
Successfully Managed Respiratory Insufficiency in a Patient with a Novel Pathogenic Variant of the BMPER Gene: A Case Report



Abstract
:1. Introduction
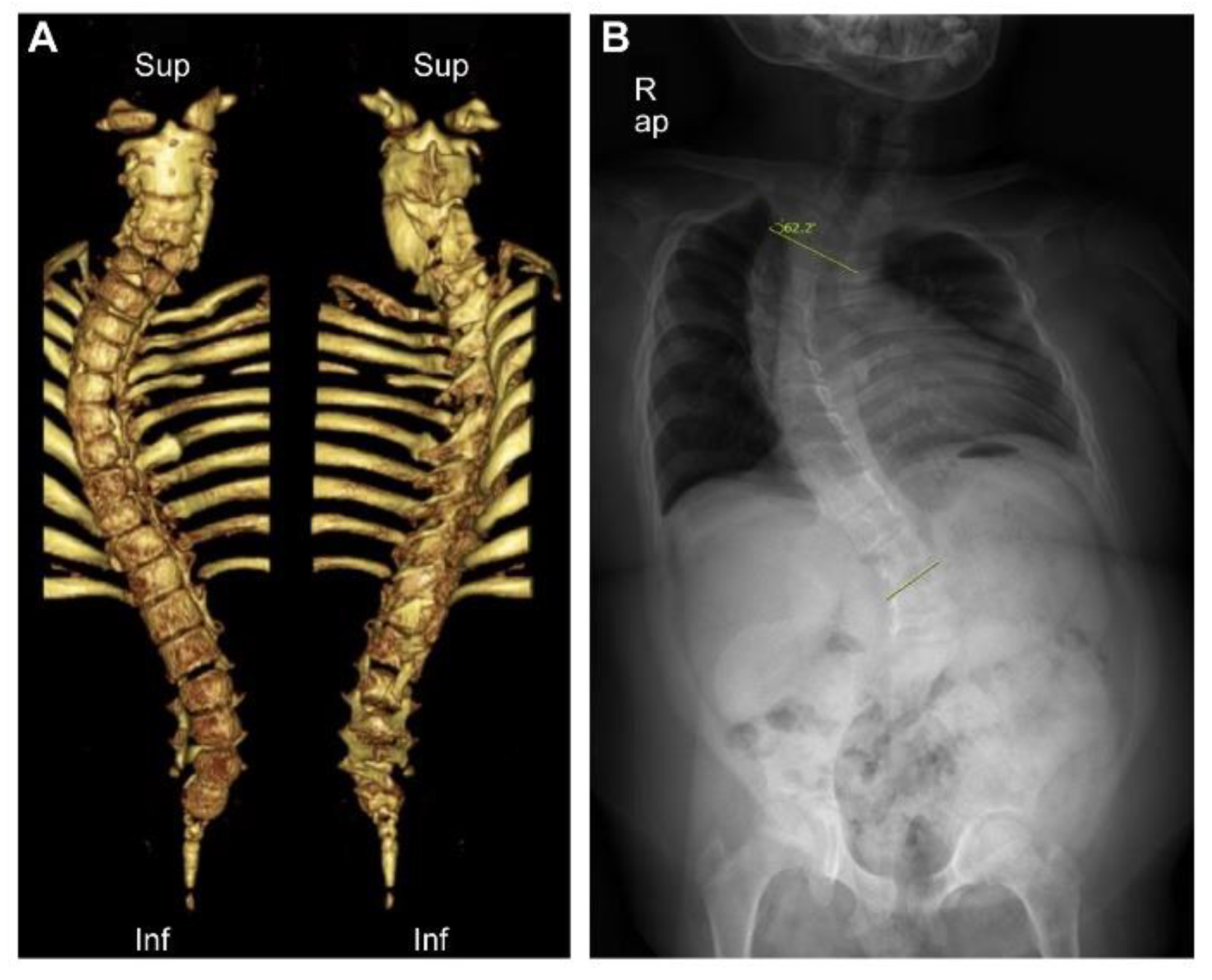
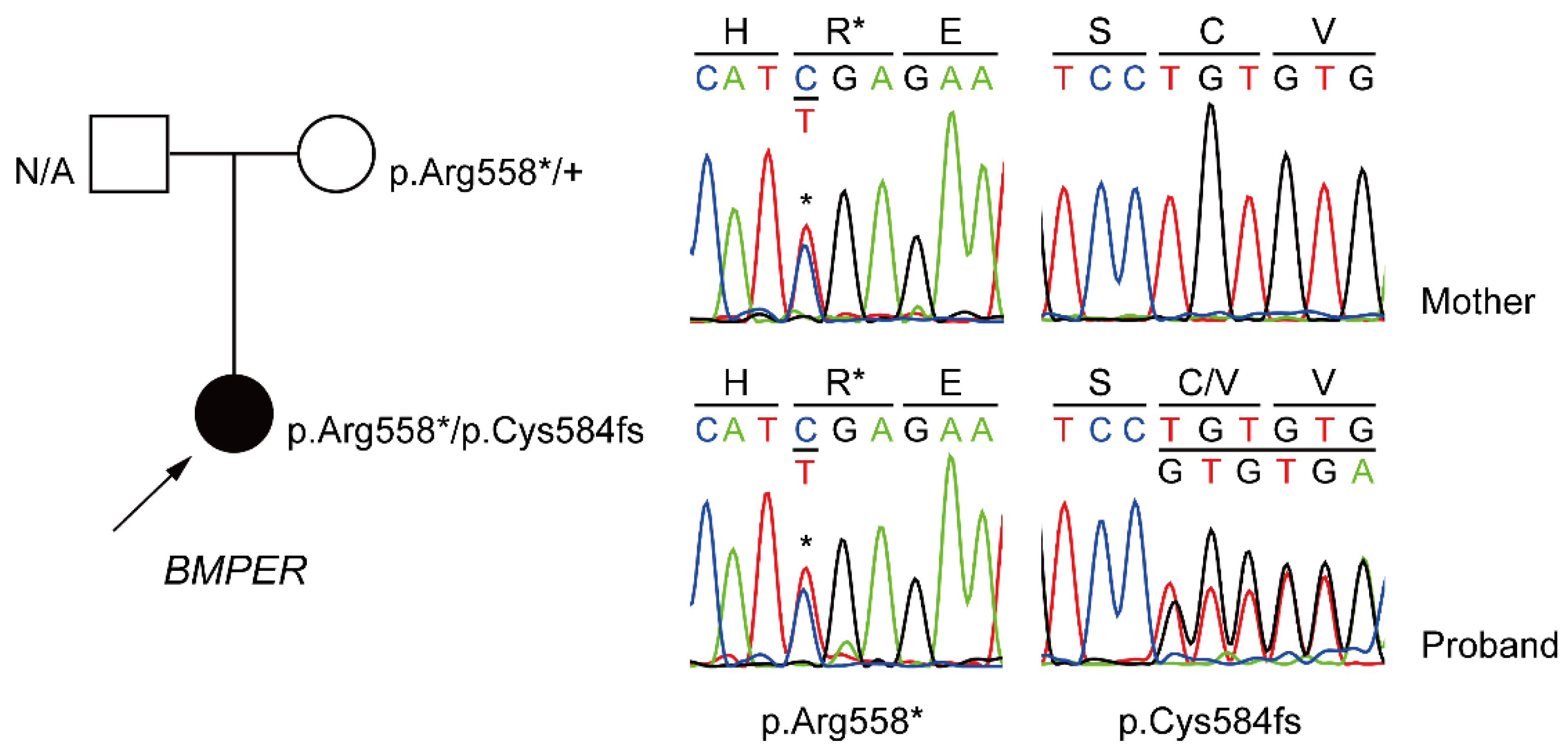
2. Case Report

{kind=link}
{kind=link}
| Patient Number | Gender | Survival | Consanguinity | Ethnicity | Respiratory Distress at Birth | Kidney Pathology | Other Abnormalities | Axial Skeletal Anomalies | cDNA | Protein | Clinical Diagnosis | Reported Year [Reference Number] | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Female | >33 years | No | Japanese | None | NB | Developmental delay, Paraparesis, feet deformities | Vertebral segmentation defects, rib anomalies, ischial hypoplasia, sacral hypoplasia | NA | NA | ISD | 1999 [10] | |
| 2 | Male | >7 years | No | Japanese | None | NoM | Developmental delay, Facial dysmorphism, Paraparesis, feet deformities | Vertebral segmentation defects, ischial hypoplasia, sacral hypoplasia | NA | NA | ISD | 1999 [10] | |
| 3 | Female | >38 years | 1st degree | Japanese | None | NoM | Paraparesis, feet deformities | Vertebral segmentation defects, rib anomalies, ischial hypoplasia, sacral hypoplasia | NA | NA | ISD | 1999 [10] | |
| 4 | Male | >3 months | No | Japanese | Yes, intubation | NoM | Coxa valga | Vertebral segmentation defects, rib anomalies, ischial hypoplasia, sacral hypoplasia | NA | NA | ISD | 1999 [10] | |
| 5 | Male | >6 months | No | Japanese | None | NoM | Ichthyosis, cryptorchidism, inguinal hernia, heart anomaly, alopecia | Vertebral segmentation defects, rib anomalies, ischial hypoplasia | NA | NA | ISD | 1999 [10] | |
| 6 | Male | >10 months | No | Caucasian | None | Polycystic, NB | Facial dysmorphism, macrocephaly | Vertebral segmentation defects, rib anomalies, ischial hypoplasia | NA | NA | ISD | 2001 [11] | |
| 7 | Male | >2 years | No | Korean | None | Polycystic | Developmental delay, dislocated knee, and hip joints | Vertebral segmentation defects, rib anomalies, ischial hypoplasia, sacral hypoplasia | NA | NA | ISD | 2003 [12] | |
| 8 | Female | >5 years | No | Japanese | None | Monocystic | Cleft palate, Developmental delay, brachymesophalangia | Vertebral segmentation defects, rib anomalies, ischial hypoplasia | NA | NA | ISD | 2003 [12] | |
| 9 | Male | Stillborn | Yes | Mali | - | Polycystic, NB | Feet deformities, Nail hypoplasia | Vertebral segmentation defects, rib anomalies, ischial hypoplasia | NA | NA | DSD | 2005 [1] | |
| 10 | Female | Stillborn | Yes | Mali | - | Polycystic, NB | Genu recurvatum | Vertebral agenesis | NA | NA | DSD | 2005 [1] | |
| 11 | Male | Neonatal death | No | NoM | Yes, fatal | NoM | Facial dysmorphism, Nail hypoplasia | Vertebral ossification defects | NA | NA | DSD | 2005 [1] | |
| 12 | Male | Neonatal death or stillborn | No | NoM | NoM | NoM | Cleft palate | Vertebral ossification defects | NA | NA | DSD | 2005 [1] | |
| 13 | Male | Neonatal death | No | Hispanic | Yes, fatal | Polycystic, NB | Facial dysmorphism | Vertebral ossification and segmentation defects, rib anomalies, ischial hypoplasia, sacral agenesis | NA | NA | DSD | 2007 [13] | |
| 14 | Male | Neonatal death | No | European | Yes, fatal | NoM | Facial dysmorphism, Inguinal hernia | Vertebral ossification defects, sacral agenesis | NA | NA | DSD | 2007 [13] | |
| 15 | Female | Neonatal death | NoM | Caucasian | Yes, intubation | Polycystic | Tracheomalacia | Vertebral ossification defects, rib anomalies, sacral agenesis | NA | NA | DSD | 2007 [13] | |
| 16 | Male | 5 years | No | European | Yes, intubation | Polycystic, Wilms tumor | Facial dysmorphism, Hearing loss, macrocephaly, Peripheral neuropathy due to spinal cord anomaly | Vertebral ossification defects, rib anomalies, sacral agenesis | c.26_35del10ins14 c.1032+5G>A | p.Ala9Glufs*4 | DSD | 2007 [13], 2010 [2], 2012 [14,15] | |
| 17 | Female | 15 months | 2nd degree | Arabic | Yes, intubation | Polycystic | Facial dysmorphism | Vertebral ossification and segmentation defects, rib anomalies, sacral agenesis | c.310C>T | p.Gln104* | DSD | 2011 [3] | |
| 18 | Female | 4 months | No | Arabic | Yes, NICU care | Normal | Nail hypoplasia | Vertebral ossification and segmentation defects, rib anomalies, sacral agenesis | c.310C>T | p.Gln104* | DSD | 2011 [3] | |
| 19 | Male | >13 years | No | British-European | None | Normal | Cleft palate, Facial dysmorphism | Scoliosis, Vertebral ossification defects, rib anomalies | c.251G>T c.1078+5G>A | p.Cys84Phe | Attenuated DSD | 2015 [9] | |
| 20 | Male | >6~13 years | No | British-European | None | Normal | Cleft palate, Facial dysmorphism | Scoliosis, Vertebral ossification defects, rib anomalies | c.251G>T c.1078+5G>A | p.Cys84Phe | Attenuated DSD | 2015 [9] | |
| 21 | Male | >6 years | No | British-European | None | Normal | Cleft palate, Facial dysmorphism | Scoliosis, Vertebral ossification defects, rib anomalies | c.251G>T c.1078+5G>A | p.Cys84Phe | Attenuated DSD | 2015 [9] | |
| 22 | Female | >2 years | No | Swedish | None | Normal | Facial dysmorphism, Hearing loss | Vertebral segmentation defects, rib anomalies, ischial hypoplasia, sacral hypoplasia | c.416C>G c.942G>A | p.Thr139Arg p.Trp314* | ISD | 2016 [16] | |
| 23 | Male | >19 years | No | Korean | Yes, oxygen | Hydronephrosis | Facial dysmorphism, Paraparesis, feet deformities, neurogenic bladder, flat acetabulum, coxa valga | Vertebral segmentation defects, rib anomalies, ischial hypoplasia | c.1672C>T c.1988G>A | p.Arg558* p.Cys663Tyr | ISD | 2016 [16] | |
| 24 | Male | >9 years | No | NoM | Yes | Renal caliectasis | Facial dysmorphism, Muscle wasting, hypertension, hypercalcinuria, coxa valga | Vertebral segmentation defects, rib anomalies, ischial hypoplasia, sacral hypoplasia | c.322T>C | p.Cys108Arg/7p14.3p14.2del | DSD | 2017 [7] | |
| 25 | Male | Fetus (terminate) | No | NoM | - | Nephroblastomatosis | Facial dysmorphism | Vertebral ossification defects, rib anomalies | c.496T>A c.501_502delGT | p.Cys166Ser p.Phe168* | DSD | 2018 [17] | |
| 26 | Male | >2 years | 3rd degree | NoM | NoM | Renal calculus, hydronephrosis, congenital megaureter | Microcephaly, developmental delay, flat acetabulum, coxa valga | Vertebral segmentation defects, rib anomalies, ischial hypoplasia, sacral hypoplasia | c.314G>A | p.Cys105Tyr | ISD | 2019 [8] | |
| 27 | Male | Fetus (terminate) | No | Jewish | - | Cystic | Skull malformation | Spine, chest malformation | NA | NA | DSD (pressumed) | 2019 [18] | |
| 28 | Male | Fetus (terminate) | No | Jewish | - | Normal | Feet deformities | Spine malformation | NA | NA | DSD (pressumed) | 2019 [18] | |
| 29 | Female | Fetus (terminate) | No | Jewish | - | Normal | Short trunk, distended abdomen, skull bone ossification defects | Vertebral ossification defects | c.410T>A | p.Val137Asp | DSD | 2019 [18] | |
| 30 | Female | >17 years | No | Korean | None | Polycystic | Facial dysmorphism | Vertebral segmentation defects, rib anomalies, ischial hypoplasia, sacral hypoplasia | c.1672C>T c.1750delT | p.Arg558* p.Cys584fs | Attenuated DSD | Current study | |
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gonzales, M.; Verloes, A.; Saint Frison, M.H.; Perrotez, C.; Bourdet, O.; Encha-Razavi, F.; Joye, N.; Taillemite, J.L.; Walbaum, R.; Pfeiffer, R.; et al. Diaphanospondylodysostosis (DSD): Confirmation of a recessive disorder with abnormal vertebral ossification and nephroblastomatosis. Am. J. Med. Genet. Part A 2005, 136a, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Funari, V.A.; Krakow, D.; Nevarez, L.; Chen, Z.; Funari, T.L.; Vatanavicharn, N.; Wilcox, W.R.; Rimoin, D.L.; Nelson, S.F.; Cohn, D.H. BMPER mutation in diaphanospondylodysostosis identified by ancestral autozygosity mapping and targeted high-throughput sequencing. Am. J. Hum. Genet. 2010, 87, 532–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Neriah, Z.; Michaelson-Cohen, R.; Inbar-Feigenberg, M.; Nadjari, M.; Zeligson, S.; Shaag, A.; Zenvirt, S.; Elpeleg, O.; Levy-Lahad, E. A deleterious founder mutation in the BMPER gene causes diaphanospondylodysostosis (DSD). Am. J. Med. Genet. Part A 2011, 155, 2801–2806. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, T.; Watabe, T. Bone Morphogenetic Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, M.; Binder, O.; Wu, Y.; Aitsebaomo, J.; Ren, R.; Bode, C.; Bautch, V.L.; Conlon, F.L.; Patterson, C. BMPER, a novel endothelial cell precursor-derived protein, antagonizes bone morphogenetic protein signaling and endothelial cell differentiation. Mol. Cell Biol. 2003, 23, 5664–5679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, F.; Wang, C.; Wang, C.; Gao, Y.; Zhang, X.; Chen, X. BMPER Enhances Bone Formation by Promoting the Osteogenesis-Angiogenesis Coupling Process in Mesenchymal Stem Cells. Cell Physiol. Biochem. 2018, 45, 1927–1939. [Google Scholar] [CrossRef] [PubMed]
- Legare, J.M.; Seaborg, K.; Laffin, J.; Giampietro, P.F. Diaphanospondylodysostosis and ischiospinal dysostosis, evidence for one disorder with variable expression in a patient who has survived to age 9 years. Am. J. Med. Genet. Part A 2017, 173, 2808–2813. [Google Scholar] [CrossRef]
- Salian, S.; Nampoothiri, S.; Shukla, A.; Girisha, K.M. Further evidence for causation of ischiospinal dysostosis by a pathogenic variant in BMPER and expansion of the phenotype. Congenit. Anom. 2019, 59, 26–27. [Google Scholar] [CrossRef]
- Zong, Z.; Tees, S.; Miyanji, F.; Fauth, C.; Reilly, C.; Lopez, E.; Tredwell, S.; Goldberg, Y.P.; Delaney, A.; Eydoux, P.; et al. BMPER variants associated with a novel, attenuated subtype of diaphanospondylodysostosis. J. Hum. Genet. 2015, 60, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, G.; Kimizuka, M.; Shiro, R.; Nii, E.; Nishiyama, M.; Kawano, T.; Kaku, T.; Kawada, Y. Ischio-spinal dysostosis: A previously unrecognised combination of malformations. Pediatr. Radiol. 1999, 29, 212–217. [Google Scholar] [CrossRef]
- Spranger, J.; Self, S.; Clarkson, K.; Pai, S. Ischiospinal dysostosis with rib gaps and nephroblastomatosis. Clin. dysmorphol. 2001, 10, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, G.; Kim, O.H.; Sato, S.; Hasegawa, T. Ischiospinal dysostosis with cystic kidney disease: Report of two cases. Clin. Dysmorphol. 2003, 12, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Vatanavicharn, N.; Graham, J.M., Jr.; Curry, C.J.; Pepkowitz, S.; Lachman, R.S.; Rimoin, D.L.; Wilcox, W.R. Diaphanospondylodysostosis: Six new cases and exclusion of the candidate genes, PAX1 and MEOX1. Am. J. Med. Genet. Part A 2007, 143, 2292–2302. [Google Scholar] [CrossRef] [PubMed]
- Scottoline, B.; Rosenthal, S.; Keisari, R.; Kirpekar, R.; Angell, C.; Wallerstein, R. Long-term survival with diaphanospondylodysostosis (DSD): Survival to 5 years and further phenotypic characteristics. Am. J. Med. Genet. Part A 2012, 158, 1447–1451. [Google Scholar] [CrossRef]
- Tasian, S.K.; Kim, G.E.; Miniati, D.N.; DuBois, S.G. Development of anaplastic Wilms tumor and subsequent relapse in a child with diaphanospondylodysostosis. J. Pediatr. Hematol. Oncol. 2012, 34, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Kuchinskaya, E.; Grigelioniene, G.; Hammarsjö, A.; Lee, H.R.; Högberg, L.; Grigelionis, G.; Kim, O.H.; Nishimura, G.; Cho, T.J. Extending the phenotype of BMPER-related skeletal dysplasias to ischiospinal dysostosis. Orphanet J. Rare Dis. 2016, 11, 1. [Google Scholar] [CrossRef] [Green Version]
- Hofstaetter, C.; Courage, C.; Bartholdi, D.; Biskup, S.; Raio, L. Prenatal diagnosis of diaphanospondylodysostosis (DSD): A case report. Clin. Case Rep. 2018, 6, 420–425. [Google Scholar] [CrossRef]
- Lior, G.; Yinon, G.; Annick, R.R.; Ortal, B.; Nitzan, K.; Haike, R.W.; Ben, P.S.; Yael, F.; Baruch, M.; Michal, B. Diaphanospondylodysostosis: Refining the prenatal diagnosis of a rare skeletal disorder. Eur. J. Med. Genet. 2019, 62, 167–171. [Google Scholar] [CrossRef]
- Stanojevic, S.; Wade, A.; Stocks, J.; Hankinson, J.; Coates, A.L.; Pan, H.; Rosenthal, M.; Corey, M.; Lebecque, P.; Cole, T.J. Reference ranges for spirometry across all ages: A new approach. Am. J. Respir. Crit. Care Med. 2008, 177, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Annane, D.; Orlikowski, D.; Chevret, S. Nocturnal mechanical ventilation for chronic hypoventilation in patients with neuromuscular and chest wall disorders. Cochrane Database Syst. Rev. 2014, 12, Cd001941. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef] [Green Version]
| Age of the Patient (Years) | ||||||
|---|---|---|---|---|---|---|
| 12 | 13 | 14 | 15 | 16 | 17 | |
| FVC † (mL) | 320 | 500 | 390 | 280 | 420 | 550 |
| FVC (% reference) | 28 | 25.02 | 18.68 | 13.39 | 20.09 | 25.98 |
| FEV1 † (mL) | 300 | 400 | 330 | 220 | 340 | 420 |
| FEV1 (% reference) | 30 | 22.57 | 18.13 | 12.12 | 19.05 | 23.30 |
| FEV1/FVC | 93.75 | 80 | 84.61 | 78.57 | 80.95 | 76.36 |
| PCF † (L/min) | 60 | 70 | 60 | 60 | 90 | 70 |
| MIP † (cmH2O) | 45 | 24 | 42 | 30 | 40 | 40 |
| MEP † (cmH2O) | 40 | 36 | 36 | 60 | 63 | 65 |
| Height (cm) | 111 | 118 | 120 | 120 | 120 | 120 |
| Weight (kg) | 38 | 49.2 | 54 | 55 | 59.5 | 60 |
| BMI (kg/cm2) | 30.84 | 35.33 | 37.5 | 38.20 | 41.32 | 41.67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.E.; Yoon, J.A.; Shin, Y.B. Successfully Managed Respiratory Insufficiency in a Patient with a Novel Pathogenic Variant of the BMPER Gene: A Case Report. Diagnostics 2022, 12, 626. https://doi.org/10.3390/diagnostics12030626
Park HE, Yoon JA, Shin YB. Successfully Managed Respiratory Insufficiency in a Patient with a Novel Pathogenic Variant of the BMPER Gene: A Case Report. Diagnostics. 2022; 12(3):626. https://doi.org/10.3390/diagnostics12030626
Chicago/Turabian StylePark, Ho Eun, Jin A. Yoon, and Yong Beom Shin. 2022. "Successfully Managed Respiratory Insufficiency in a Patient with a Novel Pathogenic Variant of the BMPER Gene: A Case Report" Diagnostics 12, no. 3: 626. https://doi.org/10.3390/diagnostics12030626