Abstract
Head and neck cancer has poor overall survival. Patients with head and neck cancer more frequently develop second primary tumors than do patients with other cancers, leading to a poor prognosis. In this study, we used next-generation sequencing to analyze and compare mutations between first tumors and second tumors in oral cancer. We retrieved tumor tissues collected from 13 patients who were diagnosed twice as having cancer. We used driver gene and trunk mutations to distinguish between recurrent cancer and primary cancer in oral cancer. We observed unique driver gene mutations in three patients with an initial clinical diagnosis of recurrent cancer; hence, we believe that the corresponding patients had primary cancer. Four patients with an initial clinical diagnosis of primary cancer were found to actually have recurrent cancer according to our results. Genetic testing can be used to enhance the accuracy of clinical diagnosis.
1. Introduction
Head and neck cancers (HNCs) constitute a group of cancers that occur in the mouth, nose, throat, larynx, sinuses, or salivary glands. The symptoms of HNC vary depending on the cancer type [1], with some patients presenting with a nonhealing lump or sore in the mouth and others presenting with a persistent sore throat. Other patients experience trouble swallowing or a change in voice [2]. Nearly 75% of HNCs are caused by the use of alcohol or tobacco [3,4]. However, in Taiwan, such cancers are often caused by areca. Studies have revealed that areca is an essential risk factor for HNC development [5,6]. This trend is different from those reported in Western countries because genomic alterations in HNC differ between the West and the East.
HNC is considered the sixth most common cancer worldwide and constitutes 6% and 3% of cancer-related deaths in men and women, respectively [7,8]. In Taiwan, approximately 7000 people are diagnosed as having HNC, and this cancer causes more than 3000 deaths each year (https://www.hpa.gov.tw/, 24 December 2019). A critical reason for the poor overall survival is that patients with HNC more frequently develop second primary tumors (SPTs) than do patients with other cancers, leading to poor prognosis [9,10]. They are defined as second tumors (STs) that manifest either simultaneously or after the diagnosis of the first tumor (FT). SPTs must be differentiated from local recurrences or primary tumor metastases [11]. Patients with HNC have a high lifetime risk of developing SPTs; the incidence of SPTs in such patients is 2% to 3% annually [8,10,12,13]. SPT diagnostic criteria were first presented by Warren and Gates in 1932; in these criteria, an SPT is defined as a new malignant tumor that is located at a new anatomic side and is adequately separated from the original lesion [14]. On the basis of recent molecular analysis results, the SPT criteria have been modified; the modified criteria suggest that individual tumors arising in the same field as premalignant lesions with different genomic alterations might be regarded as SPTs.
To explain the development of multiple primary tumors in HNC, Slaughter et al. proposed the concept of “field cancerization,” which indicates that, when large areas of mucosa are exposed to carcinogens for a prolonged period, a variety of precancerous lesions are formed, which eventually develop into several independent primary tumors [15]. These events involve multistep processes, including genetic alterations; damage induced by carcinogens, such as tobacco and alcohol; and human papillomavirus (HPV) infection. The discovery of genetic changes appears to support this concept of the origin of independent tumors [16,17]. SPTs constitute the second leading cause of death in patients with HNC [18]. In this study, we identified mutant verifications as markers of SPTs.
HNCs are highly related to lifestyle risk factors, and different forms and levels of exposure to the etiological agents are reflected in different parts of the world. Studies have reported that cancer development involves the accumulation of mutations in oncogenes or tumor suppression genes [19,20,21]; most of such studies have focused on tobacco- and alcohol-related HNCs and rarely on betel quid (BQ)-related HNC. To address this gap in the literature, we collected cancer tissues from 15 patients with SPTs and used next-generation sequencing (NGS) to analyze the mutations in first primary tumors (FPTs) and SPTs to explore the possible signaling pathways between them.
2. Materials and Methods
2.1. Patients and Samples
Tumor samples collected from 13 patients with SPT were retrieved from the human biobank of China Medical University Hospital, Taiwan. DNA was extracted using the QIAamp DNA Micro kit (Qiagen, Heidelberg, Germany) according to the manufacturer’s protocol. The extracted DNA samples were then quantified using a NanoDrop 2000 spectrophotometer (Thermal Fisher Scientific, Waltham, MA, USA) and Qubit fluorometer (Invitrogen, Carlsbad, CA, USA). The Institutional Review Board of China Medical University Hospital (CMUH102-REC1-015, 13 March 2013 approved and CMUH102-REC1-073, 23 September 2013 approved) approved our study.
2.2. Exome Capture and Massively Parallel Sequencing
A TruSeq DNA Sample Preparation Kit (Illumina, San Diego, CA, USA) was used to create the DNA library in accordance with the manufacturer’s guidelines. Genomic DNA (5 μg) was fragmented using a Covaris sonicator (Covaris, Woburn, MA, USA) to a size of 300–500 bps. The library preparation process involved the following steps: enzyme-mediated end repair, adenine addition a-tailing, adapter oligonucleotide ligation, and adapter-ligated fragment enrichment through a limited-cycle polymerase chain reaction (PCR). Human exome capture was performed according to the Illumina TruSeq Exome Enrichment Kit protocol. The DNA library was subjected to denaturation at 95 °C for 10 min and was subjected to hybridization at 58 °C for 16 h using captured probes. Subsequently, streptavidin beads were used to bind biotin-labeled probes that contained the targeted regions of interest. Three washing steps were performed to inhibit nonspecific binding to the beads. The hybridization and washing steps were then repeated. Next, PCR was executed to amplify the enriched DNA library for sequencing, after which the enriched DNA library was purified using an AMPure XP purification system (Agencourt, Beckman Coulter, Brea, CA, USA). Final quantification of the libraries was performed using a Qubit 2.0 Fluorometer high-sensitivity DNA assay (Invitrogen) and an Experion Automated Electrophoresis System (Bio-Rad, Hercules, CA, USA) to ensure sufficient product availability for sample normalization and pooling. Library-prepared samples were sequenced using a HiSeq platform (Illumina, San Diego, CA, USA) to produce 100-bps paired-end sequencing reads.
2.3. Data Analysis
We performed base calling and quality scoring using an updated implementation of real-time analysis on the aforementioned HiSeq platform. Data were demultiplexed, and BCL files were converted to FASTQ files through Bcl2fastq conversion software. Subsequently, the sequenced reads for low-quality sequences were trimmed, after which they were aligned to the human reference genome (hg19) using the Burrows-Wheeler Alignment tool [22]. Small insertions, deletions, or both, and single-nucleotide polymorphisms (SNPs) were next identified in each sample through the Genome Analysis Toolkit and VarScan under their default settings [23,24]. We then applied ANNOVAR [25] and household script to perform gene-based, region-based, and filter-based annotation to functionally annotate variants. Finally, the variants were annotated using several databases and tools, including dbSNP (build 147), ClinVar, COSMIC (ver. 70), TCGA, Polyphen-2, SIFT, and CADD [20,21,22,23,24,25,26].
2.4. Variant Validation through Sanger Sequencing
Primer3 software (Supplementary Table S1) was used to design the PCR primers in silico. We used a Verity 96-well thermal cycler (Applied Biosystems, Foster City, CA, USA) to perform PCR including specific primers, after which we executed conventional PCR-based Sanger sequencing using an ABI 3130 DNA analyzer (Applied Biosystems).
3. Results
3.1. Population Description and Clinical Information in SPT
We retrieved tumor samples collected from 13 patients with SPTs. Of these patients, 12 had FPTs and SPTs in HNCs (Table 1), and one had an FPT in HNC and an SPT in the esophagus, and one had an FPT in the ureter and an SPT in HNC. Ten (77%) patients had habits of smoking, BQ chewing, and drinking; two had habits of BQ chewing and drinking, and one (7%) had none of the aforementioned habits. Table 1 presents the clinical features of SPTs. All cancers were of the squamous cell carcinoma (SSC) type. The pathological tumor–node–metastasis (pTNM) classification system was established by the American Joint Committee on Cancer (AJCC) and the International Union against Cancer to avoid heterogeneity in prognostic classification schemes used for differentiated cancers. The AJCC has created a set of resource materials that provide in-depth information to medical professionals and cancer registrars for staging cancer patients and abstracting cancer cases, respectively. In this study, we used the pTNM classification system to classify tumors and the AJCC staging system to determine tumor stages and evaluate tumor size; the results are presented in Table 1. We divided patients into two groups (Table 1): the upper group, comprising patients clinically diagnosed twice as having primary cancer; and bottom group, comprising patients clinically diagnosed as having recurrent cancers; We recorded the treatment policy for each cancer, including chemotherapy (CT) and radiation therapy (RT).

Table 1.
Description and clinical information of patients included in the study.
3.2. Identifying Variants in SPTs
We performed massive parallel sequencing by using the HiSeq platform. We generated nearly 160 M raw reads per sample, on average; these reads were aligned with the human reference genome (hg19; Supplementary Table S2). The target regions of the 26 samples exhibited a mean depth and coverage of 141 (range: 92.37–175.21) and 99.19% (range: 98.95–99.35%), respectively. Supplementary Figure S1 illustrates a schematic of our variant identification approach. We executed whole-exon sequencing to collect data of variants from our patients’ DNA. The ratio of variant reads to total reads must be greater than 10%. The ratio of variant reads less than 10% may be mistake by amplification or NGS. Subsequently, we used the dbSNP and genome-wide association study (GWAS) database to annotate variants with global minor allele frequencies of more than 1%. Moreover, we used the ClinVar database to annotate the remaining variants. Variants were divided into the following categories: pathogenic, benign, and uncertain. Benign variants were annotated using the dbSNP, COSMIC, and HGVS databases. The pathogenicity of uncertain variants was predicted using the SIFT, PolyPhen, and Combined Annotation Dependent Depletion (CADD) tools.
3.3. Cancer-Related Gene Mutational Status in HNC
This study included 756 canonical cancer-related genes. We detected 297 mutations in 190 of these genes, namely seven frameshift deletions, three frameshift insertions, 261 missense mutations, five non-frameshift deletions, two non-frameshift insertions, and 19 stop-gains. SYNE1 (56%; 15/27), TP53 (52%; 14/27), and CDKN2A (41%; 11/27) were the most frequently mutated genes in HNC. A total of 215 variants were identified in the COSMIC, dbSNP, and TCGA databases, but 75 variants in 80 genes were not identified in these databases (Supplementary Table S1). We executed Sanger sequencing to verify these driver gene variants (Figure 1) and nondriver gene variants (Figure S2).
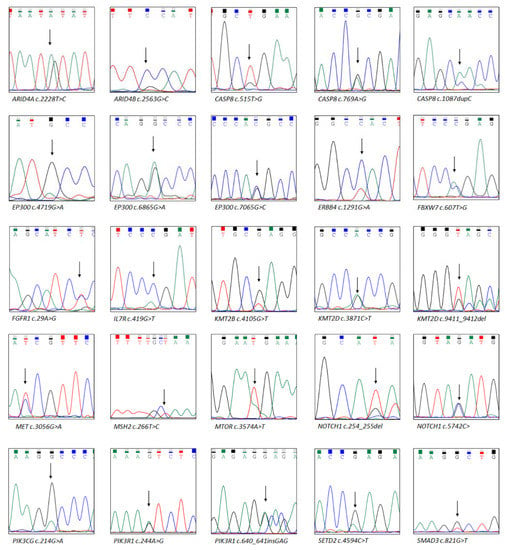
Figure 1.
Driver gene mutations confirmed using Sanger sequencing. Next-generation sequencing data provided these driver gene mutations. The arrow indicates the location of the mutation.
3.4. Mutations Analysis in FPT, SPT, and Intersection Parts
To explore differences in mutations between the FT and the ST in each patient, we divided the observed mutations into three categories: those found only in the FT (oFTp), those found only in the ST (oSTp), and those found in the intersection between these tumors (Figure 2A). NGS revealed nine identical mutations in PA46 patients and nine unique mutations in oSTp mutations. In PA50, PA53, and PA55, the mutations in the FT and ST were the same; PA54 and oFTp had 11 identical and seven unique mutations, respectively; in PA47, PA49, PA52, PA56, PA57, PA58, PA59, and PA60, the mutations in the FT and ST were different (Figure 2B). These classification theories can thus be used to distinguish a second primary oral cancer from other cancers.
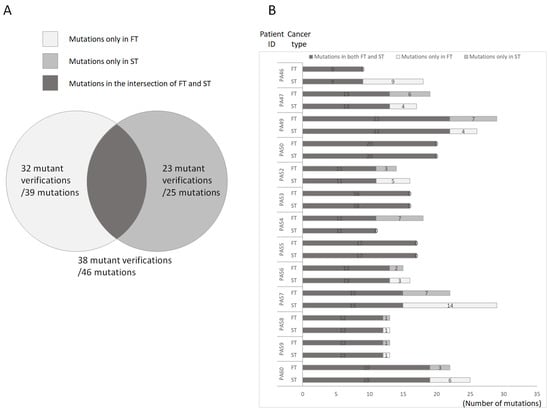
Figure 2.
Schematic of the distribution of mutations in first and second primary tumors. (A). Overview of mutations in first and second tumors. Only first primary tumor showed 39 mutations (White). We verified 32 of the 39 mutations. Only second primary tumor showed 25 mutations (gray). We verified 23 of the 25 mutations. Both first and second primary tumors showed 46 mutations (black). We verified 38 of the 46 mutations. (B). Distribution of mutations in patients. This bar chart shows mutations in first primary tumor (white), second primary tumor (gray), and first and second primary tumors (black). The number in the bar shows the number of mutations.
3.5. Molecular Diagnosis according to Driver Gene Mutations for Differentiating between the FT and ST
As revealed by the results in the preceding section, we explored differences in mutations between the FT and ST in each patient. We also determined each patient’s clinical diagnosis, as presented in Table 1. For further exploration, we used molecular diagnoses made according to driver gene mutations to distinguish between the FT and ST in each patient. Driver genes are necessary for cancer to become malignant. We used 299 driver cancer genes to distinguish between the FT and ST [26,27]. Cancer is a microevolutionary process that originates from a single cell [28,29,30]. The classification of trunk and branch mutations can elucidate the microevolution of cancer [31]. Therefore, we distinguished between trunk and branch mutations in each cancer. A total of 297 genes with verified mutations were classified into driver and nondriver categories (Figure S3). Of the, 83 and 214 were driver and nondriver genes, respectively (Figure 3A). We derived representative results of trunk and branch mutations in primary (Figure 3B) and recurrent (Figure 3C) cancers. We present representative patients in Figure 3 and the remaining results are presented in Figure S4. We sorted the unique mutations in each cancer (Table 2). As mentioned, we divided patients into upper, and bottom groups according to clinical diagnosis. The upper group comprised nine patients. Nevertheless, we believe that three of them (PA50, PA53, and PA55, 3/9) had recurrent cancer because we did not observe a unique driver gene mutation in the ST. The bottom group comprised four patients. However, we observed unique driver gene mutations in PA52, PA57, and PA59; hence, we believe that the corresponding patients had primary cancer.
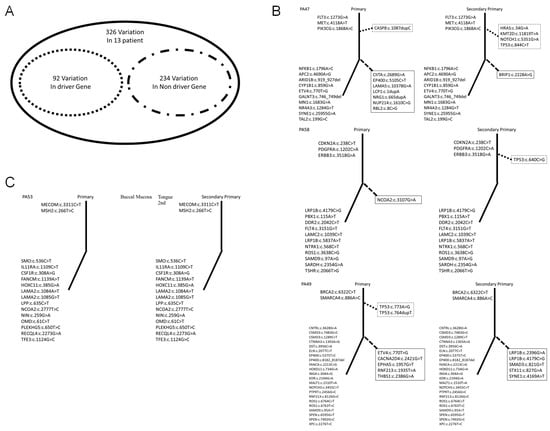
Figure 3.
Analysis of driver gene and trunk mutations in first and second primary tumors. (A). For the 15 patients, we observed 297 mutations. The corresponding genes could be divided into driver genes (83 genes) and nondriver genes (214 genes). (B). First and second primary tumors as categorized according to our results. The solid and dotted lines show trunk and branch mutations, respectively. The upper and lower parts of the graph show driver and nondriver gene mutations, respectively. The dotted line box shows driver gene mutations and trunk mutations. The solid line box shows nondriver gene mutations and branch mutations. (C). Recurrent cancers as categorized in this study.
Table 2.
Unique mutations in first and second primary tumors.
4. Discussion
In this study, we retrieved tissue samples collected from 15 patients who were diagnosed twice as having cancer. We used whole exome sequencing to analyze genetic changes in these cancers. Furthermore, we verified these driver gene variants and applied molecular diagnosis according to driver gene and trunk mutations in recurrent cancer to distinguish between first and recurrent cancers.
In clinical practice, cancer recurrence is diagnosed according to physicians’ judgment and pathological biopsy findings. In the literature, recurrence is defined as a reemergence of cancer at the same location or a nearby site within a short interval after the first diagnosis. It is also defined as pathological biopsy findings revealing the same morphology and malignancy for both occurrences of cancer [32]. The aim of the present study was to describe the differences in mutations between the FT and ST in cancer and evaluate whether the ST is an SPT or recurrence of the FT using molecular diagnosis. Liu conducted a molecular diagnosis for recurrent cancer according to driver gene and trunk mutations [33]. Our results reveal that some patients who were initially diagnosed as having primary cancer actually had recurrent cancer. These recurrent cancers did not show unique driver gene mutations (Table 2). Driver gene and trunk mutations may become a new diagnostic biomarker for distinguishing between recurrent cancer and primary cancer.
In our study, four patients were determined to have recurrent cancer according to our results (Table 2). In one patient (PA53), we did not observe any unique gene mutation between both occurrences of cancer; therefore, the cancer was clearly recurrent. Two patients (PA50, and PA55) showed unique nondriver gene mutations in the second diagnosis of cancer. These patients had undergone either CT or RT after the first cancer diagnosed (Table 1). There may be two groups of cancer cells in such patients, with part of the cancer cells dying after the treatment [34]. These patients also had recurrent cancer. Notably, for the four patients who were clinically diagnosed as having recurrent cancer, our results indicated that they had primary cancer. This result shows the inaccuracy of clinical interpretation.
We analyzed 13 patients who were diagnosed twice as having cancer. Using driver gene and trunk mutations, we distinguished between recurrent and primary cancer oral cancers. These findings may require further research for confirmation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/diagnostics12040951/s1, Figure S1: Overview of our approach to identifying variants in SPTs; Figure S2: Overview of our approach to identifying variants in SPTs; Figure S3: A total of 297 genes with verified mutations were classified into driver and nondriver categories; Figure S4: Analysis of driver gene and trunk mutations in first and second primary tumors; Table S1. Primer list; Table S2. Overview of reads count and quality control from NGS; Table S3. Overview of 297 verified mutations; Data S1: OralCancer_Raw_Data.
Author Contributions
Study design: T.-Y.L., C.-C.L., J.-G.C.; Acquisition and analysis of data: T.-Y.L., C.-C.L., Y.-C.C., Y.-S.C., Y.-T.L., J.-C.Y., H.-Y.H., D.C.; Interpretation of data: T.-Y.L., Y.-S.C., C.-C.L., Y.-C.C., J.-G.C.; Drafting of manuscript: T.-Y.L., C.-C.L., J.-G.C.; Critical revision of manuscript: J.-G.C. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported in part by grants from China Medical University Hospital, Taiwan to T.-Y.L. (DMR-108-167) and from China Medical University to J.-G.C. (CMU103-BC-7).
Institutional Review Board Statement
The Institutional Review Board of China Medical University Hospital (CMUH102-REC1-015, 13/03/2013 approved and CMUH102-REC1-073, 09/23/2013 approved) approved our study.
Informed Consent Statement
Patient consent was waived due to REASON. All of sample collected from biobanks in CMUH.
Data Availability Statement
The data used in this paper is in Supplementary Information.
Acknowledgments
We are grateful to the Human Biobank (China Medical University Hospital, Taiwan) for providing human samples.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| American Joint Committee on Cancer | AJCC |
| betel quid | BQ |
| chemotherapy | CT |
| Combined Annotation Dependent Depletion | CADD |
| first primary tumors | FPTs |
| first tumor | FT |
| genome-wide association study | GWAS |
| Head and neck cancers | HNCs |
| human papillomavirus | HPV |
| human reference genome | hg19 |
| next-generation sequencing | NGS |
| only in the FT | oFTp |
| only in the ST | oSTp |
| pathological tumor–node–metastasis | pTNM |
| polymerase chain reaction | PCR |
| radiation therapy | RT |
| second primary tumors | SPTs |
| Second tumors | STs |
| single-nucleotide polymorphisms | SNPs |
| squamous cell carcinoma | SSC |
| transitional cell carcinoma | TCC |
References
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of Human Papillomavirus-Positive Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 3235–3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zecha, J.A.; Raber-Durlacher, J.E.; Nair, R.G.; Epstein, J.B.; Elad, S.; Hamblin, M.R.; Barasch, A.; Migliorati, C.A.; Milstein, D.M.; Genot, M.T.; et al. Low-level laser therapy/photobiomodulation in the management of side effects of chemoradiation therapy in head and neck cancer: Part 2: Proposed applications and treatment protocols. Support. Care Cancer 2016, 24, 2793–2805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.F.; Chiang, S.L.; Lin, C.Y.; Chang, J.G.; Chung, C.M.; Ko, A.M.; Lin, Y.Z.; Lee, C.H.; Lee, K.W.; Chen, M.K.; et al. Corrigendum: Somatic Mutations and Genetic Variants of NOTCH1 in Head and Neck Squamous Cell Carcinoma Occurrence and Development. Sci. Rep. 2016, 6, 28409. [Google Scholar] [CrossRef] [Green Version]
- Shield, K.D.; Ferlay, J.; Jemal, A.; Sankaranarayanan, R.; Chaturvedi, A.K.; Bray, F.; Soerjomataram, I. The global incidence of lip, oral cavity, and pharyngeal cancers by subsite in 2012. CA Cancer J. Clin. 2017, 67, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.F.; Liu, T.Y.; Liu, S.T. Arecoline inhibits and destabilizes agrin-induced acetylcholine receptor cluster formation in C2C12 myotubes. Food Chem. Toxicol. 2013, 60, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.C.; Huang, Y.L.; Lee, C.H.; Chen, M.J.; Lin, L.M.; Tsai, C.C. Betel quid chewing, cigarette smoking and alcohol consumption related to oral cancer in Taiwan. J. Oral Pathol. Med. Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 1995, 24, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar]
- Chuang, S.C.; Scelo, G.; Tonita, J.M.; Tamaro, S.; Jonasson, J.G.; Kliewer, E.V.; Hemminki, K.; Weiderpass, E.; Pukkala, E.; Tracey, E.; et al. Risk of second primary cancer among patients with head and neck cancers: A pooled analysis of 13 cancer registries. Int. J. Cancer 2008, 123, 2390–2396. [Google Scholar] [CrossRef]
- Leon, X.; Quer, M.; Diez, S.; Orus, C.; Lopez-Pousa, A.; Burgues, J. Second neoplasm in patients with head and neck cancer. Head Neck 1999, 21, 204–210. [Google Scholar]
- Giefing, M.; Rydzanicz, M.; Szukala, K.; Wozniak, A.; Wierzbicka, M.; Szyfter, K.; Kujawski, M. Second primary tumors (SPT) of head and neck: Distinguishing of “true” SPT from micrometastasis by LOH analysis of selected chromosome regions. Neoplasma 2005, 52, 374–380. [Google Scholar]
- Adjei Boakye, E.; Buchanan, P.; Hinyard, L.; Osazuwa-Peters, N.; Schootman, M.; Piccirillo, J.F. Incidence and Risk of Second Primary Malignant Neoplasm After a First Head and Neck Squamous Cell Carcinoma. JAMA Otolaryngol. Head Neck Surg. 2018, 144, 727–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jegu, J.; Binder-Foucard, F.; Borel, C.; Velten, M. Trends over three decades of the risk of second primary cancer among patients with head and neck cancer. Oral Oncol. 2013, 49, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Morris, L.G.; Sikora, A.G.; Patel, S.G.; Hayes, R.B.; Ganly, I. Second primary cancers after an index head and neck cancer: Subsite-specific trends in the era of human papillomavirus-associated oropharyngeal cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, S.; Gates, O. Multiple primary malignant tumors: A survey of the literature and statistical study. Am. J. Cancer 1932, 16, 1358–1414. [Google Scholar]
- Slaughter, D.P.; Southwick, H.W.; Smejkal, W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 1953, 6, 963–968. [Google Scholar]
- Huber, M.H.; Hong, W.K. Biology and chemoprevention of head and neck cancer. Curr. Probl. Cancer 1994, 18, 81–140. [Google Scholar] [CrossRef]
- Braakhuis, B.J.; Tabor, M.P.; Kummer, J.A.; Leemans, C.R.; Brakenhoff, R.H. A genetic explanation of Slaughter’s concept of field cancerization: Evidence and clinical implications. Cancer Res. 2003, 63, 1727–1730. [Google Scholar]
- Gan, S.J.; Dahlstrom, K.R.; Peck, B.W.; Caywood, W.; Li, G.; Wei, Q.; Zafereo, M.E.; Sturgis, E.M. Incidence and pattern of second primary malignancies in patients with index oropharyngeal cancers versus index nonoropharyngeal head and neck cancers. Cancer 2013, 119, 2593–2601. [Google Scholar] [CrossRef]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef]
- Rothenberg, S.M.; Ellisen, L.W. The molecular pathogenesis of head and neck squamous cell carcinoma. J. Clin. Investig. 2012, 122, 1951–1957. [Google Scholar] [CrossRef] [Green Version]
- Pickering, C.R.; Zhang, J.; Yoo, S.Y.; Bengtsson, L.; Moorthy, S.; Neskey, D.M.; Zhao, M.; Ortega Alves, M.V.; Chang, K.; Drummond, J.; et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013, 3, 770–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 174, 1034–1035. [Google Scholar] [CrossRef] [Green Version]
- Ogi, K.; Kobayashi, J.; Nakagaki, T.; Okamoto, J.; Koike, K.; Hirokawa, N.; Someya, M.; Sakamoto, H.; Takada, K.; Tokino, T.; et al. Chemotherapy after progression on nivolumab is essential for responders with genetic alterations of driver gene: Review of two recurrent/metastatic oral squamous cell carcinoma patients. Oral Oncol. 2020, 102, 104509. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Machnik, M.; Oleksiewicz, U. Dynamic Signatures of the Epigenome: Friend or Foe? Cells 2020, 9, 653. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Wu, S.; Lai, J.; Shi, Y.; Qiu, C.; Chen, Z.; Wang, Y.; Gu, X.; Zhou, J.; Chen, S. Identification of trunk mutations in gastric carcinoma: A case study. BMC Med. Genom. 2017, 10, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, N.V.; Kang, E.T.B.; Senger, C.; Poh, C.F. Oral cancer in a 5-year-old boy: A rare case report and review of literature. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2020, 130, e10–e19. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, M.; Ying, S.; Zhang, C.; Lin, R.; Zheng, J.; Zhang, G.; Tian, D.; Guo, Y.; Du, C.; et al. Genetic Alterations in Esophageal Tissues From Squamous Dysplasia to Carcinoma. Gastroenterology 2017, 153, 166–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szturz, P.; Vermorken, J.B. Management of recurrent and metastatic oral cavity cancer: Raising the bar a step higher. Oral Oncol. 2020, 101, 104492. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).