
A Real-Life Reproducibility Assessment for NMR Metabolomics †

 , , , , ,
, , , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
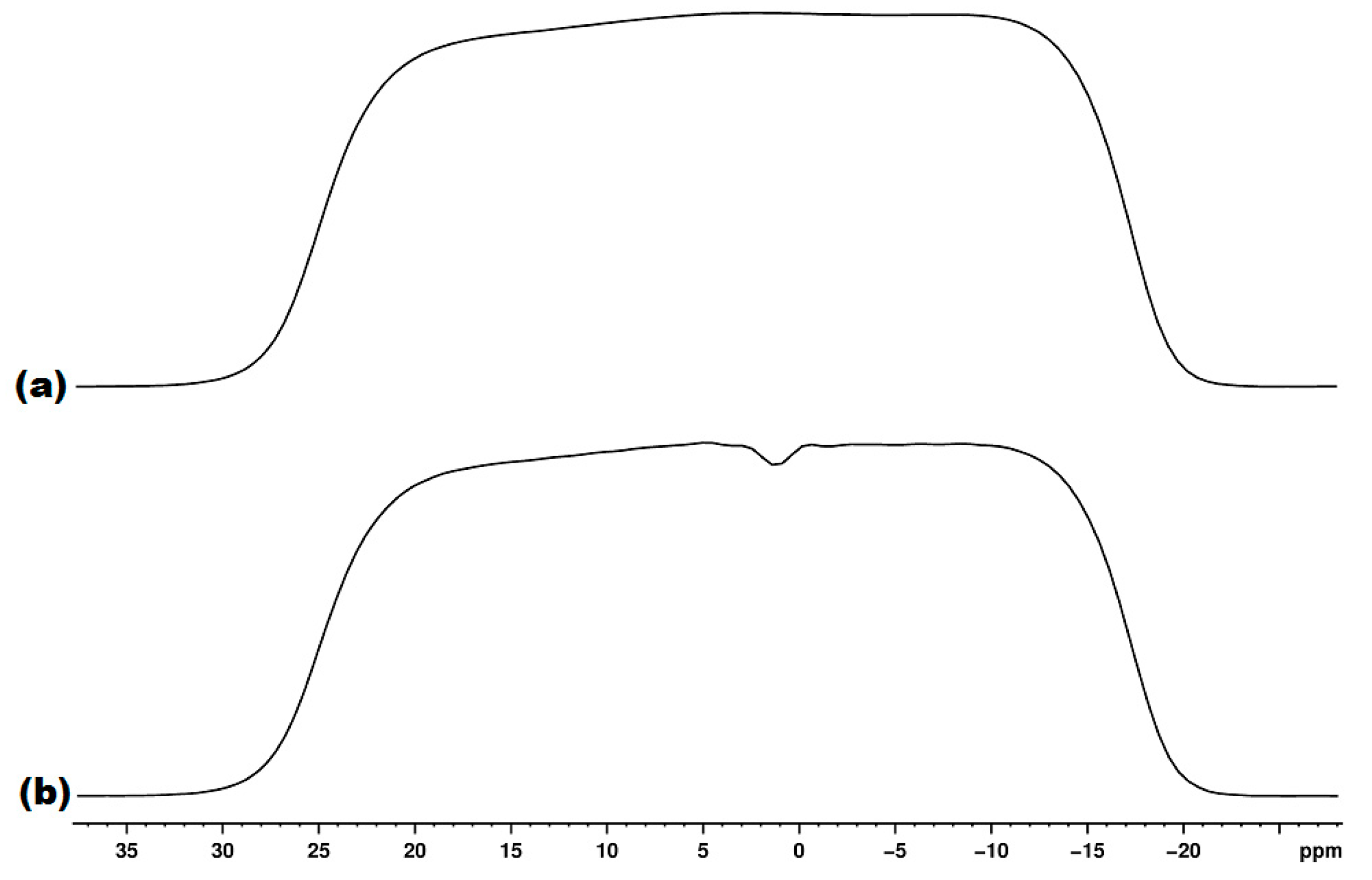
2. Materials and Methods
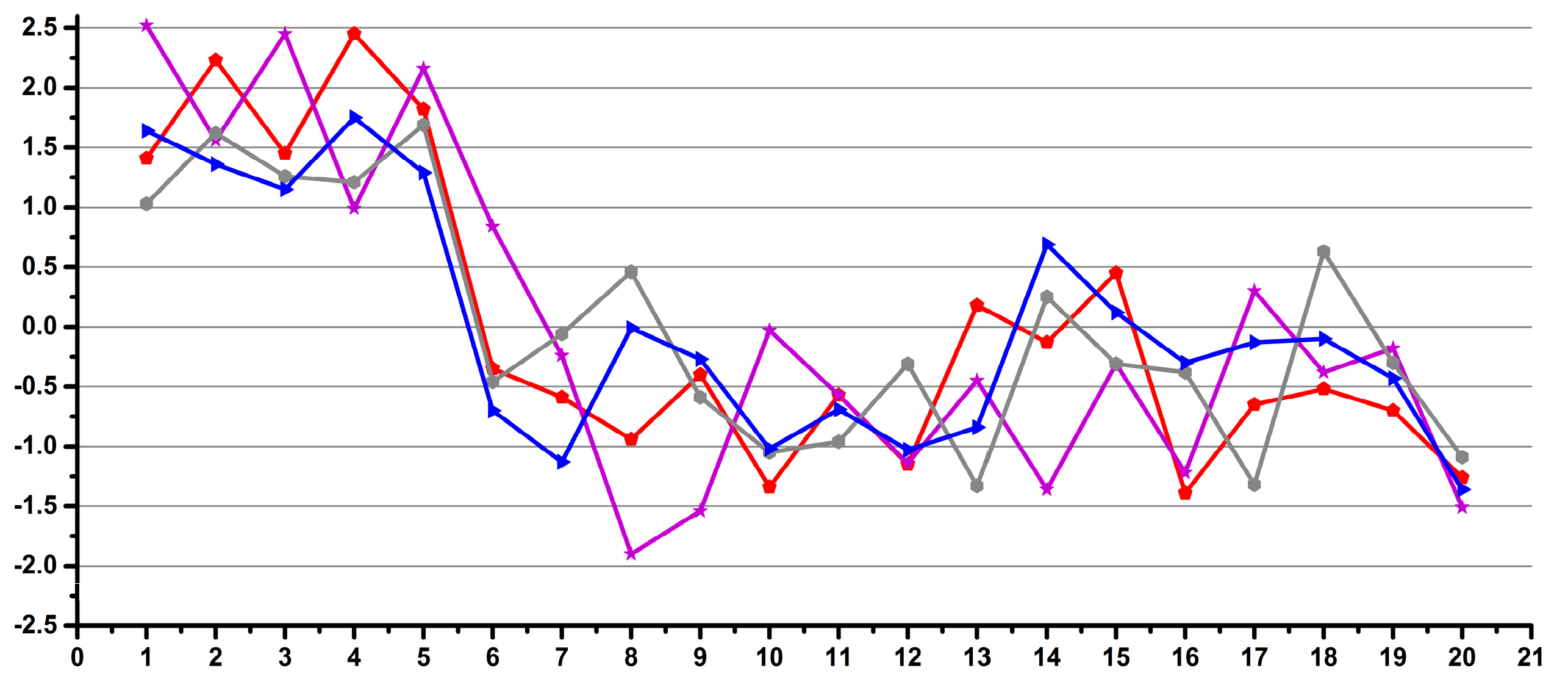
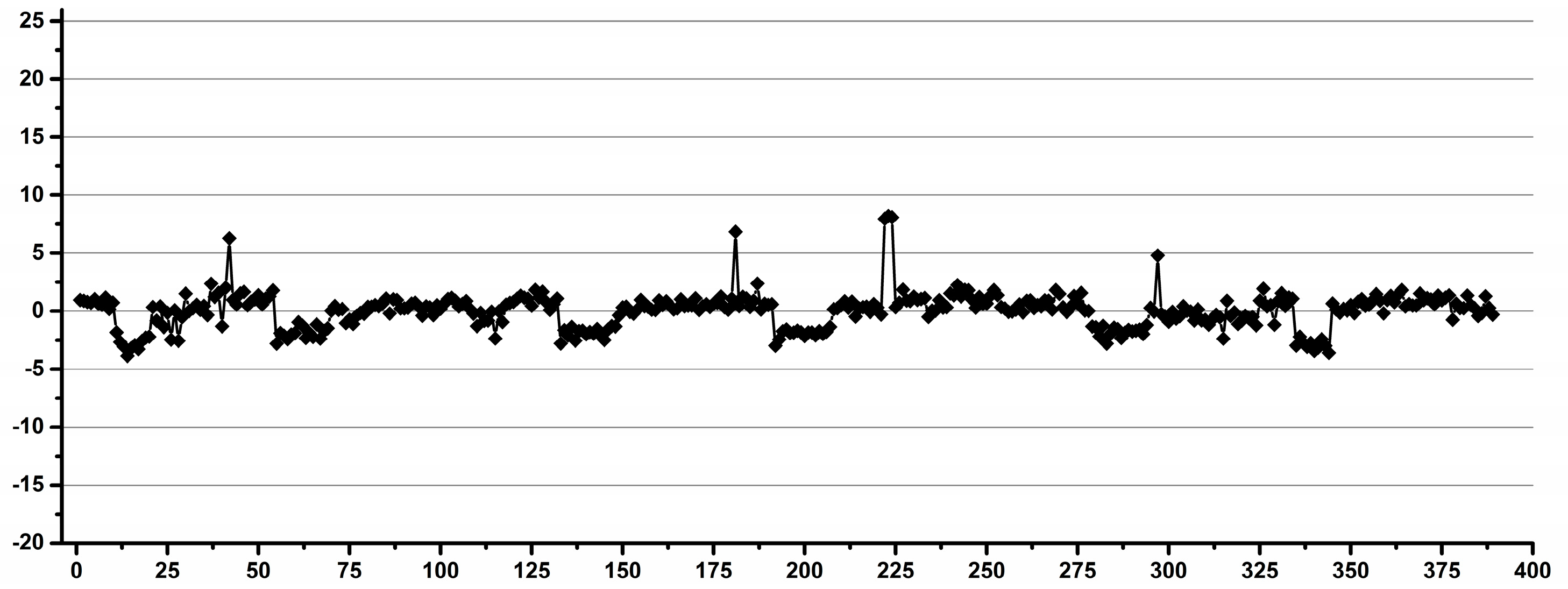
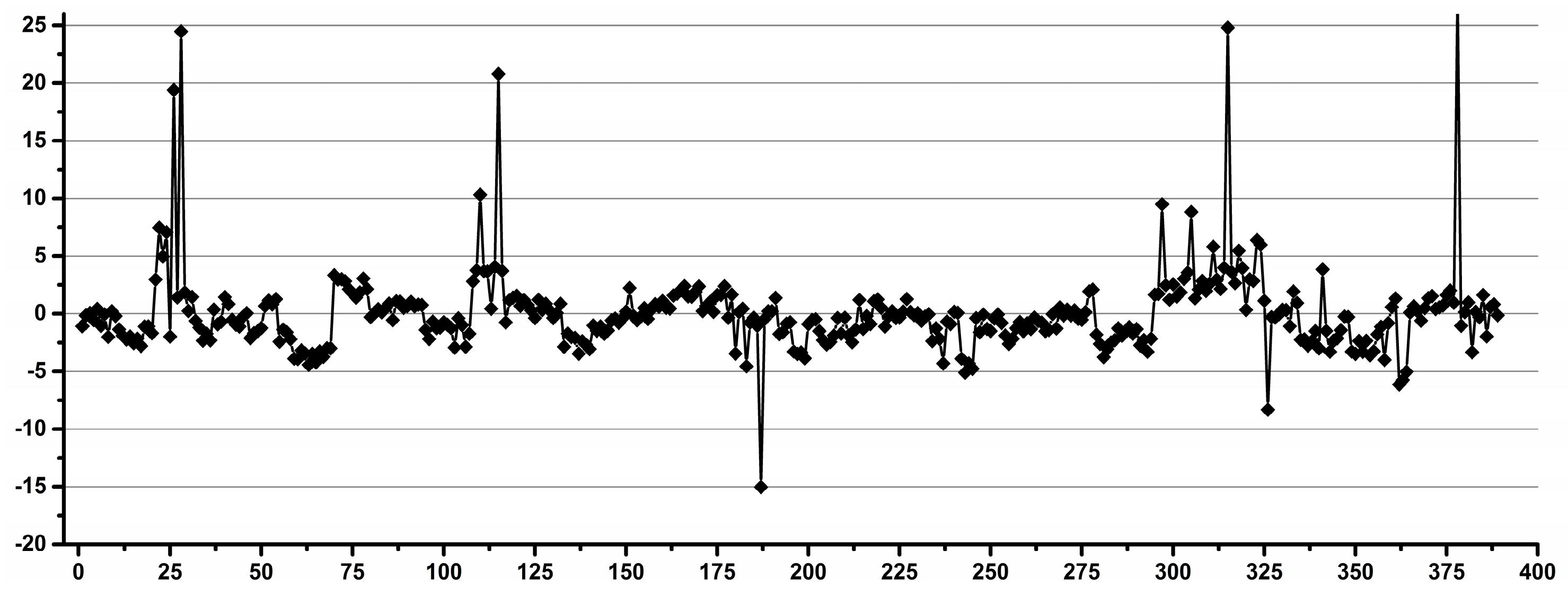
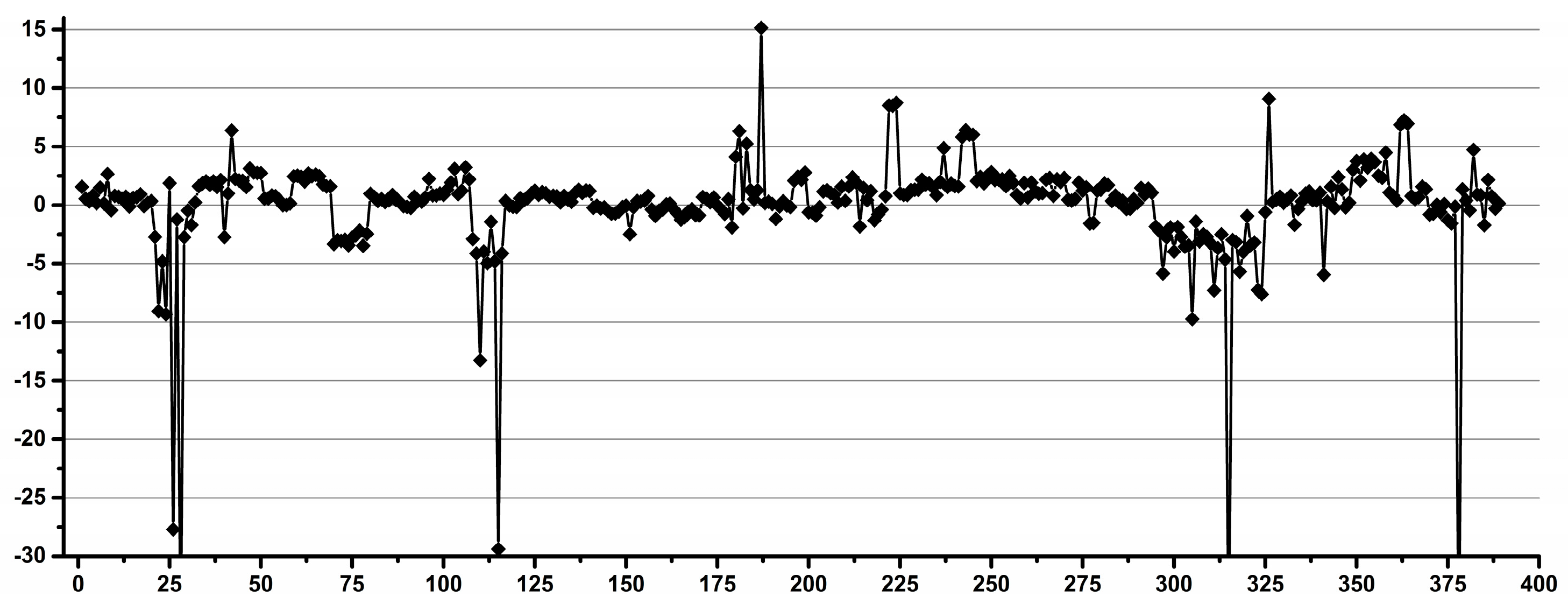
3. Results and Discussion
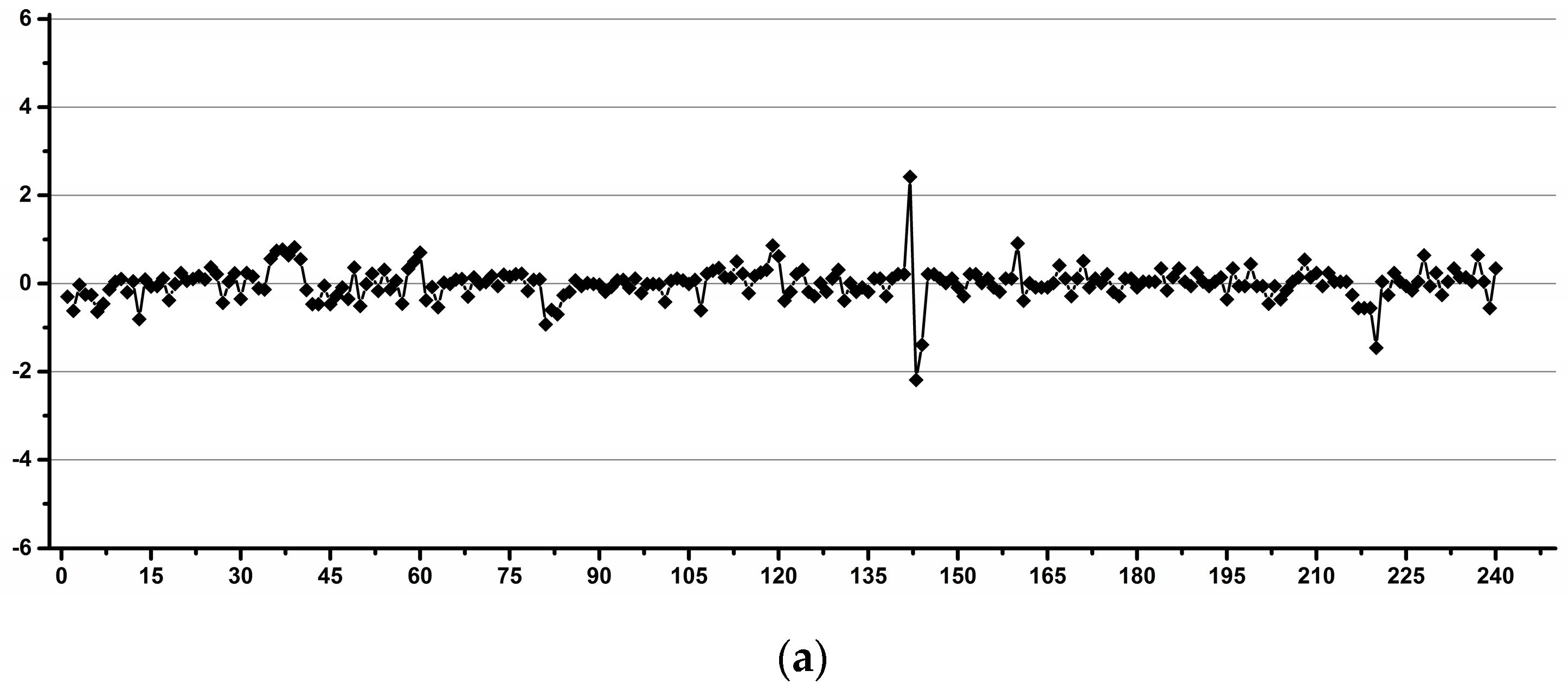
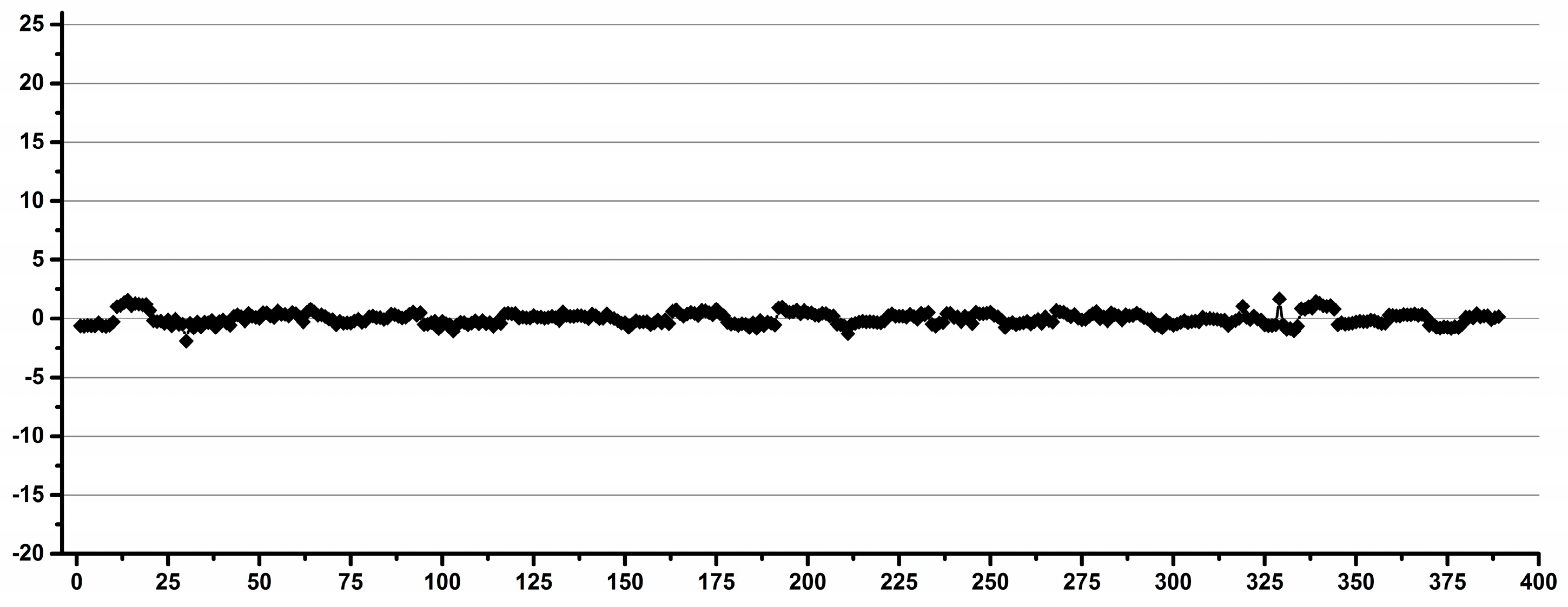
3.1. Estimation of the Operator Induced Variability
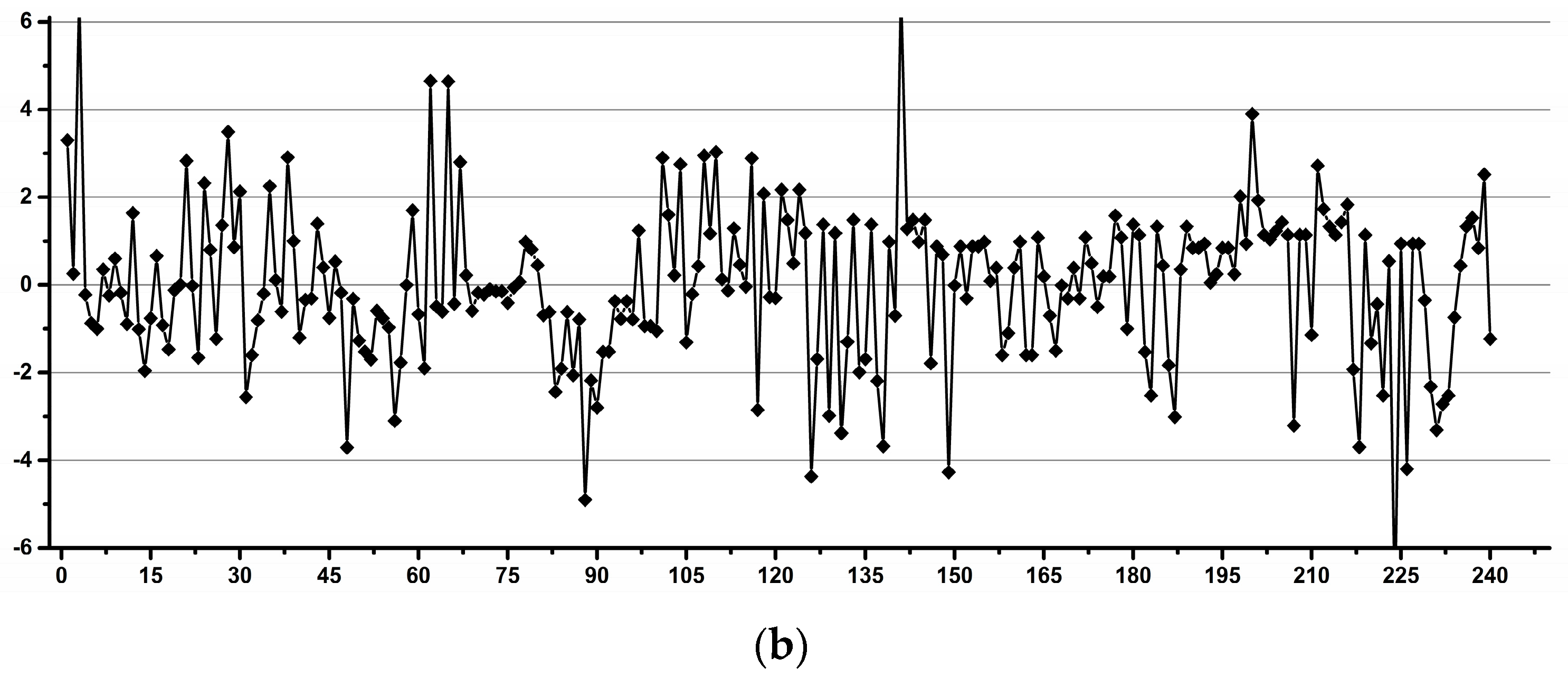
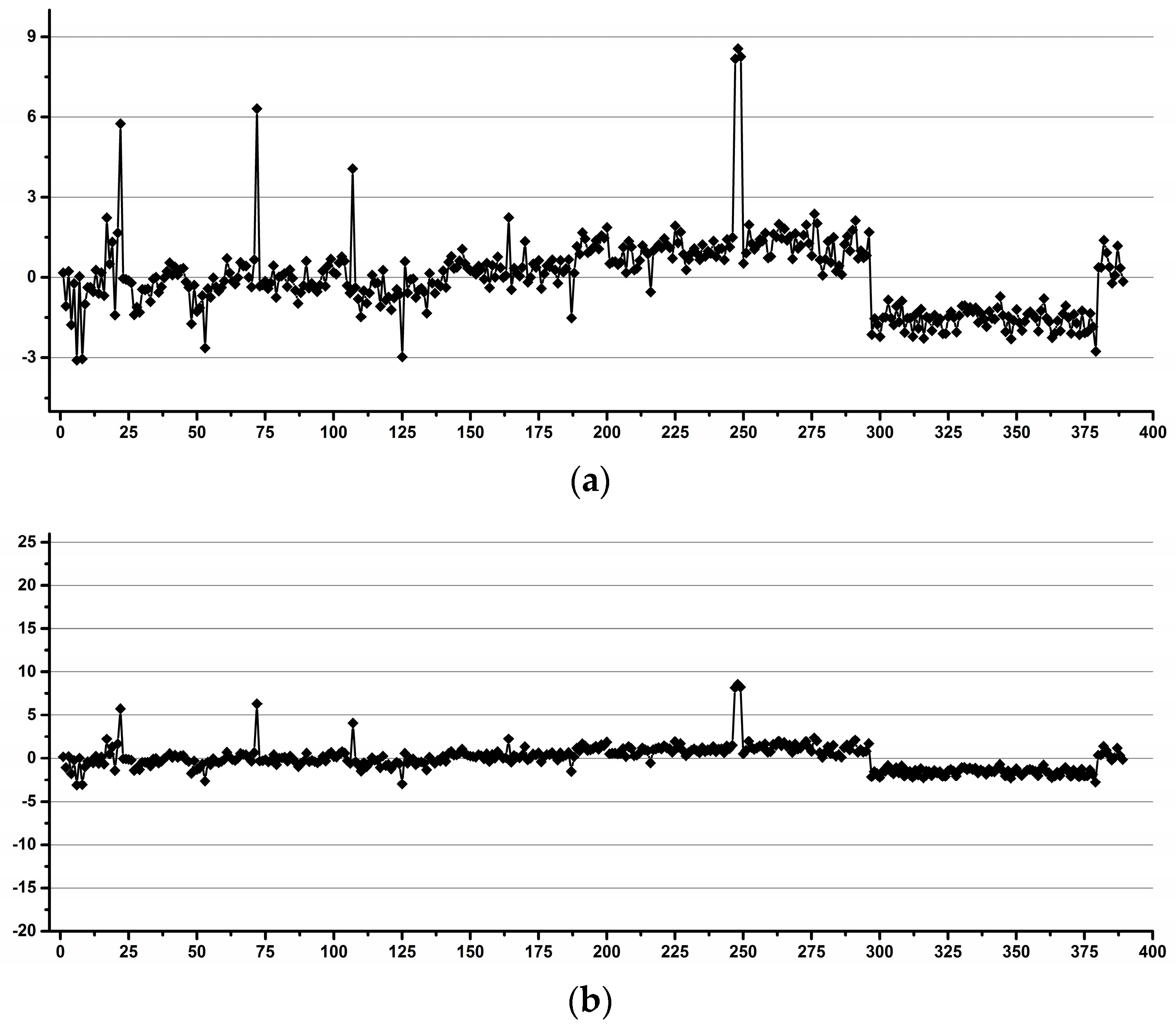
3.2. Estimation of the Experimental Induced Variability
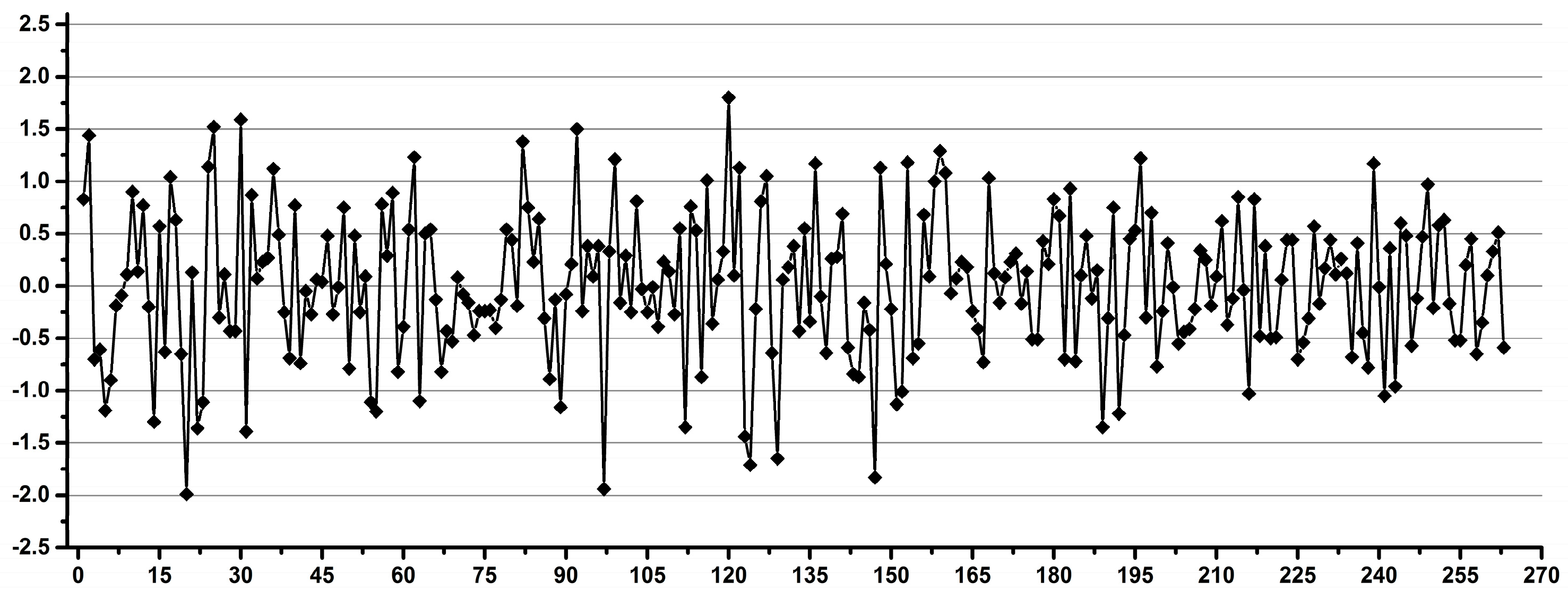
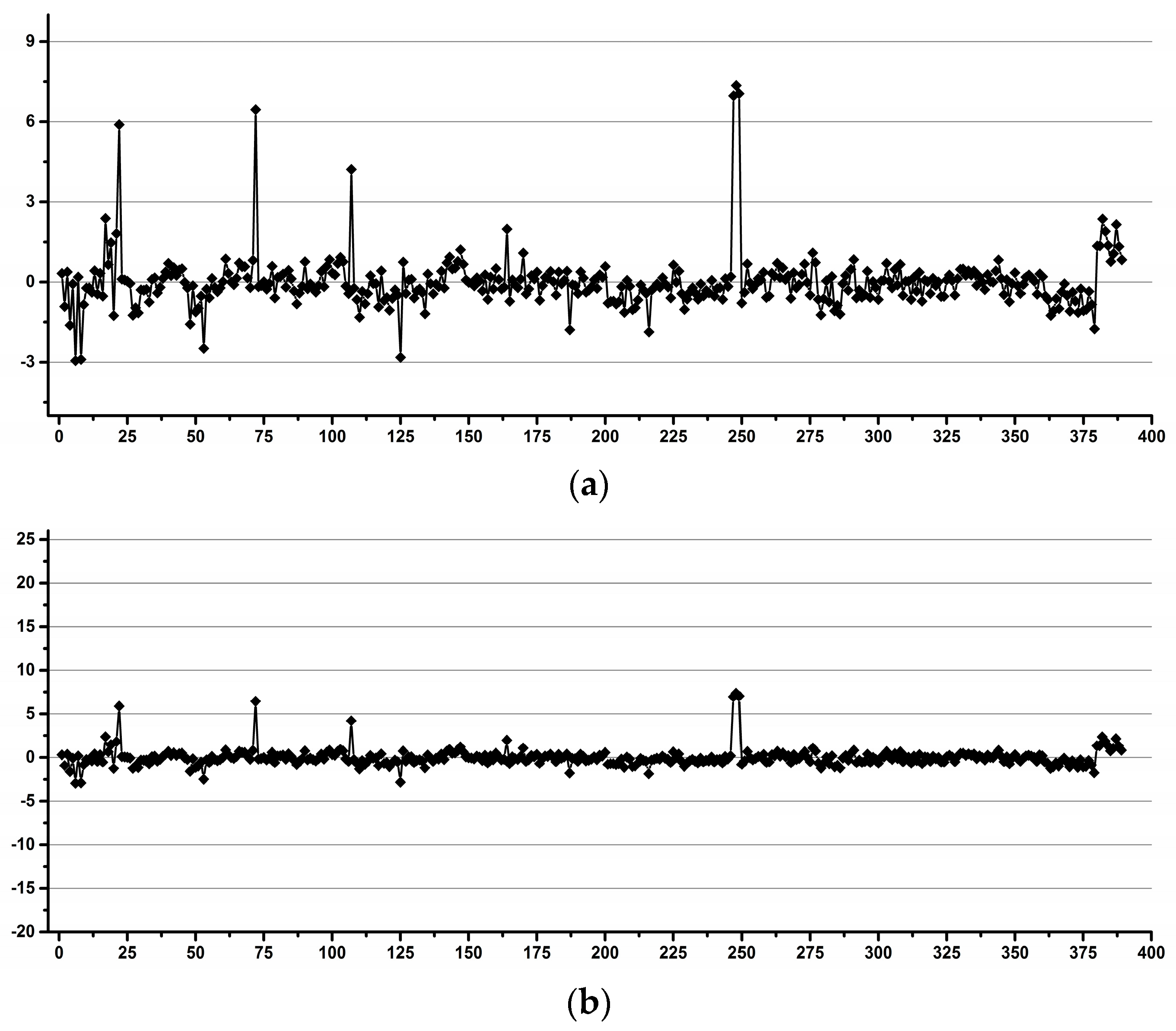
3.3. Estimation of the Overall Induced Variability for NMR Metabolomics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hollis, D.P. Quantitative analysis of aspirin, phenacetin, and caffeine mixtures by nuclear magnetic resonance spectrometry. Anal. Chem. 1963, 35, 1682–1684. [Google Scholar] [CrossRef]
- Jungnickel, J.L.; Forbes, J.W. Quantitative measurement of hydrogen types by integrated nuclear magnetic resonance intensities. Anal. Chem. 1963, 35, 938–942. [Google Scholar] [CrossRef]
- Bauer, M.; Bertario, A.; Boccardi, G.; Fontaine, X.; Rao, R.; Verrier, D. Reproducibility of 1H-NMR integrals: A collaborative study. J. Pharm. Biomed. Anal. 1998, 17, 419–425. [Google Scholar] [CrossRef]
- Guillou, C.; Trierweiler, M.; Martin, G.J. Repeatability and reproducibility of Site-Specific Isotope Ratios in quantitative 2H NMR. Magn. Reson. Chem. 1988, 26, 491–496. [Google Scholar] [CrossRef]
- Miura, T.; Sugimoto, N.; Bhavaraju, S.; Yamazaki, T.; Nishizaki, Y.; Liu, Y.; Bzhelyansky, A.; Amezcua, C.; Ray, J.; Zailer, E.; et al. Collaborative study to validate purity determination by 1H quantitative NMR spectroscopy by using internal calibration methodology. Chem. Pharm. Bull. 2020, 68, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Holzgrabe, U. Quantitative NMR spectroscopy in pharmaceutical applications. Prog. Nucl. Magn. Reason. Spectrosc. 2010, 57, 229–240. [Google Scholar] [CrossRef]
- Gallo, V.; Intini, N.; Mastrorilli, P.; Latronico, M.; Scapicchio, P.; Triggiani, M.; Bevilacqua, V.; Fanizzi, P.; Acquotti, D.; Airoldi, C.; et al. Performance assessment in fingerprinting and multi component quantitative NMR analyses. Anal. Chem. 2015, 87, 6709–6717. [Google Scholar] [CrossRef]
- Bharti, S.K.; Roy, R. Quantitative 1H NMR spectroscopy. Trends Anal. Chem. 2012, 35, 5–26. [Google Scholar] [CrossRef]
- Craig, A.; Cloarec, O.; Holmes, E.; Nicholson, J.K.; Lindon, J.C. Scaling and normalization effects in NMR spectroscopic metabonomic data sets. Anal. Chem. 2006, 78, 2262–2267. [Google Scholar] [CrossRef]
- Van der Kloet, F.M.; Bobeldijk, I.; Verheij, E.R.; Jellema, R.H. Analytical error reduction using single point calibration for accurate and precise metabolomic phenotyping. J. Proteom. Res. 2009, 8, 5132–5141. [Google Scholar] [CrossRef]
- Kriat, M.; Confort-Gouny, S.; Vion-Dury, J.; Sciaky, M.; Viout, P.; Cozzone, P.J. Quantitation of metabolites in human blood serum by proton magnetic resonance spectroscopy. A comparative study of the use of formate and TSP as concentration standards. NMR Biomed. 1992, 5, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Alum, M.F.; Shaw, P.A.; Sweatman, B.C.; Ubhi, B.K.; Haselden, J.N.; Connor, S.C. 4,4-Dimethyl-4-silapentane-1-ammonium trifluoroacetate (DSA), a promising universal internal standard for NMR-based metabolic profiling studies of biofluids, including blood plasma and serum. Metabolomics 2008, 4, 122–127. [Google Scholar] [CrossRef] [Green Version]
- Mo, H.; Harwooda, J.S.; Rafteryc, D. Receiver gain function: The actual NMR receiver gain. Magn. Reson. Chem. 2010, 48, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zheng, C.; Lanza, I.R.; Nair, K.S.; Raftery, D.; Vitek, O. Interdependence of signal processing and analysis of urine 1H NMR spectra for metabolic profiling. Anal. Chem. 2009, 81, 6080–6088. [Google Scholar] [CrossRef] [Green Version]
- Crook, A.A.; Powers, R. Quantitative NMR-based biomedical metabolomics: Current status and applications. Molecules 2020, 25, 5128. [Google Scholar] [CrossRef]
- Centelles, S.M.; Hoefsloot, H.C.J.; Khakimov, B.; Ebrahimi, P.; Lind, M.V.; Kristensen, M.; de Roo, N.; Jacobs, D.M.; van Duynhoven, J.; Cannet, C.; et al. Toward reliable lipoprotein particle predictions from NMR spectra of human blood: An interlaboratory ring test. Anal. Chem. 2017, 89, 8004–8012. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, B.; Holmes, E.; Heude, C.; Tolson, R.F.M.; Harvey, N.; Lodge, S.L.; Chetwynd, A.J.; Cannet, C.; Fang, F.; Pearce, J.T.M.; et al. Quantitative lipoprotein subclass and low molecular weight metabolite analysis in human serum and plasma by 1H NMR spectroscopy in a multilaboratory trial. Anal. Chem. 2018, 90, 11962–11971. [Google Scholar] [CrossRef]
- Da Silva, L.; Godejohann, M.; Martin, F.-P.J.; Collino, S.; Bürkle, A.; Moreno-Villanueva, M.; Bernhardt, J.; Toussaint, O.; Grubeck-Loebenstein, B.; Gonos, E.S.; et al. High-resolution quantitative metabolome analysis of urine by automated flow injection NMR. Anal. Chem. 2013, 85, 5801–5809. [Google Scholar] [CrossRef]
- Tynkkynen, T.; Wang, Q.; Ekholm, J.; Anufrieva, O.; Ohukainen, P.; Vepsalainen, J.; Mannikko, M.; Keinanen-Kiukaanniemi, S.; Holmes, M.V.; Goodwin, M.; et al. Proof of concept for quantitative urine NMR metabolomics pipeline for large-scale epidemiology and genetics. Int. J. Epidemiol. 2019, 48, 978–993. [Google Scholar] [CrossRef] [Green Version]
- Bearden, D.W.; Sheen, D.A.; Simón-Manso, Y.; Benner, B.A., Jr.; Rocha, W.F.C.; Blonder, N.; Lippa, K.A.; Beger, R.D.; Schnackenberg, L.K.; Sun, J.; et al. Metabolomics test materials for quality control: A study of a urine materials suite. Metabolites 2019, 9, 270. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, T.; Aursand, M.; Sacchi, R.; Paolillo, L.; Nonaka, M.; Shunwada, S. Determination of docosahexaenoic acid and n-3 fatty acids in refined fish oils by 1H-NMR spectroscopy: IUPAC interlaboratory study. J. AOAC Int. 2002, 85, 1341–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zailer, E.; Holzgrabe, U.; Diehl, B.W.K. Interlaboratory comparison test as an evaluation of applicability of an alternative edible oil analysis by 1H NMR spectroscopy. J. AOAC Int. 2017, 100, 1819–1830. [Google Scholar] [CrossRef] [PubMed]
- Mannina, L.; Sobolev, A.P.; Viel, S. Liquid state 1H high field NMR in food analysis. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 66, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Maes, P.; Monakhova, Y.B.; Kuballa, T.; Reusch, H.; Lachenmeier, D.W. Qualitative and quantitative control of carbonated Cola beverages using 1H NMR spectroscopy. J. Agric. Food Chem. 2012, 60, 2778–2784. [Google Scholar] [CrossRef] [PubMed]
- Spraul, M.; Schütz, B.; Rinke, P.; Koswig, S.; Humpfer, E.; Schäfer, H.; Mörtter, M.; Fang, F.; Marx, U.C.; Minoja, A. NMR-based multi parametric quality control of fruit juices: SGF Profiling. Nutrients 2009, 1, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Musio, B.; Ragone, R.; Todisco, S.; Rizzuti, A.; Latronico, M.; Mastrorilli, P.; Pontrelli, S.; Pasquale Scapicchio, N.I.; Triggiani, M.; Di Noia, T.; et al. A community-built calibration system: The case study of quantification of metabolites in grape juice by qNMR spectroscopy. Talanta 2020, 214, 120855. [Google Scholar] [CrossRef]
- Ragone, R.; Todisco, S.; Triggiani, M.; Pontrelli, S.; Latronico, M.; Mastrorilli, P.; Intini, N.; Ferroni, C.; Musio, B.; Gallo, V. Development of a food class-discrimination system by non-targeted NMR analyses using different magnetic field strengths. Food Chem. 2020, 332, 127339. [Google Scholar] [CrossRef]
- Gallo, V.; Ragone, R.; Musio, B.; Todisco, S.; Rizzuti, A.; Mastrorilli, P.; Pontrelli, S.; Intini, N.; Scapicchio, P.; Triggiani, M.; et al. A contribution to the harmonization of non-targeted NMR methods for data-driven food authenticity assessment. Food Anal. Meth. 2020, 13, 530–541. [Google Scholar] [CrossRef]
- Magdas, D.A.; Cristea, G.; Pîrnau, A.; Feher, I.; Hategan, A.R.; Dehelean, A. Authentication of Transylvanian spirits based on isotope and elemental signatures in conjunction with statistical methods. Foods 2021, 10, 3000. [Google Scholar] [CrossRef]
- Dona, A.C.; Jimenez, B.; Schaefer, H.; Humpfer, E.; Spraul, M.; Lewis, M.R.; Pearce, J.T.M.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Precision high throughput proton NMR spectroscopy of human urine, serum and plasma for large-scale metabolic phenotyping. Anal. Chem. 2014, 86, 9887–9894. [Google Scholar] [CrossRef]
- Georgescu, E.; Nicolescu, A.; Georgescu, F.; Teodorescu, F.; Shova, S.; Marinoiu, A.T.; Dumitrascu, F.; Deleanu, C. Fine tuning the outcome of 1,3-dipolar cycloaddition reactions of benzimidazolium ylides to activated alkynes. Tetrahedron 2016, 72, 2507–2520. [Google Scholar] [CrossRef]
- Pandele, A.M.; Iovu, H.; Orbeci, C.; Tuncel, C.; Miculescu, F.; Nicolescu, A.; Deleanu, C.; Voicu, S.I. Surface modified cellulose acetate membranes for the reactive retention of tetracycline. Sep. Purif. Technol. 2020, 249, 117145. [Google Scholar] [CrossRef]
- Nicolescu, A.; Blanita, D.; Boiciuc, C.; Hlistun, V.; Cristea, M.; Rotaru, D.; Pinzari, L.; Oglinda, A.; Stamati, A.; Tarcomnicu, I.; et al. Monitoring methylmalonic aciduria by NMR urinomics. Molecules 2020, 25, 5312. [Google Scholar] [CrossRef] [PubMed]
- Vulturar, R.; Chis, A.; Baizat, M.; Cozma, A.; Suharoschi, R.; Nicolescu, A.; Deleanu, C. A severe neonatal argininosuccinic aciduria case investigated by 1H NMR spectroscopy. Rev. Chim. 2020, 71, 210–218. [Google Scholar] [CrossRef]
- Grama, A.; Blaga, L.; Nicolescu, A.; Deleanu, C.; Militaru, M.; Căinap, S.S.; Pop, I.; Tita, G.; Sîrbe, C.; Fufezan, O.; et al. Novel mutation in GALT gene in Galactosemia patient with group B Streptococcus Meningitis and acute liver failure. Medicina 2019, 55, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolescu, A.; Dolenko, B.; Bezabeh, T.; Stefan, L.-I.; Ciurtin, C.; Kovacs, E.; Smith, I.C.P.; Simionescu, B.C.; Deleanu, C. Diagnosis of type II diabetes based on non-glucose regions of 1H NMR spectra of urine: A metabonomic approach. Rev. Chim. 2011, 62, 1150–1153. [Google Scholar]
- Stefan, L.I.; Nicolescu, A.; Popa, S.; Mota, M.; Kovacs, E.; Deleanu, C. 1H-NMR urine metabolic profiling in type 1 diabetes Mellitus. Rev. Roum. Chim. 2010, 55, 1033–1037. [Google Scholar]
- Ciurtin, C.; Nicolescu, A.; Stefan, L.-I.; Kovacs, E.; Smith, I.C.P.; Deleanu, C. Metabolic profiling of urine by 1H-NMR spectroscopy. A critical assessment of interpreting metabolite concentrations for normal and diabetes groups. Rev. Chim. 2007, 52, 51–55. [Google Scholar]
- Musteata, M.; Nicolescu, A.; Solcan, G.; Deleanu, C. The 1H NMR profile of healthy dog cerebrospinal fluid. PLoS ONE 2013, 3, e81192. [Google Scholar] [CrossRef] [Green Version]
- Balan, M.; Nicolescu, A.; Stavarache, C.; Ciobanu, M.; Deleanu, C. Fast NMR juice identification based on sugars and other plant metabolites from fruits. Rev. Roum. Chim. 2013, 57, 175–182. [Google Scholar]
- Todasca, M.-C.; Fotescu, L.; Chira, N.-A.; Deleanu, C.; Rosca, S. Composition changes in wines produced by different growing techniques examined through 1H-NMR spectroscopy. Rev. Chim. 2011, 62, 131–134. [Google Scholar]
- Lindon, J.C.; Sweatman, B.C.; Arus, C.; Cerdan, S.; de Certaines, J.D.; Deleanu, C.; Farrant, R.D.; Seddon, M.J.; Griffiths, J.R.; Liebfritz, D.; et al. Multicentre assessment of small molecule quantitation in human blood plasma using 1H-NMR spectroscopy. J. Magn. Reson. Anal. 1996, 2, 66–74. [Google Scholar]
- Price, W.S. Gradient NMR. Ann. Rep. NMR Spectrosc. 1996, 32, 51–142. [Google Scholar]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stavarache, C.; Nicolescu, A.; Duduianu, C.; Ailiesei, G.L.; Balan-Porcăraşu, M.; Cristea, M.; Macsim, A.-M.; Popa, O.; Stavarache, C.; Hîrtopeanu, A.; et al. A Real-Life Reproducibility Assessment for NMR Metabolomics. Diagnostics 2022, 12, 559. https://doi.org/10.3390/diagnostics12030559
Stavarache C, Nicolescu A, Duduianu C, Ailiesei GL, Balan-Porcăraşu M, Cristea M, Macsim A-M, Popa O, Stavarache C, Hîrtopeanu A, et al. A Real-Life Reproducibility Assessment for NMR Metabolomics. Diagnostics. 2022; 12(3):559. https://doi.org/10.3390/diagnostics12030559
Chicago/Turabian StyleStavarache, Cristina, Alina Nicolescu, Cătălin Duduianu, Gabriela Liliana Ailiesei, Mihaela Balan-Porcăraşu, Mihaela Cristea, Ana-Maria Macsim, Oana Popa, Carmen Stavarache, Anca Hîrtopeanu, and et al. 2022. "A Real-Life Reproducibility Assessment for NMR Metabolomics" Diagnostics 12, no. 3: 559. https://doi.org/10.3390/diagnostics12030559