Abstract
Germline mutations in the tumor suppressor gene BRCA1-associated protein-1 (BAP1) lead to BAP1 tumor predisposition syndrome (BAP1-TPDS), characterized by high susceptibility to several tumor types, chiefly melanoma, mesothelioma, renal cell carcinoma, and basal cell carcinoma. Here, we present the results of our ten-year experience in the molecular diagnosis of BAP1-TPDS, along with a clinical update and cascade genetic testing of previously reported BAP1-TPDS patients and their relatives. Specifically, we sequenced germline DNA samples from 101 individuals with suspected BAP1-TPDS and validated pathogenic variants (PVs) by assessing BAP1 somatic loss in matching tumor specimens. Overall, we identified seven patients (7/101, 6.9%) carrying six different germline BAP1 PVs, including one novel variant. Consistently, cascade testing revealed a total of seven BAP1 PV carriers. In addition, we explored the mutational burden of BAP1-TPDS tumors by targeted next-generation sequencing. Lastly, we found that certain tumors present in PV carriers retain a wild-type BAP1 allele, suggesting a sporadic origin of these tumors or a functional role of heterozygous BAP1 in neoplastic development. Altogether, our findings have important clinical implications for therapeutic response of BAP1-TPDS patients.
1. Introduction
The BRCA1-associated protein-1 (BAP1) gene, composed of 17 exons located on chromosome 3p21, encodes for a ubiquitin carboxy-terminal hydrolase that modulates multiple cellular activities, such as transcription control, DNA repair, chromatin modification, mitochondrial function, and cell death [1].
Carriers of heterozygous germline pathogenic variants (PVs) of BAP1 are affected by a hereditary condition called BAP1 tumor predisposition syndrome (BAP1-TPDS), characterized by predisposition to a wide range of tumors, whose core spectrum includes mesothelioma, cutaneous and uveal melanoma (CM and UM, respectively), renal cell carcinoma (RCC), and melanocytic BAP1-mutated atypical intradermal tumor (MBAIT). More recently, other tumors, such as basal cell carcinoma (BCC), cholangiocarcinoma, and meningioma, have been associated with BAP1-TPDS, albeit characterized by a much lower incidence [2,3,4]. Tumors not yet officially included in the BAP1-TPDS spectrum, but found in carriers of BAP1 PVs, are breast cancer, non-small cell lung adenocarcinoma, and neuroendocrine carcinoma [5,6,7].
BAP1-TPDS is inherited in an autosomal dominant fashion [2,5,8,9,10,11] with a penetrance close to 100% in older age individuals [7,12]. In general, carriers of germline BAP1 PVs develop tumors at a younger age than that of sporadic patients [4,7,12,13].
Several studies have shown that many—but not all—mesothelioma patients with germline BAP1-TPDS are characterized by prolonged survival compared to wild-type (wt) BAP1 patients [8,9,12,14,15,16,17,18]. In contrast, BAP1 PV carriers affected by melanoma—both cutaneous and uveal—exhibit increased risk of metastasis, suggesting a shorter survival rate [19]. A similar outcome is also observed in RCC patients with germline BAP1 mutations [11]. On the other hand, long-term follow-up data are not available for patients with BCC, most likely owing to the fact that these tumors are generally not aggressive [6,20].
Among BAP1-TPDS-associated tumors, MBAITs are regarded as benign skin lesions characterized by BAP1 inactivation. Since they arise during the first two decades of life and tend to increase in number with age, their detection can be useful for early cancer diagnosis as they help identify BAP1 PV carriers several years before the development of more aggressive tumors belonging to the BAP1-TPDS spectrum [6,9,21].
BAP1-TPDS individuals may also develop meningiomas, which are primary central nervous system (CNS) tumors of the meninges. Even though these tumors are generally slow-growing, highly aggressive grade III rhabdoid meningiomas can sometimes be found in TPDS patients [22,23].
BAP1-TPDS should be suspected if an individual has two or more tumors of the BAP1-TPDS spectrum (including MBAITs) or has one BAP1-TPDS malignancy and a first- or second-degree relative with a tumor included in the BAP1-TPDS spectrum, excluding cases or families with two BCCs and/or CMs because of their high frequency in the general population [6]. In this regard, Walpole and colleagues have recently shown that it is essential to test and identify germline BAP1 carriers in order to implement surveillance, which ultimately leads to improved survival and cost savings for the healthcare system [4]. Indeed, cascade genetic screening in patients’ relatives carrying germline mutations can be critical for timely initiation of therapies and/or preventive measures (e.g., limiting sunlight exposure) [6].
BAP1 expression is frequently lost in mesothelioma, UM, and RCC due to somatic inactivation [7,18,24]. More specifically, this inactivation has been observed in 30–60% of sporadic mesotheliomas [7,25,26,27], a characteristic that has been used in diagnostics [28].
Although the tumor genome of sporadic mesothelioma patients has been extensively investigated, the tumor genome of patients affected by BAP1-TPDS has yet to be systematically characterized. A single paper analyzed the genome of several metachronous tumors in a BAP1-TPDS patient [29]. In a recent study involving 17,152 patients with different tumor types, Srinivasan et al. reported some information of the somatic genome of six patients with TPDS [30].
We report here the results of our 10-year-long clinical diagnostics of BAP1-TPDS. Overall, we performed BAP1 Sanger sequencing and multiplex ligation-dependent probe amplification (MLPA) assay on germline DNA samples from 101 suspected BAP1-TPDS individuals. To validate the functional role of the identified variants, we searched for BAP1 loss in the tumor tissues from the patients harboring mutations. When possible, germline genetic testing and tumor analyses were performed on the patients’ relatives, as well.
The diagnostic and prognostic significance of our integrated analysis of germline and somatic alterations in BAP1-TPDS individuals will be discussed in the context of precision medicine therapies and preventive strategies.
2. Materials and Methods
2.1. Patients
BAP1-TPDS individuals were enrolled in the study in the following Italian healthcare settings: Antonio e Biagio e Cesare Arrigo Hospital (Alessandria); Pathology Unit at San Luigi Gonzaga Hospital (Orbassano, Turin); Molecular Genetics and Biology Unit at Santa Croce e Carle Hospital (Cuneo); Division of Dermatology at Maggiore della Carità Hospital (Novara); Genetics and Pathology Units at City of Health and Science Hospital (Turin); Immunology and Diagnostics Molecular Oncology Unit of Veneto Institute of Oncology IOV-IRCCS (Padua).
Patients were selected according to the following criteria: a family history of cancers and the presence of one tumor of the BAP1-TPDS spectrum affecting the proband or a family member. The clinical features of the 101 patients are reported in Table S1. Thirty-nine out of 101 patients with CM or multiple CMs with a family history of melanoma were previously reported [31].
2.2. Germline Genome Analyses
Genomic DNA from whole peripheral blood of 98 patients was analyzed by Sanger sequencing of the 17 exons, intron–exon boundaries, and promoter region (~1000 bp upstream of the ATG) of BAP1 (NM_004656.2) as previously described [32]. The germline DNA extracted from the peripheral blood of patients MM981, MM1012, and MM400 was analyzed by NGS technique using a customized gene panel (kit QIAseq™ Targeted DNA Panels, Qiagen, Hilden, Germany) based on amplicon technology, which targets the exonic regions and the splice junctions of five known familial melanoma susceptibility genes (i.e., BAP1, CDKN2A, CDK4, POT1, and MITF). Briefly, genomic dsDNAs were subjected to fragmentation, adapter ligation with unique molecular indices, target enrichment, and library amplification according to the manufacturer’s protocol (Qiagen, Hilden, Germany). The library pool was sequenced with the MiSeq platform (Illumina, San Diego, CA, USA) using the reagents of the v3-600 flow-cell (Illumina, San Diego, CA). The FASTQ files were imported into the CLC Genomic Workbench program (Qiagen, Hilden, Germany) and analyzed through a pipeline optimized for the identification of germline variants (Identify QIAseq DNA Germline Variants).
Variants were classified as pathogenic according to the American College of Medical Genetics (ACMG) guidelines [33,34]. The identified PVs were also evaluated on tumor DNA by Sanger sequencing. The germline copy number variation (CNV) was assessed by MLPA assay using SALSA MLPA P417 BAP1 probemix (MRC-Holland, Amsterdam, The Netherlands).
The MSH6 variant identified by NGS in the tumor samples from proband HO19.01 (II-3) and her brother (II-2) was validated by Sanger sequencing on the germline DNAs of these two subjects. Detailed protocols for amplification, sequencing, and tumor analyses are reported in the Supplementary Data.
2.3. Immunohistochemical and LOH Analyses
To determine the functional role of the variants in carcinogenesis, different analyses were performed on formalin-fixed paraffin-embedded (FFPE) tumor tissues or pleural effusion specimens. Immunohistochemistry (IHC) was performed on FFPE tissue sections using an anti-human BAP1 primary antibody (rabbit monoclonal, clone C-4, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). To determine BAP1 LOH, microsatellite analysis on tumor DNA extracted from FFPE specimens was performed. MLPA assays on DNA extracted from the pleural effusion samples were also performed. MLPA, IHC, and microsatellite analyses were performed as described previously [32,35].
2.4. RT-PCR Analysis of Splice Variant Effects
Total RNA was extracted from the peripheral blood mononuclear cells after Ficoll-Paque density gradient centrifugation and reverse-transcribed into cDNA using the High Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific, Waltham, MA, USA) (HO19.01 case) or, alternatively, extracted by the Maxwell® RSC Whole Blood DNA method (Promega, WI, USA) and transcribed using the SuperScript® III First-Strand Synthesis System (Invitrogen, Thermo Fisher Scientific, CA, USA) (MM400 and MM1012 cases).
The cDNA was amplified using the following primer pairs:
- -
- forward BAP1-exon 1 (F 5′-ATGAATAAGGGCTGGCTGGAGCT-3′)–reverse BAP1-exon 4 (R 5′-CTGGTGGGCAAAGAACATG-3′) for the HO19.01 patient;
- -
- forward BAP1-exon 5 (F 5′-CCCTGAGTCGCATGAAGGA-3′)–reverse BAP1-exon 7 (R 5′-GTAGACCTTCAGCCCATCCA-3′) for the MM400 patient;
- -
- forward BAP1-exon 8 (F 5′-CGAGGAGTGGACAGACAAG-3′)–reverse BAP1-exon 10 (R 5′-ACTTGTTGCTGGCTGACTTG-3′) for the MM1012 patient.
The PCR products were loaded in a 2% agarose gel to detect the transcript alterations. Sanger sequencing was performed directly on PCR products or on abnormal-size products extracted from the gel.
2.5. Somatic NGS-Targeted Sequencing
DNA-based NGS analysis was performed on tumor specimens using a multigene panel (Oncomine Comprehensive Assay v3, OCAv3) (Thermo Fisher Scientific, Waltham, MA, USA), which targets 161 genes (including BAP1) following the manufacturer’s instructions (for details, see: https://www.thermofisher.com/order/catalog/product/A35805 (accessed on 10 January 2022)). Briefly, the tumor areas were selected by a pathologist (LR) by means of focal assistant dissection at the microscope in order to ensure adequate content of tumor cells. Somatic DNA was extracted using a Maxwell® RSC DNA FFPE Kit (Promega, Madison, WI, USA) following the manufacturer’s instructions. DNA concentration was quantified using Qubit™ dsDNA High Sensitivity Assay kit (Thermo Fisher Scientific, Waltham, MA, USA) on the Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). NGS analysis was based on Ion AmpliSeq technology, which requires 20 ng of input for high-quality FFPE DNA to interrogate the 161 genes (including BAP1). Deamination reaction was conducted using uracil-DNA glycosylase—heat labile (Thermo Fisher Scientific, Waltham, MA, USA). All library preparations for Oncomine Comprehensive Assay v3 (OCAv3) were manually performed according to the manufacturer’s instructions. Multiplex PCR amplification was carried out using a DNA concentration of approximately 20 ng as input for both assays. Libraries were loaded on Ion 540™ Chips using the Ion Chef™ System and sequenced using Ion Torrent GeneStudio™ S5 Prime (all Thermo Fisher Scientific, Waltham, MA, USA).
The data were mapped to the human genome assembly 19, loaded as a standard reference genome into Ion Reporter™ Software (v. 5.16) (Thermo Fisher Scientific, Waltham, MA, USA). Torrent Suite Software (TSS) vs 5.12.2 generates BAM files, which were uploaded on the Ion Reporter (IR) server used for initial automated analysis. The quality of the sequencing reaction was verified by filtering the coverage analysis, mapped reads, mean depth, uniformity, and alignment of the target region according to the established p-value set from 0.0 to 1.0 [36,37]. Subsequently, visual inspection of BAMs was carried out by graphic alignment using Ion Reporter™ Genomic Viewer (IRGV) (Thermo Fisher Scientific, Waltham, MA, USA). Variants of unknown significance (VUSs) were evaluated using MetaLR and MetaSVM, which combine 10 in silico prediction tools (i.e., SIFT, PolyPhen-2 HDIV, PolyPhen-2 HVAR, GERP++, Mutation Taster, Mutation Assessor, FATHMM, LRT, and SiPhy) [38]. Variants described in ClinVar (www.clinvar.org (accessed on 18 January 2022)) as benign or likely benign were discarded. All samples were sequenced with a mean coverage of 2000×, with ≥95% of uniformity of amplicon target that had at least 500 reads. The variant allele frequency (VAF) was calculated as the percentage of sequence reads observed matching a specific DNA variant divided by the overall coverage at that locus. VAF is thus a surrogate measure of the proportion of DNA molecules in the original specimen carrying the variant. Copy number (CN) value was set at 2 for autosomal amplicons, and copy gains (≥3) or losses (≤1) were detected.
2.6. Cascade Testing and Follow-Up
Cascade genetic screening was performed on 9 relatives of the identified PV carriers. For 8 relatives, the search for the specific germline PV was performed by Sanger sequencing on germline DNA, whereas for one (MPM_HO1901 II-2) on the available tumor tissue. The tumor tissue of carriers was also analyzed when available. An update of the clinical and tumor molecular features of three carriers we previously described—i.e., the proband’s daughter in family A (III-2), the proband’s brother in family A1 (II-2), and the proband’s daughter in family A1 (III-2) [31,35]—is reported in Table 1.

Table 1.
Germline BAP1 PVs identified in the families analyzed in the present study.
3. Results
3.1. Identification of Families with BAP1-TPDS
Upon sequencing germline DNAs from 101 Italian patients with suspected BAP1-TPDS (Table S1), we found six different germline BAP1 PVs harbored by patients from seven families (Table 1) (Figure S1). Four of these families (carriers of the PVs c.46_47insA, c.1153C>T p.Arg385*, c.783+2T>C, and c.38-1G>T) had already been described by our group [31,32,35,38]. Table 1 provides an update on the clinical features of these probands and/or their relatives. The three previously unreported families carry the following BAP1 PVs: c.783+2T>C (the same as ID_5), c.605G>A p.Trp202*, and c.376-2A>G.
Of the six aforementioned PVs, which appear to be very rare in the general population (MAF < 0.0001) [35], two are nonsense, three are splicing, and one is a frameshift mutation.
MLPA analysis of germline DNA did not reveal any deletions or duplications of BAP1.
3.2. Family A
Family A was one of the first families to be tested for BAP1 mutations in 2015 [35] due to a known familial history of mesothelioma. Specifically, three members of this family (i.e., the proband, her sister, and her mother) were all affected by mesothelioma—either pleural or peritoneal—and four of them (i.e., the proband, her daughter and son, and the mother of the proband) carried the c.46_47insA PV (Figure S2). The other family members were not tested. As the proband’s daughter (III-2) had been diagnosed with meningioma—in 2017, at the age of 48—we sought to determine whether this tumor also displayed loss of BAP1 protein expression. IHC analysis of FFPE tissue revealed robust BAP1 protein expression in the nuclei of the stromal cells, but it failed to detect BAP1 expression in the tumor cells (Figure 1), suggesting a functional role of the variant in this meningioma.
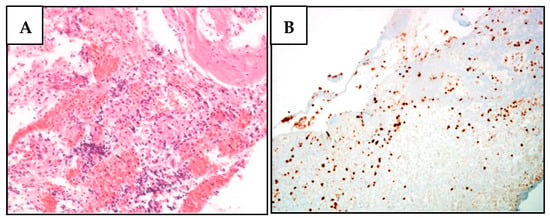
Figure 1.
BAP1 nuclear expression is lost in meningioma cells of the proband’s daughter (III-2) (family A). (A) H&E staining of a relapsed meningioma section characterized by the presence of rare and minute fragments of a highly vascular round and spindle cell proliferation, with scant atypia and no evident mitoses. Tumor cells expressed EMA and smooth muscle actin, in the absence of synaptophysin, S100, CD34, and HMB45. (B) No BAP1 expression was observed in meningioma cells, while it was maintained in stromal cells.
3.3. Family A1
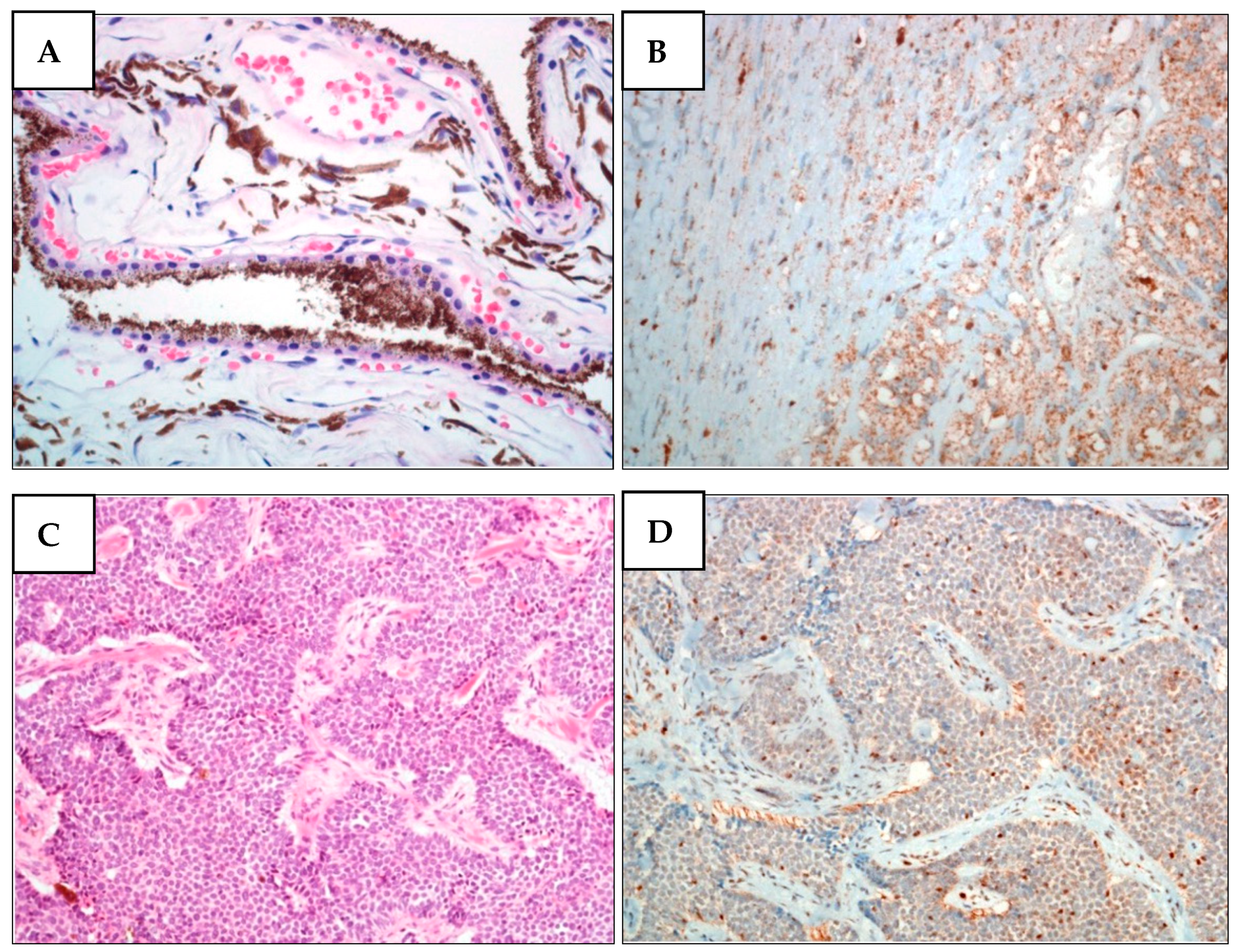
Similarly to family A, family A1 had also already been reported by our group [31]. Proband II-5 presented with the following metachronous tumors: pleural mesothelioma (PlM), multiple melanoma, meningioma, and basal cell carcinoma (BCC), and carried the c.1153C>T p.Arg385* PV. The follow-up showed that her carrier daughter (III-2) had developed a CM in 2015, at the age of 33 [31], whereas her brother (II-2) had developed a UM in 2014 and a PlM in 2019, at 59 and 64 years old, respectively. Upon genome sequencing, II-2 was found to harbor the nonsense c.1153C>T p.Arg385* variant (Figure S3). No other family members were available for BAP1 testing. IHC analyses performed on the proband’s BCC and on her brother’s UM revealed loss of BAP1 expression (Figure 2). Proband II-5 died in 2019, nine years after being diagnosed with PlM, whereas her brother (II-2) died two years after the same diagnosis. The proband’s daughter (III-2) is still alive.
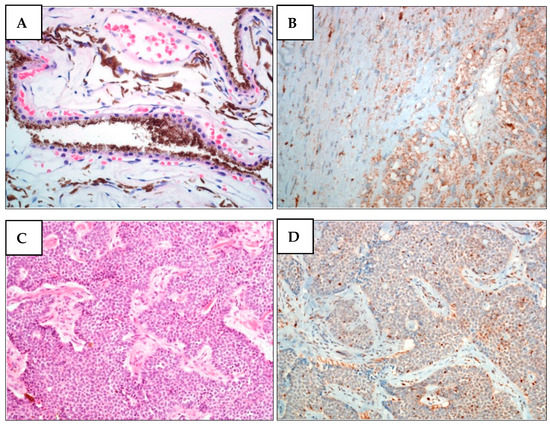
Figure 2.
BAP1 nuclear expression is lost in melanoma cells from the proband’s brother (II-2) and in BCC cells from proband (II-5) (family A1). (A) H&E staining of II-2 UM. Unevenly pigmented epithelioid cell melanoma with radial extension involving over half of the iris and infiltrating the underlying connective tissue of the ciliary body. (B) BAP1 IHC of II-2 UM. The expression of BAP1 is lost in both nucleus and cytoplasm of tumor cells. (C) H&E staining of II-5 BCC. (D) BAP1 IHC of II-5 BCC. In basal cell carcinoma, BAP1 expression was only observed in lymphocytes and stromal cells but not in tumor cells.
3.4. Family ID_5
Family ID_5 had also been previously described by our group [32]. The proband carries the canonical splice-site variant c.783+2T>C, which has led to a complete loss of BAP1 protein expression, as judged by IHC. This is consistent with MLPA analysis of the proband’s pleural effusion, showing a 50% deletion of BAP1 [32]. In the same study, we also showed by FISH analysis that loss of BAP1 expression was likely due to different chromosomal alterations, such as homozygous or heterozygous deletion or monosomy. Specifically, FISH analysis of pleural effusion, which contains a high number of normal cells, revealed that BAP1 biallelic loss was 24%, while the same analysis of FFPE showed that the loss was 73% (Table 2) [32].
Table 2.
Analyses on tumor specimens of the mutated patients and relatives.
In addition, we also performed microsatellite analysis on the DNA extracted from pleural effusion of patient ID_5 III-1, recording a 50% reduction in one allele (Table 2 and data not shown). Moreover, we sought to identify specific somatic BAP1 molecular alterations. NGS analysis of FFPE PlM tissue revealed the already known germline variant (VAF 52.96%, coverage > 500×) but failed to detect any other pathogenic mutations. However, a significant loss (p = 0.006) of the gene copy number was recorded, albeit fairly heterogeneous, confirming the previously described heterogeneous loss detected by FISH (Table 2) [32]. This mutation results in exon 9 skipping, as shown by the experiments described below.
3.5. Family PD-601
This family has never been described before (Figure S4). The MM1012 female proband had a history of CM, the first one diagnosed at 55 years. Her mother and paternal aunt had lung cancer. A cousin—daughter of one of her maternal aunts—had been diagnosed with CM, while another one—a daughter of another maternal aunt with leukemia—had an unspecified womb cancer. Moreover, her paternal grandmother had suffered from liver cancer.
The proband germline DNA was tested for BAP1 and revealed the canonical splice-site variant c.783+2T>C, also found in patient ID_5 III-1. Although the melanoma cells showed a weak focal BAP1 nuclear expression at IHC (Figure S5A), the DNA extracted from the FFPE tumor specimens revealed LOH in the tumor (Figure S5B). Cascade testing for this family was not performed. We tested the effect of the variant on BAP1 splicing by analyzing the transcript of the proband. cDNA amplification revealed two BAP1 PCR products of different sizes, consistent with exon 9 skipping (Figure S6). Sequencing of the fragments confirmed exon 9 skipping, supporting a pathogenic role of this variant.
3.6. Family ID MPM_HO1901
The proband (II-3) (Figure S7) had developed PlM (in 2020, age 58), RCC (in 2007, age 46), and lung adenocarcinoma (LUAD) (in 2008, age 47) and carried the canonical splice-site variant c.38-1G>T [39].
To demonstrate the effect of this variant on BAP1 splicing, we amplified the cDNA of patient MPM_HO1901 II-3 by using primers located on exon 1 and 4, and observed, along the wt PCR product, a small fragment consistent with exon 2 skipping (Figure S8A). Sequencing of the PCR products revealed two shortened transcripts, one caused by exon 2 skipping and the other resulting from the use of a cryptic acceptor site in exon 2 (Figure S8B,C), that are expected to encode two proteins lacking 10 or eight amino acids, respectively. These mutant proteins may be unstable or may have lost their deubiquitinase activity, which requires the N-terminal of BAP1. Overall, these data are consistent with a likely pathogenic effect of this variant.
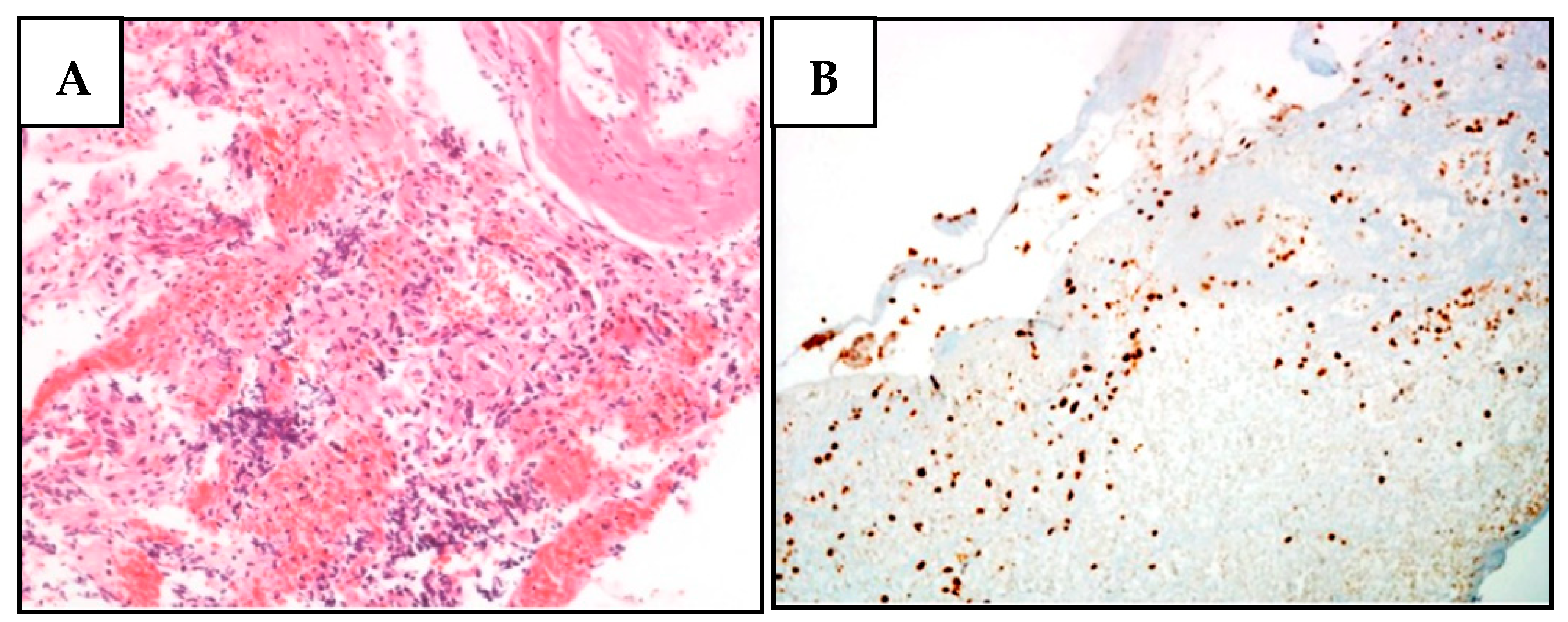
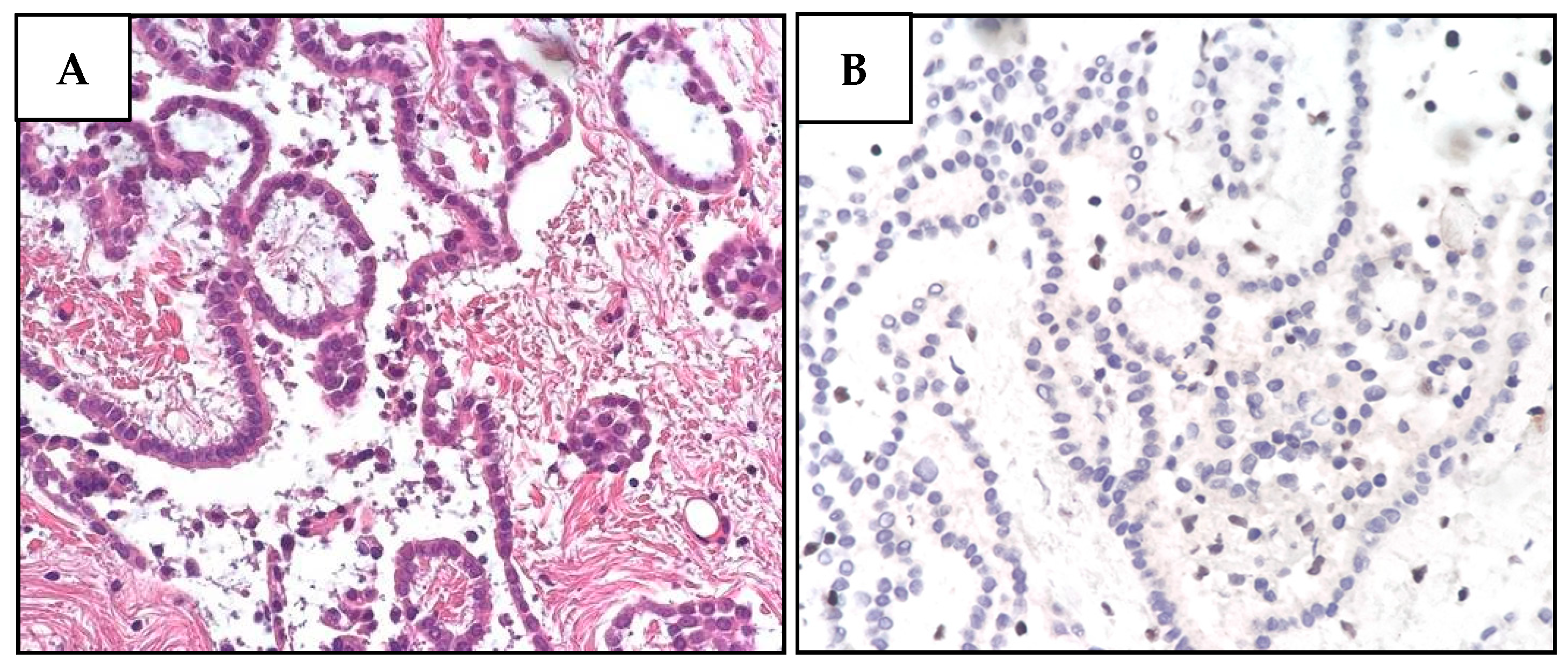
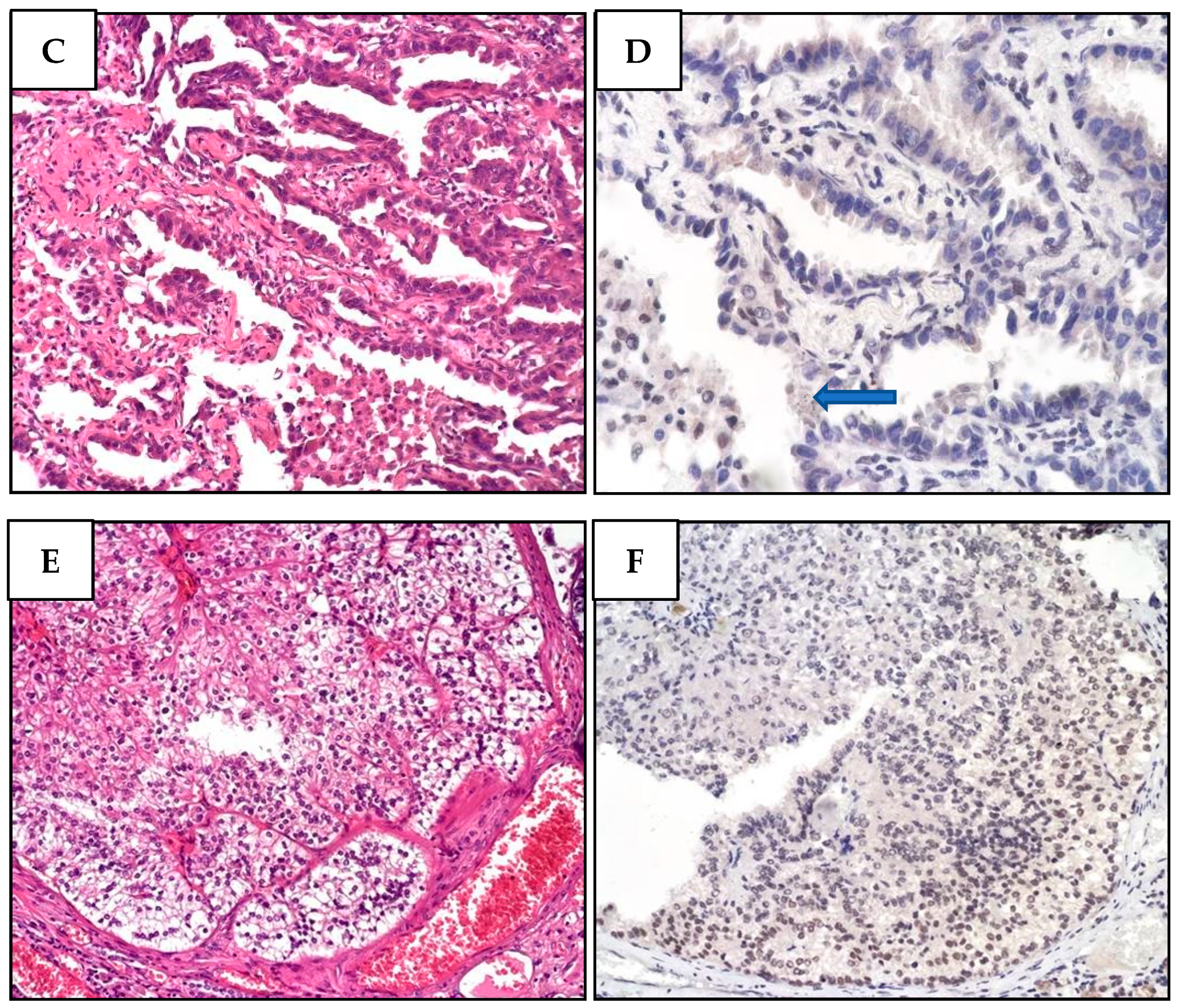
Next, we performed IHC on the proband’s tumor tissues to evaluate BAP1 status. While PlM and LUAD cells showed a complete loss of BAP1 protein expression (Figure 3), the nuclei of normal stromal cells expressed high levels of BAP1 protein. In contrast to the other tumors, RCC cells displayed a partially unreactive expression possibly due to poor tissue preservation (Figure S9).
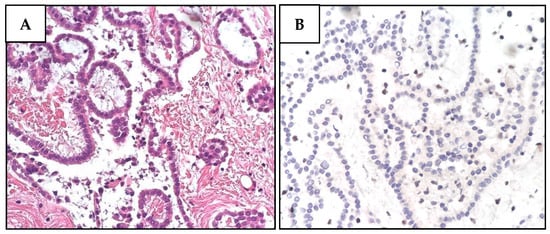
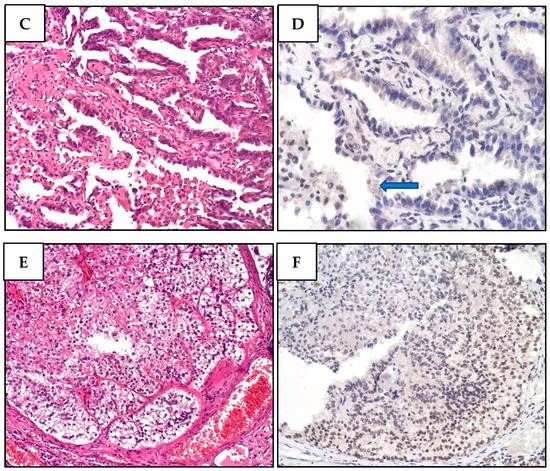
Figure 3.
Histological and immunohistochemical features of proband MPM_HO1901 (II-3) tumors. (A) H&E staining of pleural mesothelioma (PlM), showing a diffuse, mostly superficial, tubular-papillary proliferation of well-differentiated neoplastic mesothelial cells with focal invasion of the adipose tissue. Neoplastic cells are positive for calretinin and WT1, while they are negative for TTF1 and CEA. (B) BAP1 immunohistochemical expression is lost in both the nucleus and cytoplasm of PlM cells, while it is retained in normal stromal cells. (C) Conventional LUAD reveals neoplastic glandular structures. The lesion is a well-differentiated adenocarcinoma with a predominant lepidic growth of neoplastic cells and focal areas of stromal invasion. (D) BAP1 expression is absent in adenocarcinoma cells, while it is retained in normal interalveolar histiocytes (arrow). (E) Renal cell carcinoma (RCC) showing tumor cells characterized by a distinct cell membrane and optically clear cytoplasm associated with more eosinophilic neoplastic cells. The grading score is moderately differentiated due to the presence of polymorphic nuclei, evident nucleoli, and rare mitoses. (F) BAP1 immunohistochemistry reveals a heterogeneous expression pattern including areas of nuclear retention (upper left) and areas with BAP1 nuclear and cytoplasmic loss (lower right).
NGS analysis confirmed the presence of the germline variant in all the tumor tissues analyzed, albeit with different VAFs (Table 2, Figure S10A). In particular, PlM cells harbored the germline splicing variant with a VAF of 89% (coverage 1000×), which was associated with a significant (p = 0.00004) gene copy number loss (0.9 ratio) (Figure S10B), consistent with the complete somatic deletion of the wt allele. Accordingly, MLPA on DNA extracted from fresh pleural effusion specimens showed a 50% loss of the entire BAP1 gene (Figure S10C), which was again consistent with the complete loss of the wt sequence.
The LUAD tumor tissue carried the germline BAP1 variant with a VAF of 21.2% (coverage 500×). We also found three somatic mutations with a low VAF. The DNA extracted from the FFPE specimens was partially degraded, so the results could not be adequately interpreted.
Although BAP1 loss is not often detected in LUAD [40], we did not detect BAP1 protein expression by IHC analysis. Our results shows that LUAD may be associated with BAP1-TPDS, as previously reported [29].
NGS analysis of the RCC sample identified a germline mutation with a VAF of 48.48% (coverage 1500×), associated to the pathogenic SNV p.Trp196* (2% VAF, coverage 1500×, p-value = 0.002, COSM1424466) reported in reference databases (Table 2). Hence, this tumor might not have been caused by TPDS, at least at its initiation.
Cascade genetic analyses were feasible for the proband’s brothers and daughters. One of the proband’s brothers (II-2) died of PlM in 1997. Unfortunately, after performing IHC on his FFPE samples, all tissues were unstained—even stromal cells—likely due to poor specimen condition (data not shown). Nonetheless, NGS analysis revealed the presence of the germline BAP1 variant c.38–1G>A with a VAF of 53.54% (coverage > 500×). The tumor also showed the pathogenic somatic variant p.Trp196* (3.36% VAF, coverage 500×, p-value = 0.0004) (Table 2). Interestingly, the same somatic variant was harbored by the proband’s RCC.
Upon somatic NGS analysis of both the proband’s (II-3) and her brother’s (II-2) tumor samples, we identified a second variant: p.Arg1076His in MSH6 (48% VAF in the proband’s mesothelioma and RCC; 25% VAF in her brother’s mesothelioma) (Table 2). This gene (MIM#600678) is responsible for Lynch syndrome (OMIM#614350). No further driver mutations were detected in any of the other genes tested. We also searched for PVs in the other brother of the proband (II-1), who was found mutated at the germline level in MSH6 but not in BAP1. We also performed mutational analyses on the germline DNA of the proband’s healthy daughters, uncovering that one of them carried the PV in BAP1 (age 23, III-2), while the other one carried the PV in MSH6 (age 26, III-1).
To deepen the role of this variant, we analyzed the mesothelioma tissue of proband II-3. Since MSH6 was normally expressed, as judged by IHC, and the mismatch repair (MMR) status was not modified (data not shown), we deemed this variant to be benign. Of note, while VarSome classifies this variant as likely pathogenic, ClinVar reports conflicting interpretations (https://varsome.com/variant/hg19/MSH6%3AR1076H?annotation-mode=germline (accessed on 4 March 2022); https://www.ncbi.nlm.nih.gov/clinvar/variation/186361/?oq=((182140[AlleleID]))&m=NM_000179.3(MSH6):c.3227G%3EA%20(p.Arg1076His) (accessed on 4 March 2022)). However, we cannot rule out a tissue-specific effect in the colonocytes.
3.7. Family PD-578
Like family PD-601, this family has never been reported (Figure S11). Proband MM981 had multiple CMs (i.e., two melanomas at 68, one at 73, and one at 77 years old) and prostate cancer at the age of 60. He has three sisters: one is healthy; the second one has pancreatic cancer; the third one was diagnosed with breast cancer at 56 years old and died at the age of 60. Her daughter died of mesothelioma at the age of 56. Moreover, the proband’s paternal uncle developed prostate cancer. In this family, we only tested the proband’s germline DNA, identifying a previously unknown BAP1 stop-gain variant: c.605G>A p.Trp202*.
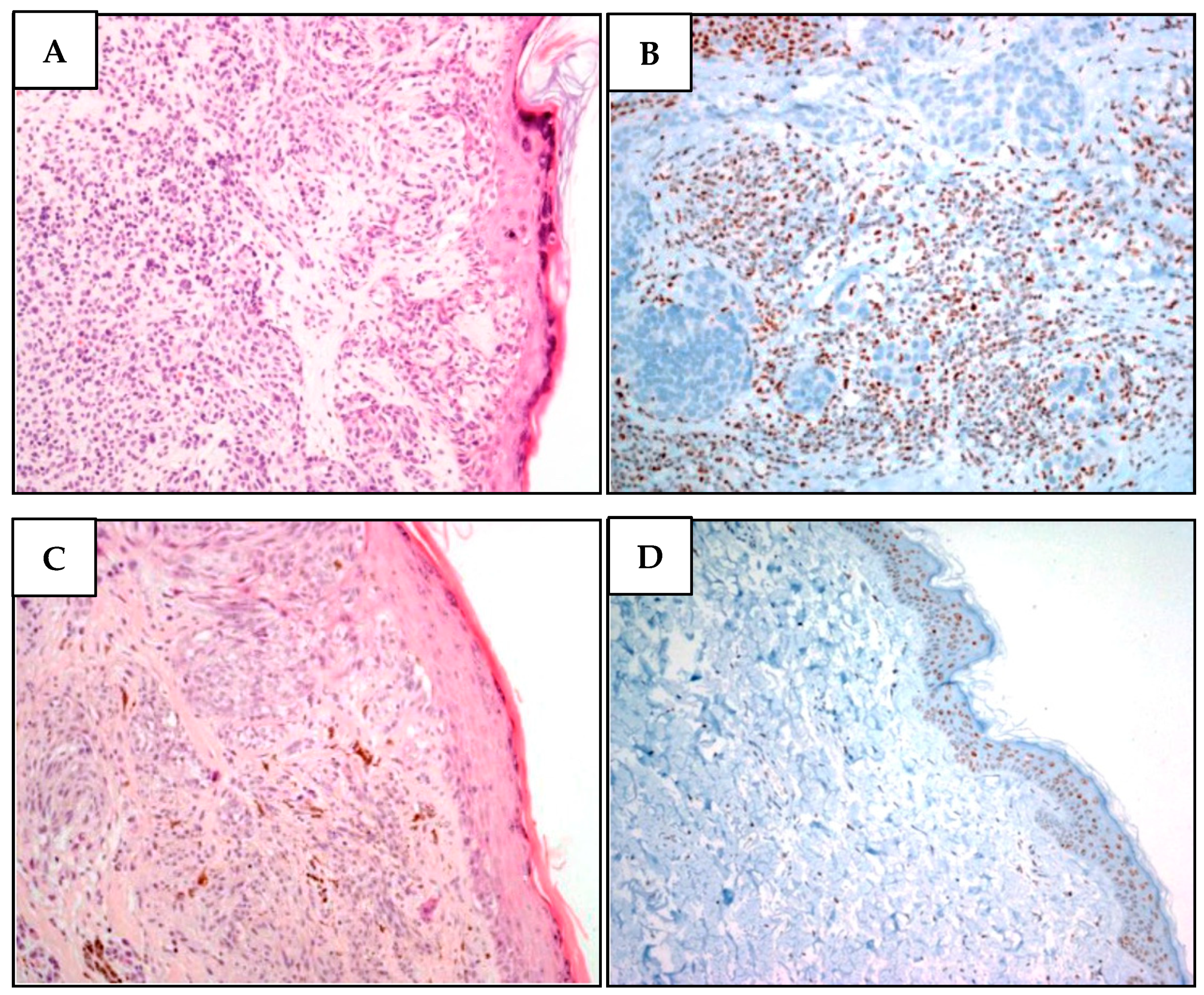
IHC of two different melanomas of the proband showed loss of nuclear BAP1 protein expression (Figure 4). Cascade genetic analyses were unfeasible.
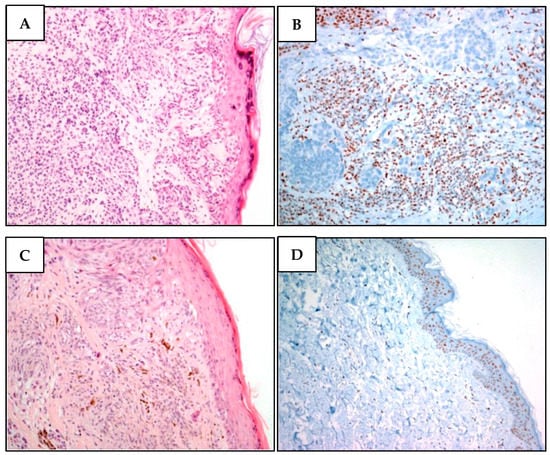
Figure 4.
Two different melanomas of proband MM981. (A). Malignant melanoma cells showing superficial extension, vertical growth, absence of ulceration, and nevus residual with a bland lymphocytic infiltration. (B) While BAP1 is undetectable in neoplastic atypical melanoma cells, it is present in stromal cells. (C) Malignant melanoma section denoting superficial extension, vertical growth phase, absence of ulceration, with spindle cell appearance and poor lymphocyte infiltration. There are signs of regression in the absence of angiolymphatic and perineural invasion. (D) Lack of BAP1 immunohistochemical loss in atypical nevus cells, while the epidermal cells regularly express BAP1.
3.8. Family PD-238
This family has never been described before (Figure S12). The MM400 proband was diagnosed at 52 with bladder cancer and at 58 with UM. Her father and one of her paternal aunts (or: distal relative of fifth degree) had also UM. The other paternal aunt developed a brain tumor, whereas the two paternal uncles were diagnosed with lung and stomach cancer, respectively. We only tested the germline DNA from the proband and found the splice-site variant c.376-2A>G in BAP1, which is predicted to cause exon 6 skipping, leading to the formation of a premature stop codon after eight amino acids. This variant was previously reported in an Australian family [7], but, to our knowledge, its effect on splicing has not been functionally evaluated. To investigate the effect of the variant on BAP1 transcript, we evaluated total RNA extracted from whole blood of the PV carrier. cDNA amplification and sequencing revealed exon 6 skipping (Figure S13), supporting a pathogenic role of this variant. Cascade genetic and tumor analyses were not feasible.
4. Discussion
The present study reports the results of a ten-year program of genetic testing of BAP1-TPDS individuals performed at a single reference center in Italy. Among our study panel of 101 suspected BAP1-TPDS individuals, we successfully identified seven patients carrying six different germline BAP1 PVs, one of which never described before (Table 1). Importantly, we show that certain tumors present in PV carriers retain a wt BAP1 allele, which implies a sporadic origin of these tumors or a functional role of heterozygous BAP1 in cancer development. Besides having important clinical implications for BAP1-TPDS patients, our findings highlight a number of important issues that still need to be fully addressed. These are discussed below.
4.1. Should Testing Criteria Be More Stringent?
Currently, BAP1 genetic testing is recommended for patients who develop two or more tumors of the BAP1-TPDS spectrum or for those affected by only one of those tumors provided they have a first- or second-degree relative with a confirmed BAP1-TPDS tumor [6]. Individuals or families with two BCCs and/or CMs should be excluded from BAP1 genetic testing due to the high frequency of these tumors in the general population [6,11]. On the other hand, families with multiple melanomas should first be checked for the presence of the most common high-penetrance melanoma predisposing genes or subjected to NGS panel analysis. In our study, we adopted less stringent criteria to select patients for germline BAP1 mutation analysis. We feel that less stringent criteria allowed us to identify BAP1 carriers from non-typical TPDS families, when family history is incomplete (e.g., ID_5 III-1).
Among our study panel, we found a germline BAP1 PV prevalence of 6.9% (7/101) (CI95% (2.8–13.8)), which is in agreement with that reported by other studies for familial cases (7.7% (3/39) [32], 6% (9/150) [41]). In sporadic patients with mesothelioma, the prevalence of BAP1-TPDS is about 1% [6], which is higher than what we found in our previous study performed on patients highly exposed to asbestos (<0.001%, [35]). For familial melanomas, previous reports showed a prevalence of germline BAP1 PVs ranging from 20–30% for UM [11,42] to <1% for CM [43,44,45].
4.2. The Importance of Detecting Secondary Somatic Mutations of BAP1 in Tumor Tissues
Emerging evidence from prospective clinical trials on BAP1-TPDS patients suggests that an early diagnosis of TPDS may be crucial for shaping the personalized therapeutic option offered to these patients [46]. In this regard, Srinivasan et al. [30] have recently proposed that the identification of biallelic mutations of genes involved in DNA repair in cancer tissues, such as BAP1, should be routinely performed for all patients so as to prospectively evaluate the effectiveness of personalized therapies. Noteworthily, this study showed that 27% of the tumors arising in individuals with inherited cancer syndromes did not display somatic loss of the second allele, which is a necessary tumor-promoting event according to Knudson’s theory. As a partial explanation for this phenomenon, the authors hypothesize a tissue-specific role of the tumor suppressor or a pathogenic effect of the mutation even when it is in its heterozygous form. Alternatively, they also propose that the bioinformatic technique they used to analyze the tumor might not have been sensitive enough to detect all the mutational alterations [30]. Overall, the study shows that the rate of somatic biallelic inactivation of BAP1 in tumors developed in carriers of germline PVs is 80–90%, and that the second hit is mostly acquired through LOH [30].
4.3. Which Is the Best Strategy to Detect a Secondary Somatic Mutation?
Among the methods used to address this issue, FISH is particularly useful for detecting chromosomal deletions, whereas CNV analysis of NGS sequencing data or MLPA, which can only be performed using fresh tissues, is instrumental for revealing both short and whole gene deletions. In contrast, microsatellite-based analysis is only effective in identifying large deletions. The loss of the second allele of the gene of interest may be carried throughout the entire specimen, representing the major tumor clone—see mesothelioma of proband MPM_HO1901 in this study—or it may only be present in a minor clone—as in the case of the RCC and LUAD of the same proband. Thus, the fact that we cannot rule out complete biallelic loss of a gene of interest, such as BAP1, due to technical limitations and/or tumor heterogeneity, raises the possibility that, in the same patient, some tumors may be responsive to treatments designed to kill cells lacking BAP1, whereas others might only be partially responsive or totally unresponsive to such treatment.
4.4. Tumor Genome in BAP1-TPDS
Analysis of BAP1-TPDS tumor genomes has never been thoroughly performed.
In 2020, Shinozaki-Ushiku and colleagues reported the genome analyses of several metachronous tumors in a patient with TPDS [29]. They found loss of BAP1 protein expression in all the tested tumors (6/7). Somatic loss of BAP1 was due to different mutations in PlM of the right thoracic cavity, peritoneal mesothelioma, lung adenocarcinoma, and bladder cancer, whereas no BAP1 somatic mutation was observed in cholangiocarcinoma and PlM of the left thoracic cavity. Moreover, they identified a low mutational burden.
Srinivasan et al. have recently shown that tumors from germline PV carriers display fewer driver events than those observed in non-carriers [30]. Fittingly, none of our tumor samples carried somatic alterations in other cancer driver genes besides BAP1. However, a limitation of our analysis is that we did not perform a whole-genome study or use techniques able to detect complex rearrangements.
4.5. Redefining the Pathogenicity of a Splice Mutation
Another controversial issue in the characterization of BAP1-TPDS—and any other genetic disease—is the definition of the pathogenicity of a certain mutation. An intriguing case in this regard is that represented by mutation c.783+2T>C (III-1 in family ID_5 and III-5 in family PD-601). This mutation affects the second base (T) of the canonical GT splice site donor of BAP1 intron 9. Even though this mutation should in theory be pathogenic because it leads to skipping of BAP1 exon 9, it has been recently reclassified as a variant of unknown significance (VUS) by Goldberg and co-workers [47]. However, this reclassification may have been biased by the use of a forward primer mapping on exon 9—which is therefore not suitable to identify exon 9 skipping—when sequencing BAP1 transcript from tumor specimens. Indeed, when we repeated the same experiment, but this time using a forward primer designed on exon 8, we were able to detect an alternative BAP1 splicing consistent with exon 9 skipping, supporting a pathogenic role of the c.783+2T>C variant. Interestingly, it has been estimated that GT>GC substitution within the canonical 5′ splice site can sometimes be partially tolerated, leading to variable amounts of canonical transcripts (1–84%) [48]. Thus, it is possible that this abnormal splicing does not occur in 100% of mutated transcripts. It should be nevertheless pointed out that we eventually observed LOH in the tumor tissue.
Taken all together, these data indicate that the variant c.783+2T>C leads to altered BAP1 transcript, but given that this effect may be incomplete, it should be regarded as likely pathogenic.
4.6. Cascade Genetic Testing
Guidelines published in Gene Reviews [6] recommend that carriers of BAP1 PVs should undergo surveillance for BAP1-TPDS tumors. Indeed, patients who are affected by one BAP1-related malignancy are at increased risk of developing other cancers belonging to the BAP1-TPDS spectrum, and early tumor detection may allow a more effective management of such tumors [4]. For example, dermatologic screening, combined with preventive measures (e.g., limiting sun exposure), may lower the risk of BAP1-TPDS patients developing severe forms of CM. Likewise, ocular screening for UM should be highly recommended.
With regard to mesothelioma, there is no consensus on the most effective screening modalities. Before recommending chest CT to asymptomatic individuals with previous asbestos exposure, the subsequent increased risk of cancer due to radiation exposure should be evaluated [6]. We have previously shown that the combined risk of genetic predisposition and asbestos exposure results in an increased risk of mesothelioma in the carriers of germline mutations [39]. The identification of BAP1 mutation carriers with mesothelioma can be extremely useful because these patients may potentially benefit from precision medicine, as shown for MPM_HO1901 [14,17,39,46].
In our cohort of BAP1 PV carrier families, we identified mutations in four relatives who were healthy at the genetic testing. On a recent follow-up, we found that three of them had been diagnosed with confirmed tumors of the BAP1-TPDS spectrum, while only one, the 23-year-old daughter of MPM_HO1901, was still healthy. Nevertheless, screening measures have been implemented for this subject.
Regarding associations between specific BAP1 mutations and a peculiar disease phenotype, in 2018 a comprehensive study [7] collating data from 181 BAP1-TPDS families showed that among patients who developed mesothelioma, melanoma, and other tumors, those carrying a null BAP1 mutation had an earlier age of onset than patients with missense mutations. Both null and missense carriers displayed a lower age of onset for these tumors in comparison with the general US population. The authors also reported that among carriers of BAP1 null mutations, peritoneal mesothelioma was more prevalent than pleural mesothelioma, in contrast to what was observed in the general population. No other associations were found.
4.7. What Can We Say about the TPDS Spectrum?
In this study, we performed IHC on tumor samples from patient MPM_HO1901, detecting loss of BAP1 expression in different tumor types. Of note, our NGS and IHC results on the patient’s LUAD tissue also suggest a possible functional role of BAP1 in this tumor, albeit at a later stage of tumor progression. Interestingly, we observed loss of BAP1 expression in the nucleus and in cytosol of meningioma cells as well as in BCC cells from two different germline BAP1 PV carriers belonging to two different families, confirming that meningioma and BCC are also included in the broader BAP1-TPDS spectrum [7,49,50].
4.8. New Pathogenic Mutation
In this study, we also describe three novel families, named PD-578, PD-238, and PD-601. Proband MM981 (PD-578) carries the p.Trp202* PV in BAP1, a previously unknown nonsense variant, and his melanoma shows loss of protein expression.
4.9. Survival of BAP1 Carriers
Since mean survival from diagnosis of malignant PlM (MPM) patients ranges between 9 and 17 months [51], five out of seven BAP1 PV patients with PlM analyzed in this study should be considered as long-term survivors (Table 3). Indeed, patient II-1 (family A), who developed peritoneal mesothelioma when he was 63 years old (in 2001), lived for 72 months (6 years) after diagnosis. Similarly, proband II-5 (family A1) lived 108 months (nine years) after being diagnosed with PlM, while her brother died after 24 months. ID_5 III-1 also survived 24 months after the diagnosis. Interestingly, patient MPM_HO1901 (II-3) is still alive, 27 months after the diagnosis, and, since she is positive for a mutation in BAP1, has been recruited in a trial aiming to test the combination of immunotherapy and PARP inhibitors as second-line treatment (NCT04940637) [46]. These results agree with previous studies showing prolonged survival of PlM TPDS patients compared to that of sporadic patients [8,9,12,14,15,16,17,18].
Table 3.
Survival of mesothelioma patients carrying germline BAP1 PVs.
5. Conclusions
We report here the results of our genetic screening of both germline and tumor samples from a cohort of BAP1-TPDS patients, identifying six different germline BAP1 PVs in seven families. Overall, our findings stress the importance of an appropriate surveillance program for BAP1-TPDS carriers, which should involve not only those individuals who have already developed a tumor but also their healthy relatives carrying a BAP1 PV.
Finally, we recommend that confirmation of biallelic loss in the tumor tissue should be carried out before recruiting patients for precision medicine-based clinical trials.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/diagnostics12071710/s1, Ref. [52]. Table S1. Clinical features of the patients (n = 101) analyzed in the present study. Figure S1: Lollipop plot of all variants discovered in this study mapped on the BAP1 gene. Figure S2: Pedigree of family A. Figure S3. Pedigree of family A1. Figure S4. Pedigree of family PD-601. Figure S5. IHC and LOH analyses of tumor sample MM1012. Figure S6. Functional analysis of the c.783+2G>T variant from MM1012 blood samples. Figure S7. Pedigree of family MPM_HO1901. Figure S8. Functional analysis of the c.38-1G>T variant carried by MPM_HO1901. Figure S9. Histological and immunohistochemical features of proband MPM_HO1901 (II-3) RCC. Figure S10. Molecular analyses of proband’s (MPM_HO1901 II-3) PlM tissue sample. Figure S11. Pedigree of family PD-578. Figure S12. Pedigree of family PD-238. Figure S13. Functional analysis of the variant carried by MM400. Supplemental Data.
Author Contributions
Conceptualization, I.D. and M.S. (Marika Sculco); investigation, M.S. (Marika Sculco), M.L.V., A.A., M.G.C., M.S. (Michela Salvo), G.B., A.P., G.W., F.N., A.L., L.R., C.T., L.E. and C.M. (Chiara Menin); resources, F.G., R.L., A.M. (Antonio Maconi), O.R., R.B., D.G., P.B., A.M. (Antonella Maffè), G.A., C.M. (Chiara Menin), L.R., G.V.S. and C.D.; data curation, M.S. (Marika Sculco), M.L.V., A.A., D.F. and I.D.; writing—original draft preparation, M.S. (Marika Sculco), M.L.V., A.A., A.P., G.W., F.N., A.L., L.R., C.M. (Chiara Menin) and I.D.; writing—review and editing, F.G., R.L., A.M. (Antonio Maconi), O.R., R.B., D.G., P.B., A.M. (Antonella Maffè), G.A., C.T., G.V.S., C.D., D.F., C.M. (Corrado Magnani), E.M.; D.M. and G.M.; visualization, M.S. (Marika Sculco), M.L.V., M.S. (Michela Salvo), A.A., L.R. and C.M. (Chiara Menin); supervision, I.D.; project administration, I.D.; funding acquisition, I.D., C.M. (Corrado Magnani), G.M. and G.V.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the offer of compensation to the inhabitants of Casale Monferrato deceased or affected by mesothelioma, HEreditary Risk in MESothelioma (HERMES) Project (to I.D. and C.Ma.), and received funding from AIRC under IG 2018—ID. 21390 project (to G.M.) and under IG 2019—ID. 23760 project (to G.V.S.). The APC was funded by the Italian Ministry of Education, University and Research (MIUR) program “Departments of Excellence 2018–2022”, FOHN Project—Department of Health Sciences, Università del Piemonte Orientale.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the local Ethics Committee (protocol 132/13, approved on 14 November 2013 and protocol 169/18, approved on 8 February 2019).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Acknowledgments
The authors would like to thank Marcello Arsura (Abeschool) for reviewing the English language.
Conflicts of Interest
Grosso reports, outside the submitted work, personal fees for advisory roles, speaker engagements, and travel and accommodation expenses from Merck Sharp & Dohme, Novocure, Bristol Meyer Squibb, Boehringer Ingelheim, Pharmamar, and Novartis. Bironzo reports, outside the submitted work, personal fees for lectures, speaker bureau, presentations, and educational events from Beigene, Roche, Astra Zeneca, Takeda, and BMS. Bironzo received institutional research grants outside the submitted work from Roche and Pfizer. Bironzo reports virtual meeting subscriptions from Amgen and Daiichi Sankyo. Scagliotti reports, outside the submitted work, personal fees for lectures, presentations, speaker bureaus, manuscript writing, and educational events from Eli Lilly, Roche, Pfizer, Astrazeneca, Novartis, MSD, Takeda, and Beigene. Scagliotti reports participation on a data safety monitoring board and advisory board of Beigene, Takeda, MSD, Novartis, Astrazeneca, Pfizer, Roche, and Eli Lilly outside the submitted work. Magnani received payment for participation in different trials regarding asbestos-related diseases from the public prosecution office and research funding (BRIC Project) from INAIL outside the submitted work. Dianzani has been appointed by the public prosecution office to discuss court cases with asbestos-related neoplasms. The remaining authors declare no conflicts of interest.
References
- Bononi, A.; Yang, H.; Giorgi, C.; Patergnani, S.; Pellegrini, L.; Su, M.; Xie, G.; Signorato, V.; Pastorino, S.; Morris, P.; et al. Germline BAP1 Mutations Induce a Warburg Effect. Cell Death Differ. 2017, 24, 1694–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 Mutations Predispose to Malignant Mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, M.; Yang, H. Molecular Pathways: Targeting Mechanisms of Asbestos and Erionite Carcinogenesis in Mesothelioma. Clin. Cancer Res. 2012, 18, 598–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walpole, S.; Hayward, N.K.; Pritchard, A.L.; Johansson, P.A. Microsimulation Model for Evaluating the Cost-Effectiveness of Surveillance in BAP1 Pathogenic Variant Carriers. JCO Clin. Cancer Inform. 2021, 5, 143–154. [Google Scholar] [CrossRef]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and Cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef]
- Pilarski, R.; Rai, K.; Cebulla, C.; Abdel-Rahman, M. BAP1 Tumor Predisposition Syndrome. In GeneReviews®; University of Washington: Seattle, WA, USA, 2022. [Google Scholar]
- Walpole, S.; Pritchard, A.L.; Cebulla, C.M.; Pilarski, R.; Stautberg, M.; Davidorf, F.H.; de la Fouchardière, A.; Cabaret, O.; Golmard, L.; Stoppa-Lyonnet, D.; et al. Comprehensive Study of the Clinical Phenotype of Germline BAP1 Variant-Carrying Families Worldwide. J. Natl. Cancer Inst. 2018, 110, 1328–1341. [Google Scholar] [CrossRef]
- Baumann, F.; Flores, E.; Napolitano, A.; Kanodia, S.; Taioli, E.; Pass, H.; Yang, H.; Carbone, M. Mesothelioma Patients with Germline BAP1 Mutations Have 7-Fold Improved Long-Term Survival. Carcinogenesis 2015, 36, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Carbone, M.; Harbour, J.W.; Brugarolas, J.; Bononi, A.; Pagano, I.; Dey, A.; Krausz, T.; Pass, H.I.; Yang, H.; Gaudino, G. Biological Mechanisms and Clinical Significance of BAP1 Mutations in Human Cancer. Cancer Discov. 2020, 10, 1103–1120. [Google Scholar] [CrossRef]
- Carbone, M.; Arron, S.T.; Beutler, B.; Bononi, A.; Cavenee, W.; Cleaver, J.E.; Croce, C.M.; D’Andrea, A.; Foulkes, W.D.; Gaudino, G.; et al. Tumour Predisposition and Cancer Syndromes as Models to Study Gene-Environment Interactions. Nat. Rev. Cancer 2020, 20, 533–549. [Google Scholar] [CrossRef]
- Rai, K.; Pilarski, R.; Cebulla, C.M.; Abdel-Rahman, M.H. Comprehensive Review of BAP1 Tumor Predisposition Syndrome with Report of Two New Cases. Clin. Genet. 2016, 89, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Carbone, M.; Pass, H.I.; Ak, G.; Alexander, H.R.; Baas, P.; Baumann, F.; Blakely, A.M.; Bueno, R.; Bzura, A.; Cardillo, G.; et al. Medical and Surgical Care of Mesothelioma Patients and Their Relatives Carrying Germline BAP1 Mutations. J. Thorac. Oncol. 2022, 17, 873–889. [Google Scholar] [CrossRef] [PubMed]
- Kobrinski, D.A.; Yang, H.; Kittaneh, M. BAP1: Role in Carcinogenesis and Clinical Implications. Transl. Lung Cancer Res. 2020, 9, S60–S66. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Morrow, B.; Thomas, A.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Gadiraju, M.; Panou, V.; Gao, S.; Mian, I.; et al. Inherited Predisposition to Malignant Mesothelioma and Overall Survival Following Platinum Chemotherapy. Proc. Natl. Acad. Sci. USA 2019, 116, 9008–9013. [Google Scholar] [CrossRef] [Green Version]
- Panou, V.; Gadiraju, M.; Wolin, A.; Weipert, C.M.; Skarda, E.; Husain, A.N.; Patel, J.D.; Rose, B.; Zhang, S.R.; Weatherly, M.; et al. Frequency of Germline Mutations in Cancer Susceptibility Genes in Malignant Mesothelioma. J. Clin. Oncol. 2018, 36, 2863–2871. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, S.; Yoshikawa, Y.; Pass, H.I.; Emi, M.; Nasu, M.; Pagano, I.; Takinishi, Y.; Yamamoto, R.; Minaai, M.; Hashimoto-Tamaoki, T.; et al. A Subset of Mesotheliomas with Improved Survival Occurring in Carriers of BAP1 and Other Germline Mutations. J. Clin. Oncol. 2018, 36, 3485–3494. [Google Scholar] [CrossRef]
- Forde, P.M.; Anagnostou, V.; Sun, Z.; Dahlberg, S.E.; Kindler, H.L.; Niknafs, N.; Purcell, T.; Santana-Davila, R.; Dudek, A.Z.; Borghaei, H.; et al. Durvalumab with Platinum-Pemetrexed for Unresectable Pleural Mesothelioma: Survival, Genomic and Immunologic Analyses from the Phase 2 PrE0505 Trial. Nat. Med. 2021, 27, 1910–1920. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, X.Y.; Li, J.; Zhu, W.W. Prognostic and Clinicopathological Significance of BAP1 Protein Expression in Different Types of Cancer-A Meta-Analysis. Genet. Test. Mol. Biomark. 2018, 22, 115–126. [Google Scholar] [CrossRef]
- Gupta, M.P.; Lane, A.M.; DeAngelis, M.M.; Mayne, K.; Crabtree, M.; Gragoudas, E.S.; Kim, I.K. Clinical Characteristics of Uveal Melanoma in Patients With Germline BAP1 Mutations. JAMA Ophthalmol. 2015, 133, 881–887. [Google Scholar] [CrossRef] [Green Version]
- De la Fouchardière, A.; Cabaret, O.; Savin, L.; Combemale, P.; Schvartz, H.; Penet, C.; Bonadona, V.; Soufir, N.; Bressac-de Paillerets, B. Germline BAP1 Mutations Predispose Also to Multiple Basal Cell Carcinomas. Clin. Genet. 2015, 88, 273–277. [Google Scholar] [CrossRef]
- O’Shea, S.J.; Robles-Espinoza, C.D.; McLellan, L.; Harrigan, J.; Jacq, X.; Hewinson, J.; Iyer, V.; Merchant, W.; Elliott, F.; Harland, M.; et al. A Population-Based Analysis of Germline BAP1 Mutations in Melanoma. Hum. Mol. Genet. 2017, 26, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Shankar, G.M.; Santagata, S. BAP1 Mutations in High-Grade Meningioma: Implications for Patient Care. Neuro. Oncol. 2017, 19, 1447–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, G.M.; Abedalthagafi, M.; Vaubel, R.A.; Merrill, P.H.; Nayyar, N.; Gill, C.M.; Brewster, R.; Bi, W.L.; Agarwalla, P.K.; Thorner, A.R.; et al. Germline and Somatic BAP1 Mutations in High-Grade Rhabdoid Meningiomas. Neuro. Oncol. 2017, 19, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oehl, K.; Vrugt, B.; Wagner, U.; Kirschner, M.B.; Meerang, M.; Weder, W.; Felley-Bosco, E.; Wollscheid, B.; Bankov, K.; Demes, M.C.; et al. Alterations in BAP1 Are Associated with Cisplatin Resistance through Inhibition of Apoptosis in Malignant Pleural Mesothelioma. Clin. Cancer Res. 2021, 27, 2277–2291. [Google Scholar] [CrossRef]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive Genomic Analysis of Malignant Pleural Mesothelioma Identifies Recurrent Mutations, Gene Fusions and Splicing Alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1549–1565. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, Y.; Emi, M.; Hashimoto-Tamaoki, T.; Ohmuraya, M.; Sato, A.; Tsujimura, T.; Hasegawa, S.; Nakano, T.; Nasu, M.; Pastorino, S.; et al. High-Density Array-CGH with Targeted NGS Unmask Multiple Noncontiguous Minute Deletions on Chromosome 3p21 in Mesothelioma. Proc. Natl. Acad. Sci. USA 2016, 113, 13432–13437. [Google Scholar] [CrossRef] [Green Version]
- Righi, L.; Duregon, E.; Vatrano, S.; Izzo, S.; Giorcelli, J.; Rondón-Lagos, M.; Ascoli, V.; Ruffini, E.; Ventura, L.; Volante, M.; et al. BRCA1-Associated Protein 1 (BAP1) Immunohistochemical Expression as a Diagnostic Tool in Malignant Pleural Mesothelioma Classification: A Large Retrospective Study. J. Thorac. Oncol. 2016, 11, 2006–2017. [Google Scholar] [CrossRef] [Green Version]
- Shinozaki-Ushiku, A.; Kohsaka, S.; Kage, H.; Oda, K.; Miyagawa, K.; Nakajima, J.; Aburatani, H.; Mano, H.; Ushiku, T. Genomic Profiling of Multiple Primary Cancers Including Synchronous Lung Adenocarcinoma and Bilateral Malignant Mesotheliomas: Identification of a Novel BAP1 Germline Variant. Pathol. Int. 2020, 70, 775–780. [Google Scholar] [CrossRef]
- Srinivasan, P.; Bandlamudi, C.; Jonsson, P.; Kemel, Y.; Chavan, S.S.; Richards, A.L.; Penson, A.V.; Bielski, C.M.; Fong, C.; Syed, A.; et al. The Context-Specific Role of Germline Pathogenicity in Tumorigenesis. Nat. Genet. 2021, 53, 1577–1585. [Google Scholar] [CrossRef]
- Betti, M.; Aspesi, A.; Biasi, A.; Casalone, E.; Ferrante, D.; Ogliara, P.; Gironi, L.C.; Giorgione, R.; Farinelli, P.; Grosso, F.; et al. CDKN2A and BAP1 Germline Mutations Predispose to Melanoma and Mesothelioma. Cancer Lett. 2016, 378, 120–130. [Google Scholar] [CrossRef]
- Betti, M.; Aspesi, A.; Ferrante, D.; Sculco, M.; Righi, L.; Mirabelli, D.; Napoli, F.; Rondón-Lagos, M.; Casalone, E.; Vignolo Lutati, F.; et al. Sensitivity to Asbestos Is Increased in Patients with Mesothelioma and Pathogenic Germline Variants in BAP1 or Other DNA Repair Genes. Genes Chromosomes Cancer 2018, 57, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A Comprehensive Refinement of the ACMG-AMP Variant Classification Criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Betti, M.; Casalone, E.; Ferrante, D.; Romanelli, A.; Grosso, F.; Guarrera, S.; Righi, L.; Vatrano, S.; Pelosi, G.; Libener, R.; et al. Inference on Germline BAP1 Mutations and Asbestos Exposure from the Analysis of Familial and Sporadic Mesothelioma in a High-Risk Area. Genes Chromosomes Cancer 2015, 54, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Strom, S.P. Current Practices and Guidelines for Clinical Next-Generation Sequencing Oncology Testing. Cancer Biol. Med. 2016, 13, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vestergaard, L.K.; Oliveira, D.N.P.; Poulsen, T.S.; Høgdall, C.K.; Høgdall, E.V. OncomineTM Comprehensive Assay v3 vs. OncomineTM Comprehensive Assay Plus. Cancers 2021, 13, 5230. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and Integration of Deleteriousness Prediction Methods for Nonsynonymous SNVs in Whole Exome Sequencing Studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [Green Version]
- Sculco, M.; La Vecchia, M.; Aspesi, A.; Pinton, G.; Clavenna, M.G.; Casalone, E.; Allione, A.; Grosso, F.; Libener, R.; Muzio, A.; et al. Malignant Pleural Mesothelioma: Germline Variants in DNA Repair Genes May Steer Tailored Treatment. Eur. J. Cancer 2022, 163, 44–54. [Google Scholar] [CrossRef]
- Sun, T.; Wang, X.; Wang, M.; Minerowicz, C.; Sanchez, H.; Laskin, W.; Cohen, P.; Zhong, M. Somatic Mutation of BAP1 Can Lead to Expression Loss in Non-Small Cell Lung Carcinoma: Next Generation Sequencing and IHC Analysis in a Large Single Institute Cohort. Int. J. Surg. Pathol. 2021, 30, 512–519. [Google Scholar] [CrossRef]
- Ohar, J.A.; Cheung, M.; Talarchek, J.; Howard, S.E.; Howard, T.D.; Hesdorffer, M.; Peng, H.; Rauscher, F.J.; Testa, J.R. Germline BAP1 Mutational Landscape of Asbestos-Exposed Malignant Mesothelioma Patients with Family History of Cancer. Cancer Res. 2016, 76, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Turunen, J.A.; Markkinen, S.; Wilska, R.; Saarinen, S.; Raivio, V.; Täll, M.; Lehesjoki, A.-E.; Kivelä, T.T. BAP1 Germline Mutations in Finnish Patients with Uveal Melanoma. Ophthalmology 2016, 123, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Boru, G.; Grosel, T.W.; Pilarski, R.; Stautberg, M.; Massengill, J.B.; Jeter, J.; Singh, A.; Marino, M.J.; McElroy, J.P.; Davidorf, F.H.; et al. Germline Large Deletion of BAP1 and Decreased Expression in Non-Tumor Choroid in Uveal Melanoma Patients with High Risk for Inherited Cancer. Genes Chromosomes Cancer 2019, 58, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Njauw, C.-N.J.; Kim, I.; Piris, A.; Gabree, M.; Taylor, M.; Lane, A.M.; DeAngelis, M.M.; Gragoudas, E.; Duncan, L.M.; Tsao, H. Germline BAP1 Inactivation Is Preferentially Associated with Metastatic Ocular Melanoma and Cutaneous-Ocular Melanoma Families. PLoS ONE 2012, 7, e35295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potjer, T.P.; Bollen, S.; Grimbergen, A.J.E.M.; van Doorn, R.; Gruis, N.A.; van Asperen, C.J.; Hes, F.J.; van der Stoep, N. Multigene Panel Sequencing of Established and Candidate Melanoma Susceptibility Genes in a Large Cohort of Dutch Non-CDKN2A/CDK4 Melanoma Families. Int. J. Cancer 2019, 144, 2453–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passiglia, F.; Bironzo, P.; Righi, L.; Listì, A.; Arizio, F.; Novello, S.; Volante, M.; Scagliotti, G.V. A Prospective Phase II Single-Arm Study of Niraparib Plus Dostarlimab in Patients With Advanced Non–small-Cell Lung Cancer and/or Malignant Pleural Mesothelioma, Positive for PD-L1 Expression and Germline or Somatic Mutations in the DNA Repair Genes: Rationale and Study Design. Clin. Lung Cancer 2020, 22, e63–e66. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, Y.; Laitman, Y.; Ben David, M.; Bazak, L.; Lidzbarsky, G.; Salmon, L.B.; Shkedi-Rafid, S.; Barshack, I.; Avivi, C.; Darawshe, M.; et al. Re-Evaluating the Pathogenicity of the c.783+2T>C BAP1 Germline Variant. Hum. Mutat. 2021, 42, 592–599. [Google Scholar] [CrossRef]
- Lin, J.H.; Tang, X.Y.; Boulling, A.; Zou, W.B.; Masson, E.; Fichou, Y.; Raud, L.; Le Tertre, M.; Deng, S.J.; Berlivet, I.; et al. First Estimate of the Scale of Canonical 5’ Splice Site GT>GC Variants Capable of Generating Wild-Type Transcripts. Hum. Mutat. 2019, 40, 1856–1873. [Google Scholar] [CrossRef]
- Cheung, M.; Kadariya, Y.; Talarchek, J.; Pei, J.; Ohar, J.A.; Kayaleh, O.R.; Testa, J.R. Germline BAP1 Mutation in a Family with High Incidence of Multiple Primary Cancers and a Potential Gene-Environment Interaction. Cancer Lett. 2015, 369, 261–265. [Google Scholar] [CrossRef] [Green Version]
- Landry, A.P.; Wang, J.Z.; Nassiri, F.; Patil, V.; Gao, A.; Zadeh, G. BAP1-Deficient Meningioma Presenting with Trabecular Architecture and Cytokeratin Expression: A Report of Two Cases and Review of the Literature. J. Clin. Pathol. 2021. [Google Scholar] [CrossRef]
- Novello, S.; Pinto, C.; Torri, V.; Porcu, L.; Di Maio, M.; Tiseo, M.; Ceresoli, G.; Magnani, C.; Silvestri, S.; Veltri, A.; et al. The Third Italian Consensus Conference for Malignant Pleural Mesothelioma: State of the Art and Recommendations. Crit. Rev. Oncol. Hematol. 2016, 104, 9–20. [Google Scholar] [CrossRef]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome) and Microsatellite Instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).