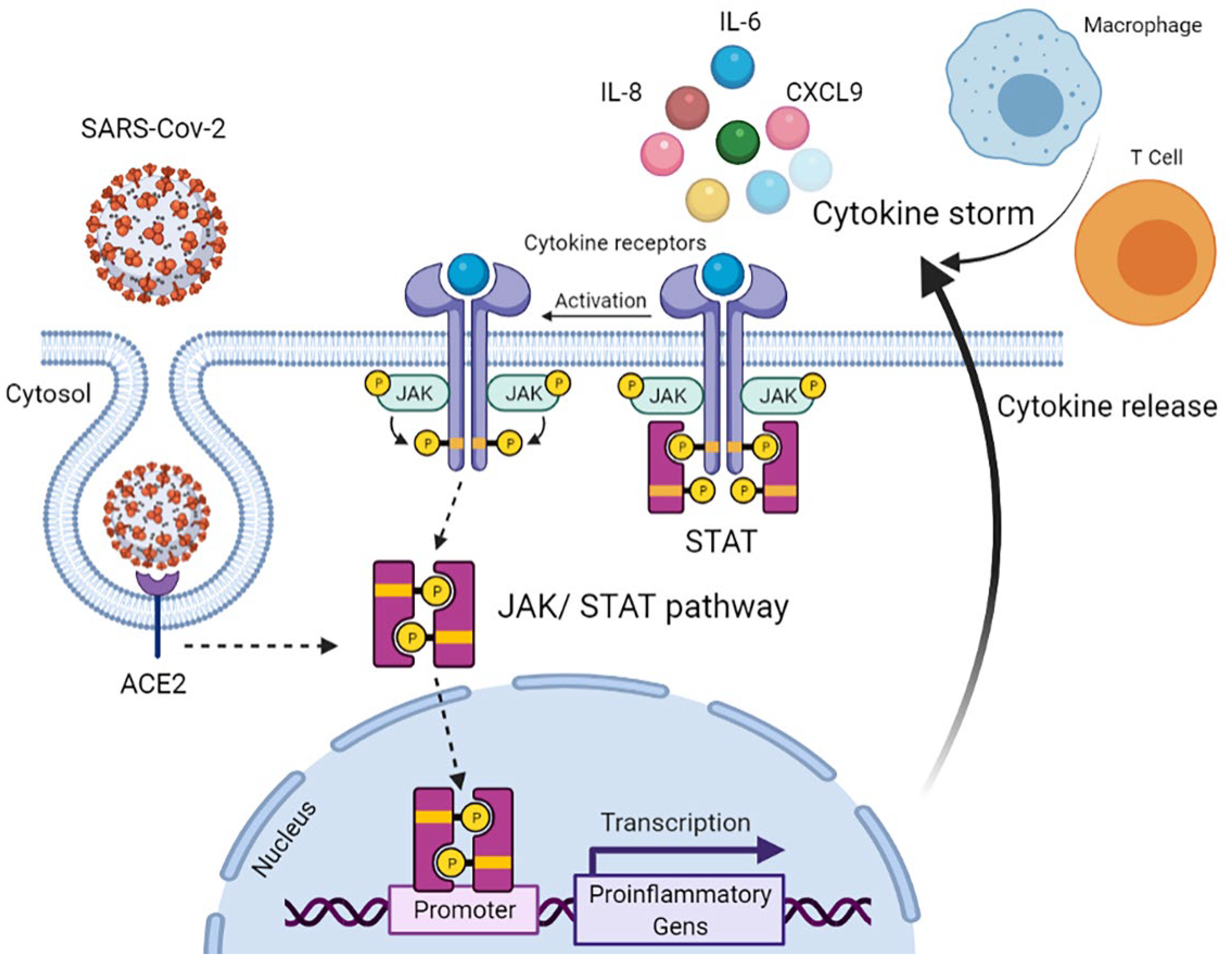
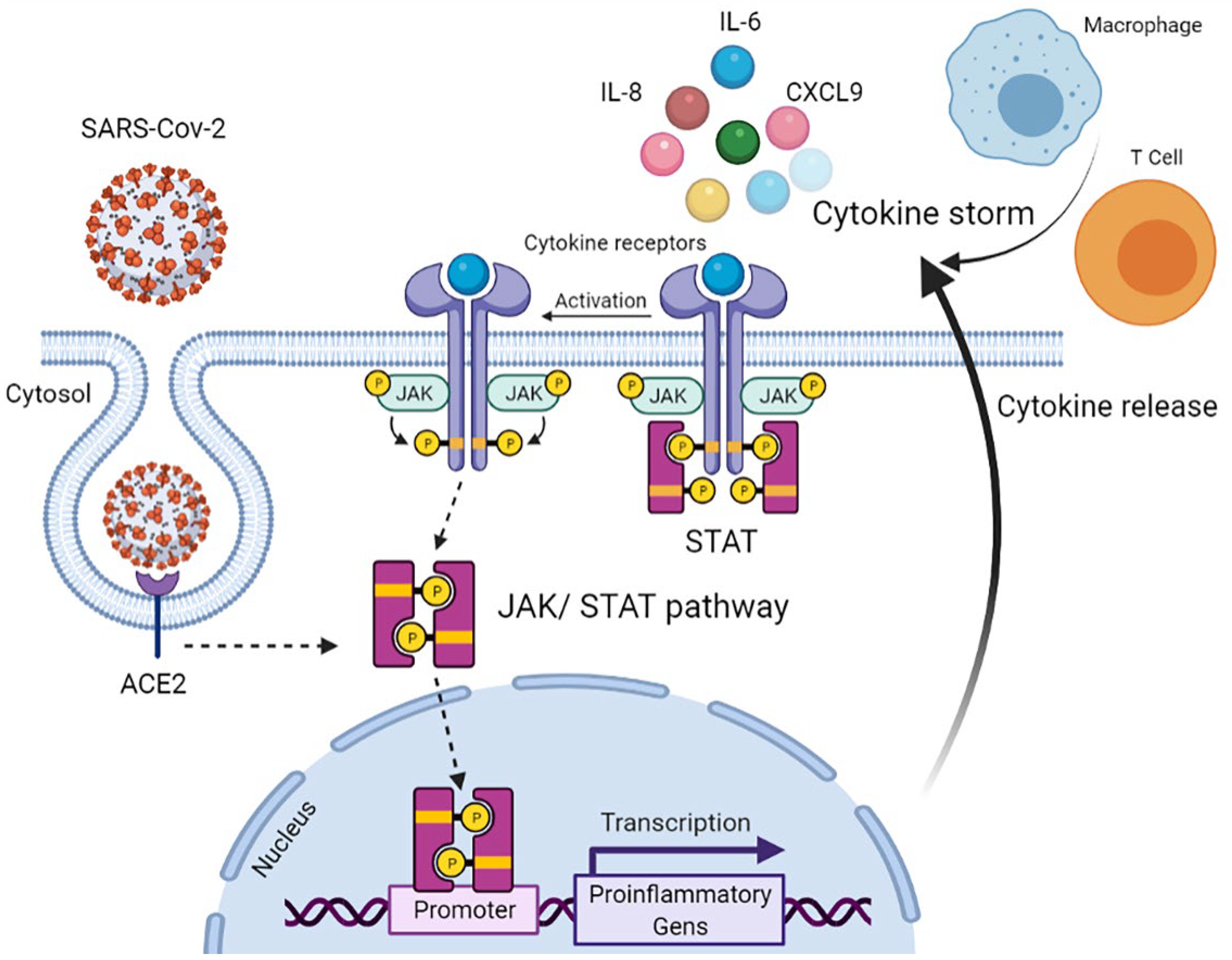
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Rapid Autopsy and Sample Preparation
2.3. Proteomic Analysis: Olink® Proximity Extension Assay (PEA)
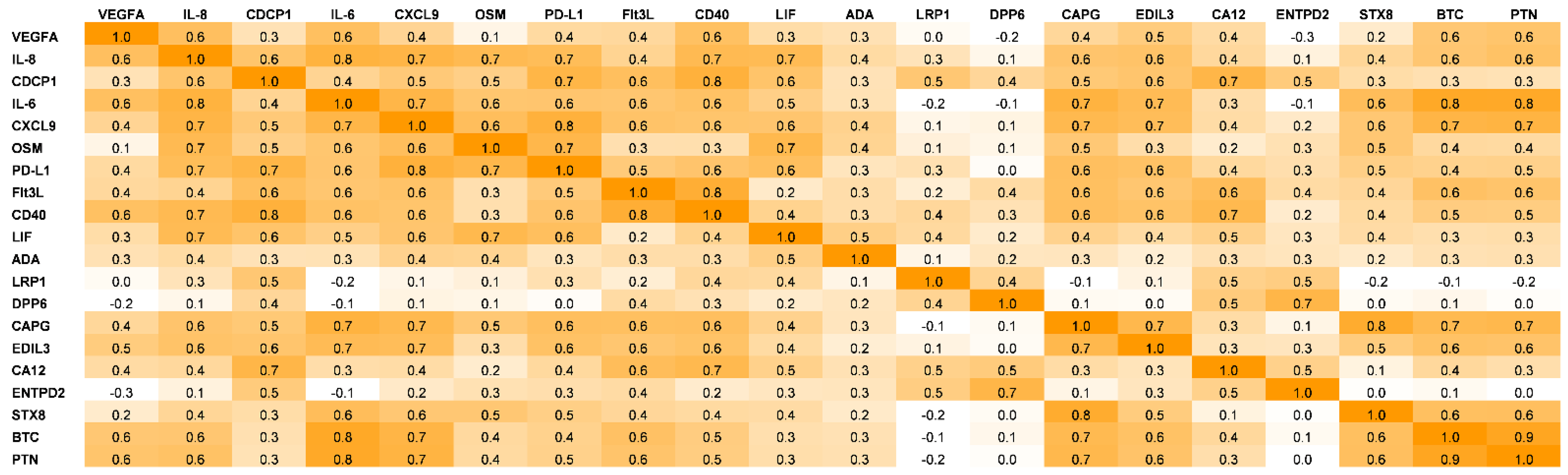
2.4. Pearson’s Correlations Coefficient
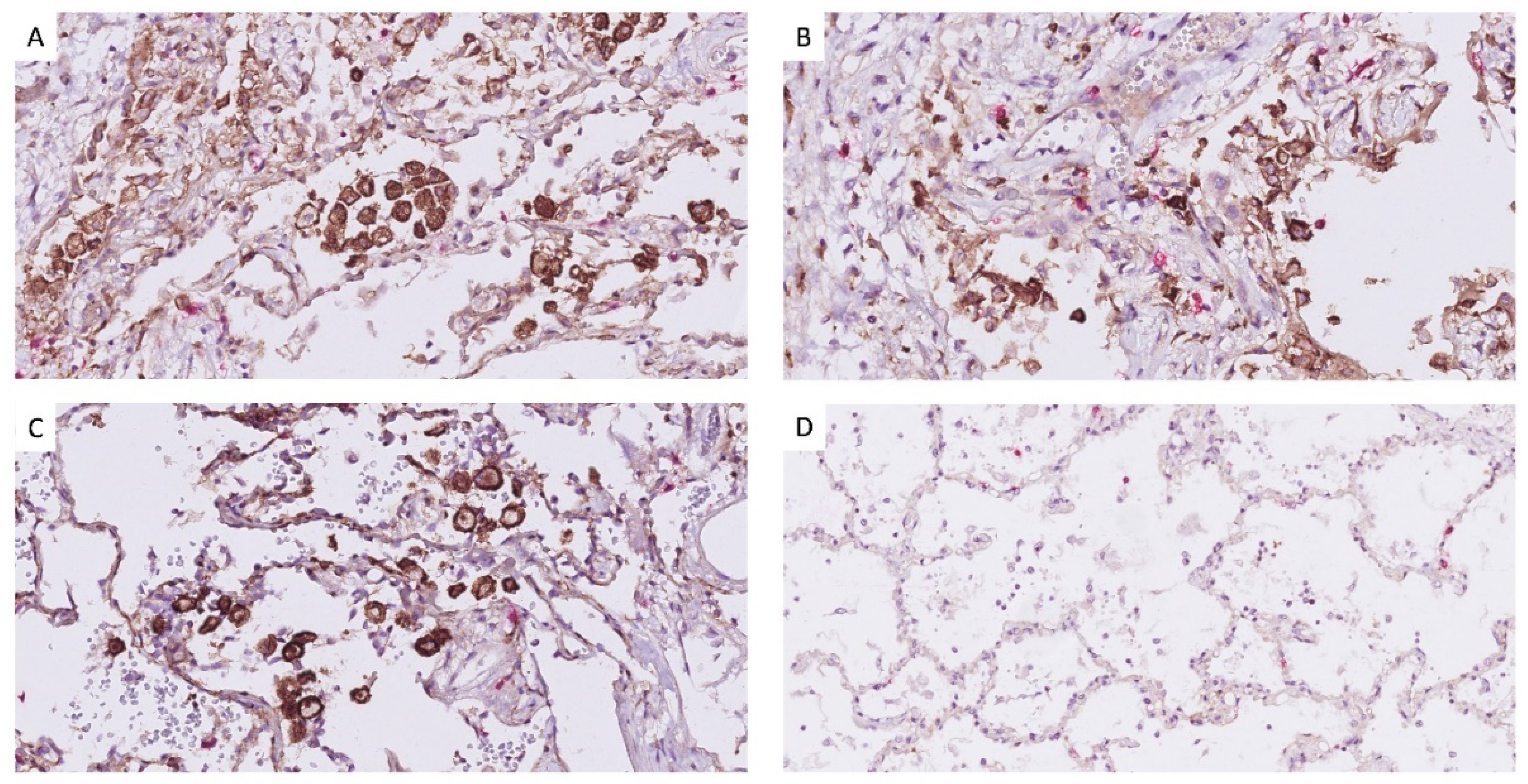
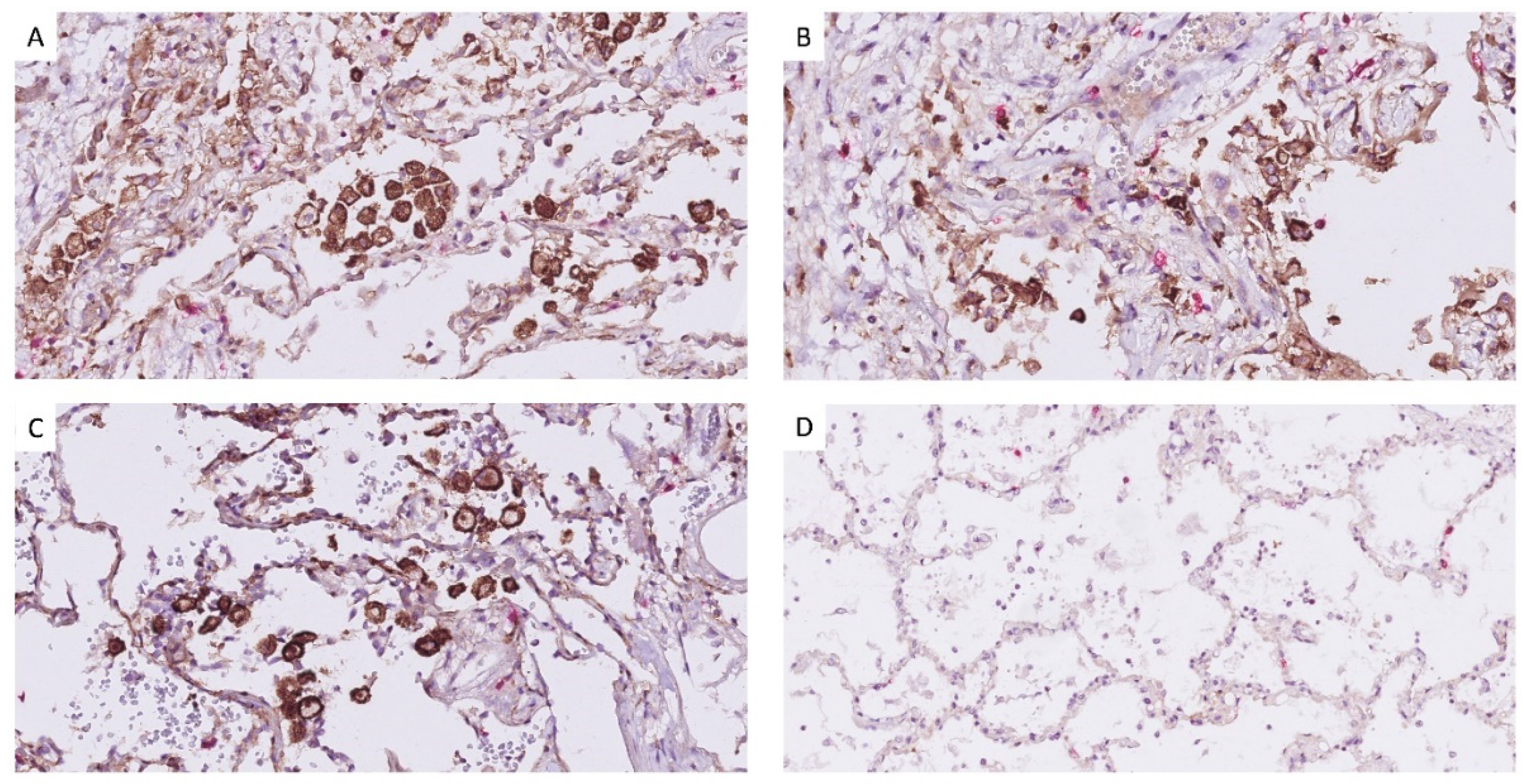
2.5. Histology Processing and Immuno-Histochemistry
2.6. Statistical Analysis
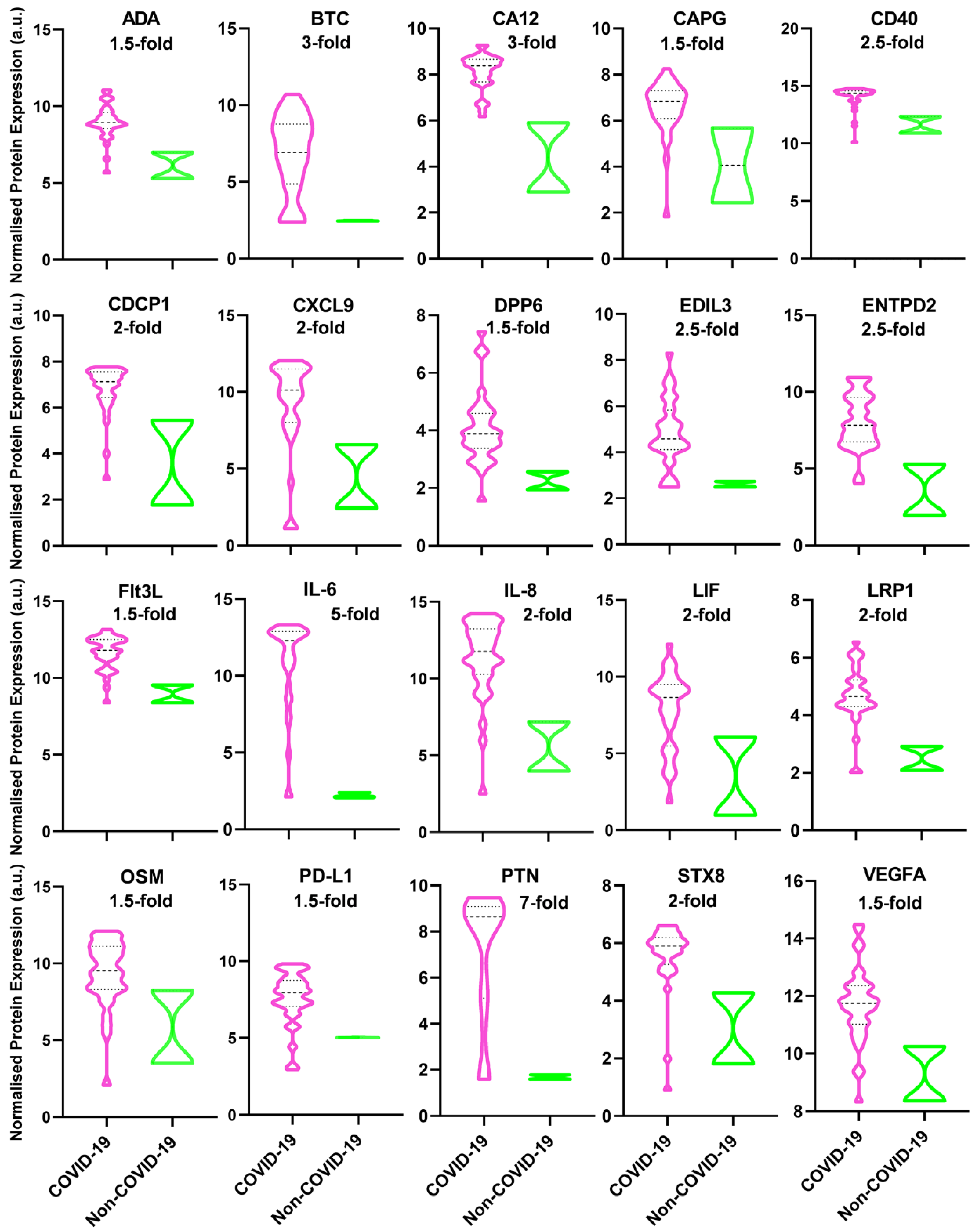
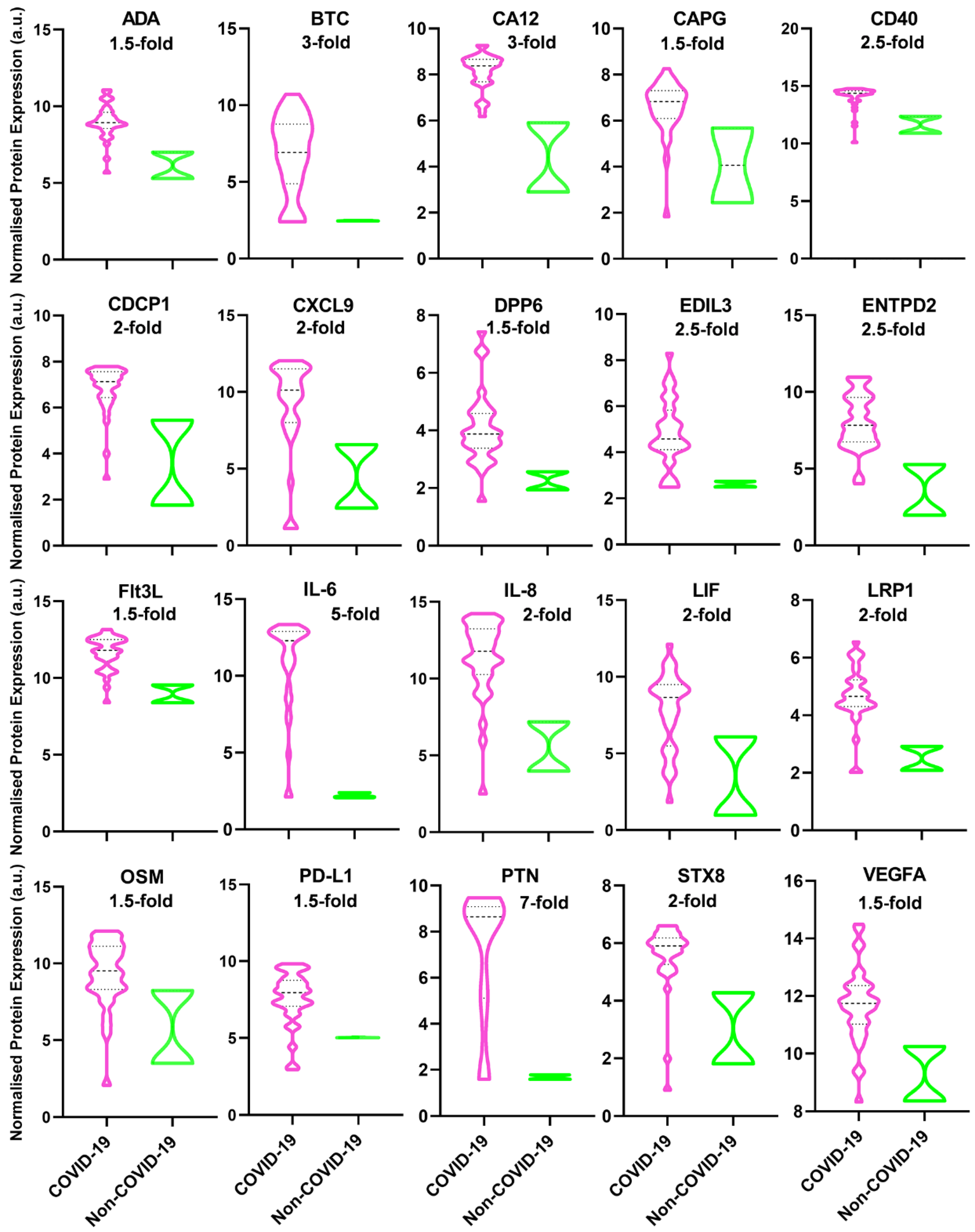
3. Results
4. Discussion
Limitations of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Forchette, L.; Sebastian, W.; Liu, T. A Comprehensive Review of COVID-19 Virology, Vaccines, Variants, and Therapeutics. Curr. Med. Sci. 2021, 41, 1037–1051. [Google Scholar] [CrossRef] [PubMed]
- de la Rica, R.; Borges, M.; Gonzalez-Freire, M. COVID-19: In the Eye of the Cytokine Storm. Front. Immunol. 2020, 11, 558898. [Google Scholar] [CrossRef] [PubMed]
- Melo, A.K.G.; Milby, K.M.; Caparroz, A.; Pinto, A.; Santos, R.R.P.; Rocha, A.P.; Ferreira, G.A.; Souza, V.A.; Valadares, L.D.A.; Vieira, R.; et al. Biomarkers of cytokine storm as red flags for severe and fatal COVID-19 cases: A living systematic review and meta-analysis. PLoS ONE 2021, 16, e0253894. [Google Scholar] [CrossRef] [PubMed]
- Mélo Silva Júnior, M.L.d.; Souza, L.M.A.d.; Dutra, R.E.M.C.; Valente, R.G.d.M.; Melo, T.S. Review on therapeutic targets for COVID-19: Insights from cytokine storm. Postgrad. Med. J. 2020, 97, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Saad, R.; Taylor, E.W.; Rayman, M.P. Selenium and selenoproteins in viral infection with potential relevance to COVID-19. Redox Biol. 2020, 37, 101715. [Google Scholar] [CrossRef] [PubMed]
- Satarker, S.; Tom, A.A.; Shaji, R.A.; Alosious, A.; Luvis, M.; Nampoothiri, M. JAK-STAT Pathway Inhibition and their Implications in COVID-19 Therapy. Postgrad. Med. 2021, 133, 489–507. [Google Scholar] [CrossRef]
- Yarmol-Matusiak, E.A.; Cipriano, L.E.; Stranges, S. A comparison of COVID-19 epidemiological indicators in Sweden, Norway, Denmark, and Finland. Scand. J. Public Health 2021, 49, 69–78. [Google Scholar] [CrossRef]
- Szekely, L.; Bozoky, B.; Bendek, M.; Ostad, M.; Lavignasse, P.; Haag, L.; Wu, J.; Jing, X.; Gupta, S.; Saccon, E.; et al. Pulmonary stromal expansion and intra-alveolar coagulation are primary causes of COVID-19 death. Heliyon 2021, 7, e07134. [Google Scholar] [CrossRef]
- Akarsu, S.; Kurt, A.N.; Dogan, Y.; Yilmaz, E.; Godekmerdan, A.; Aygun, A.D. The differential diagnostic values of cytokine levels in pleural effusions. Mediat. Inflamm. 2005, 2005, 2–8. [Google Scholar] [CrossRef]
- Wei, X.S.; Wang, X.; Ye, L.L.; Niu, Y.R.; Peng, W.B.; Wang, Z.H.; Zhang, J.C.; Zhou, Q. Pleural effusion as an indicator for the poor prognosis of COVID-19 patients. Int. J. Clin. Pract. 2021, 75, e14123. [Google Scholar] [CrossRef]
- Assarsson, E.; Lundberg, M.; Holmquist, G.; Bjorkesten, J.; Thorsen, S.B.; Ekman, D.; Eriksson, A.; Rennel Dickens, E.; Ohlsson, S.; Edfeldt, G.; et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 2014, 9, e95192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filbin, M.R.; Mehta, A.; Schneider, A.M.; Kays, K.R.; Guess, J.R.; Gentili, M.; Fenyves, B.G.; Charland, N.C.; Gonye, A.L.K.; Gushterova, I.; et al. Longitudinal proteomic analysis of severe COVID-19 reveals survival-associated signatures, tissue-specific cell death, and cell-cell interactions. Cell Rep. Med. 2021, 2, 100287. [Google Scholar] [CrossRef] [PubMed]
- Tay, T.R.; Tee, A. Factors affecting pleural fluid adenosine deaminase level and the implication on the diagnosis of tuberculous pleural effusion: A retrospective cohort study. BMC Infect. Dis. 2013, 13, 546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Wang, L.; Wang, B.; Cretoiu, S.M.; Wang, Q.; Wang, X.; Chen, C. Regulatory mechanisms of betacellulin in CXCL8 production from lung cancer cells. J. Transl. Med. 2014, 12, 70. [Google Scholar] [CrossRef] [Green Version]
- Deniz, S.; Uysal, T.K.; Capasso, C.; Supuran, C.T.; Ozensoy Guler, O. Is carbonic anhydrase inhibition useful as a complementary therapy of Covid-19 infection? J. Enzym. Inhib. Med. Chem. 2021, 36, 1230–1235. [Google Scholar] [CrossRef]
- Heng, P. Characterisation of Macrophage Capping Protein as a Novel Inflammatory Mediator. Ph.D. Thesis, University of Melbourne, Melbourne, Australia, 2017. [Google Scholar]
- Rolla, R.; Puricelli, C.; Bertoni, A.; Boggio, E.; Gigliotti, C.L.; Chiocchetti, A.; Cappellano, G.; Dianzani, U. Platelets: ‘multiple choice’ effectors in the immune response and their implication in COVID-19 thromboinflammatory process. Int. J. Lab. Hematol. 2021, 43, 895–906. [Google Scholar] [CrossRef]
- Lun, Y.; Borjini, N.; Miura, N.N.; Ohno, N.; Singer, N.G.; Lin, F. CDCP1 on Dendritic Cells Contributes to the Development of a Model of Kawasaki Disease. J. Immunol. 2021, 206, 2819–2827. [Google Scholar] [CrossRef]
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32. [Google Scholar] [CrossRef]
- Postema, P.G.; Ten Sande, J.N.; Christiaans, I.; Ling, X.; Alders, M.; Boekholdt, M.; Varro, A.; Nattel, S.; Bezzina, C.R.; Wilde, A.A.M. Characterisation of familial idiopathic ventricular fibrillation linked to DPP6. Eur. Heart J. 2013, 34, 4559. [Google Scholar] [CrossRef]
- Consiglio, C.R.; Cotugno, N.; Sardh, F.; Pou, C.; Amodio, D.; Rodriguez, L.; Tan, Z.; Zicari, S.; Ruggiero, A.; Pascucci, G.R.; et al. The Immunology of Multisystem Inflammatory Syndrome in Children with COVID-19. Cell 2020, 183, 968–981.e7. [Google Scholar] [CrossRef]
- Feldbrügge, L.; Moss, A.C.; Yee, E.U.; Csizmadia, E.; Mitsuhashi, S.; Longhi, M.S.; Sandhu, B.; Stephan, H.; Wu, Y.; Cheifetz, A.S.; et al. Expression of Ecto-nucleoside Triphosphate Diphosphohydrolases-2 and -3 in the Enteric Nervous System Affects Inflammation in Experimental Colitis and Crohn’s Disease. J. Crohn’s Colitis 2017, 11, 1113–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shortman, K.; Naik, S.H. Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 2007, 7, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Fu, B.; Wei, H. IL-6 modulation for COVID-19: The right patients at the right time? J. Immunother. Cancer 2021, 9, e002285. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, J.; Gao, M.; Fan, H.; Wang, Y.; Xu, X.; Chen, C.; Liu, J.; Kim, J.; Aliyari, R.; et al. Interleukin-8 as a Biomarker for Disease Prognosis of Coronavirus Disease-2019 Patients. Front. Immunol. 2020, 11, 602395. [Google Scholar] [CrossRef]
- Foronjy, R.F.; Dabo, A.J.; Cummins, N.; Geraghty, P. Leukemia inhibitory factor protects the lung during respiratory syncytial viral infection. BMC Immunol. 2014, 15, 41. [Google Scholar] [CrossRef] [Green Version]
- Potere, N.; Del Buono, M.G.; Mauro, A.G.; Abbate, A.; Toldo, S. Low Density Lipoprotein Receptor-Related Protein-1 in Cardiac Inflammation and Infarct Healing. Front. Cardiovasc. Med. 2019, 6, 51. [Google Scholar] [CrossRef]
- Arunachalam, P.S.; Wimmers, F.; Mok, C.K.P.; Perera, R.; Scott, M.; Hagan, T.; Sigal, N.; Feng, Y.; Bristow, L.; Tak-Yin Tsang, O.; et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 2020, 369, 1210–1220. [Google Scholar] [CrossRef]
- Chen, J.Z.; Vitetta, L. Increased PD-L1 Expression May Be Associated With the Cytokine Storm and CD8(+) T-Cell Exhaustion in Severe COVID-19. J. Infect. Dis. 2021, 223, 1659–1660. [Google Scholar] [CrossRef]
- Wang, X. Chapter Three—Pleiotrophin: Activity and mechanism. In Advances in Clinical Chemistry; Makowski, G.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 98, pp. 51–89. [Google Scholar]
- Bhat, S.S.; Friedmann, K.S.; Knorck, A.; Hoxha, C.; Leidinger, P.; Backes, C.; Meese, E.; Keller, A.; Rettig, J.; Hoth, M.; et al. Syntaxin 8 is required for efficient lytic granule trafficking in cytotoxic T lymphocytes. Biochim. Biophys. Acta 2016, 1863, 1653–1664. [Google Scholar] [CrossRef]
- Pang, J.; Xu, F.; Aondio, G.; Li, Y.; Fumagalli, A.; Lu, M.; Valmadre, G.; Wei, J.; Bian, Y.; Canesi, M.; et al. Efficacy and tolerability of bevacizumab in patients with severe Covid-19. Nat. Commun. 2021, 12, 814. [Google Scholar] [CrossRef]
- Correll, K.A.; Edeen, K.E.; Zemans, R.L.; Redente, E.F.; Serban, K.A.; Curran-Everett, D.; Edelman, B.L.; Mikels-Vigdal, A.; Mason, R.J. Transitional human alveolar type II epithelial cells suppress extracellular matrix and growth factor gene expression in lung fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L283–L294. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, S.; Xia, H.; Shi, D.; Chen, Y.; Zheng, S.; Chen, Y.; Gao, H.; Guo, F.; Ji, Z.; et al. Cytokine Signature Associated With Disease Severity in COVID-19. Front. Immunol. 2021, 12, 681516. [Google Scholar] [CrossRef]
- Metcalfe, S.M. COVID-19 lockdown: De-risking exit by protecting the lung with leukaemia inhibitory factor (LIF). Med. Drug Discov. 2020, 6, 100043. [Google Scholar] [CrossRef]
- Campo, G.; Contoli, M.; Fogagnolo, A.; Vieceli Dalla Sega, F.; Zucchetti, O.; Ronzoni, L.; Verri, M.; Fortini, F.; Pavasini, R.; Morandi, L.; et al. Over time relationship between platelet reactivity, myocardial injury and mortality in patients with SARS-CoV-2-associated respiratory failure. Platelets 2021, 32, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Sabbatino, F.; Conti, V.; Franci, G.; Sellitto, C.; Manzo, V.; Pagliano, P.; De Bellis, E.; Masullo, A.; Salzano, F.A.; Caputo, A.; et al. PD-L1 Dysregulation in COVID-19 Patients. Front. Immunol. 2021, 12, 695242. [Google Scholar] [CrossRef]
- Vivarelli, S.; Falzone, L.; Torino, F.; Scandurra, G.; Russo, G.; Bordonaro, R.; Pappalardo, F.; Spandidos, D.A.; Raciti, G.; Libra, M. Immune-checkpoint inhibitors from cancer to COVID19: A promising avenue for the treatment of patients with COVID-19 (Review). Int. J. Oncol. 2021, 58, 145–157. [Google Scholar] [CrossRef]
- Zhang, C.; Li, Z.; Xu, L.; Che, X.; Wen, T.; Fan, Y.; Li, C.; Wang, S.; Cheng, Y.; Wang, X.; et al. CXCL9/10/11, a regulator of PD-L1 expression in gastric cancer. BMC Cancer 2018, 18, 462. [Google Scholar] [CrossRef] [Green Version]
- Stroeder, R.; Walch-Ruckheim, B.; Fischbach, J.; Juhasz-Boss, I.; Rube, C.; Solomayer, E.F.; Smola, S. Oncostatin M treatment increases the responsiveness toward cisplatin-based chemoradiotherapy in cervical cancer cells in a STAT3-dependent manner. Oncol. Lett. 2018, 16, 3351–3358. [Google Scholar] [CrossRef]
- Huang, Q.; Duan, L.; Qian, X.; Fan, J.; Lv, Z.; Zhang, X.; Han, J.; Wu, F.; Guo, M.; Hu, G.; et al. IL-17 Promotes Angiogenic Factors IL-6, IL-8, and Vegf Production via Stat1 in Lung Adenocarcinoma. Sci. Rep. 2016, 6, 36551. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Li, Y.X.; Jiang, L.J.; Chen, Q.; Wang, T.; Ye, D.W. Targeting JAK-STAT Signaling to Control Cytokine Release Syndrome in COVID-19. Trends Pharmacol. Sci. 2020, 41, 531–543. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Pascale, R.M.; Feo, F. Epidermal growth factor-like repeats and discoidin I-like domains 3: A multifaceted oncoprotein at the crossroad of MAPK and TGF-beta pathways in human hepatocellular carcinoma. Transl. Cancer Res. 2016, 5, 103–109. [Google Scholar] [CrossRef]
- Solanilla, A.; Dechanet, J.; El Andaloussi, A.; Dupouy, M.; Godard, F.; Chabrol, J.; Charbord, P.; Reiffers, J.; Nurden, A.T.; Weksler, B.; et al. CD40-ligand stimulates myelopoiesis by regulating flt3-ligand and thrombopoietin production in bone marrow stromal cells. Blood 2000, 95, 3758–3764. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Protein Name | Function |
|---|---|---|
| ADA | Adenosine deaminase | An enzyme converts adenosine to inosine, vital for the differentiation of lymphoid cells while showing enhanced activity in diseases wherein immunity is stimulated [13]. |
| BTC | Betacellulin | A member of the EGF family binds to ErbB1 and ErbB4 homodimers, activating the EGFR–PI3K–Akt–Erk pathway to form an inflammatory microenvironment and produce IL-8 [14]. |
| CA12 | Carbonic anhydrase 12 | An enzyme catalyzes the hydration of carbon dioxide to bicarbonate and H+ ions, involved in many physiological processes. The similarity between high-altitude pulmonary oedema and COVID-19 suggests the possible role of CA in COVID-19, thus proposing the use of CA inhibitors for COVID-19 treatment [15]. |
| CAPG | Macrophage-capping protein | A pro-inflammatory mediator released by macrophage activation subsequently triggers pro-inflammatory cytokine release. The overexpression of CAPG also reveals the ongoing state of inflammatory diseases [16]. |
| CD40 | CD40 | CD40 and CD40L are surface receptors, members of the TNF and TNF receptor superfamilies, respectively. Inducing inflammatory and pro-thrombotic responses. CD40 is expressed on monocytes, macrophages, B, T, NK, and dendritic cells. In addition to platelet activation, CD40/CD40L interaction regulates a variety of cellular and molecular processes involved in innate and adaptive immune responses [17]. |
| CDCP1 | CUB domain-containing protein 1 | Typically, a key regulator of tumour cell survival and metastasis, affecting immune-mediated diseases; however, CDCP1 is one of the most upregulated genes in SARS-CoV-2-infected children with Kawasaki disease [18]. |
| CXCL9 | C-X-C motif chemokine ligand 9 | One of the major chemokines reported being elevated in SARS-CoV infection [19]. |
| DPP6 | Dipeptidyl aminopeptidase-like protein 6 | DPP6 overexpression in Purkinje fibres elicits short-coupled extrasystoles triggering idiopathic ventricular fibrillation [20]. |
| EDIL3 | EGF-like repeat and discoidin I-like domain-containing protein 3 | A glycoprotein in arterial vessel walls which is overexpressed in Kawasaki disease or after vascular injury; also, it inhibits the recruitment and extravasation of inflammatory cells across the endothelium [21]. |
| ENTPD2 | Ectonucleoside triphosphate diphosphohydrolase 2 | Expression of ENTPD2 in the enteric nervous system exacerbates inflammation in inflammatory bowel disease [22]. |
| Flt3L | Fms-related tyrosine kinase 3 ligand | Flt3L functions as both a cytokine and a growth factor, increasing the amount of immune cells, especially dendritic cells and lymphocytes [23]. |
| IL-6 | Interleukin 6 | High IL-6 has been linked consistently to severe COVID-19 cases [24]. |
| IL-8 | Interleukin 8 | A strong biomarker for the severity of COVID-19 and disease prognosis [25]. |
| LIF | Leukaemia inhibitory factor | In viral pneumonia, LIF opposes the cytokine storm in the lungs [26]. |
| LRP1 | Prolow-density lipoprotein receptor-related protein 1 | A modulator of tissue inflammation and organ repair, e.g., the brain, kidney, lung, vasculature, and AMI [27]. |
| OSM | Oncostatin-M | A key regulator of IL-6 and a biomarker for the clinical severity of COVID-19, suggesting the role of bacterial product infection [28]. |
| PD-L1 | Programmed cell death 1 ligand 1 | PD-L1 is overexpressed in monocytes, NK cells, and, particularly, basophils and eosinophils in severe COVID-19 patients. The cytokine storm may be led to increased PD-L1 expression, leading to CD8+ T-cell exhaustion. Blockading PD-L1/PD-1 could recover CD8+ T-cell numbers and functionalities [29]. |
| PTN | Pleiotrophin | A small cationic protein which is associated with bone development, cancer metastasis, inflammation, neural regeneration, and tissue repair [30]. |
| STX8 | Syntaxin-8 | Vesicle trafficking protein is necessary for lytic granule trafficking in cytotoxic T lymphocytes [31]. |
| VEGFA | Vascular Endothelial Growth Factor A | Hypoxia upregulates VEGF expression inducing vascular leakiness in SARS-Cov-2-infected lung tissues, resulting in plasma extravasation and pulmonary oedema, later increasing tissue hypoxia in a vicious cycle [32]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Razaghi, A.; Szakos, A.; Alouda, M.; Bozóky, B.; Björnstedt, M.; Szekely, L. Proteomic Analysis of Pleural Effusions from COVID-19 Deceased Patients: Enhanced Inflammatory Markers. Diagnostics 2022, 12, 2789. https://doi.org/10.3390/diagnostics12112789
Razaghi A, Szakos A, Alouda M, Bozóky B, Björnstedt M, Szekely L. Proteomic Analysis of Pleural Effusions from COVID-19 Deceased Patients: Enhanced Inflammatory Markers. Diagnostics. 2022; 12(11):2789. https://doi.org/10.3390/diagnostics12112789
Chicago/Turabian StyleRazaghi, Ali, Attila Szakos, Marwa Alouda, Béla Bozóky, Mikael Björnstedt, and Laszlo Szekely. 2022. "Proteomic Analysis of Pleural Effusions from COVID-19 Deceased Patients: Enhanced Inflammatory Markers" Diagnostics 12, no. 11: 2789. https://doi.org/10.3390/diagnostics12112789
APA StyleRazaghi, A., Szakos, A., Alouda, M., Bozóky, B., Björnstedt, M., & Szekely, L. (2022). Proteomic Analysis of Pleural Effusions from COVID-19 Deceased Patients: Enhanced Inflammatory Markers. Diagnostics, 12(11), 2789. https://doi.org/10.3390/diagnostics12112789