
Identification of SOX18 as a New Gene Predisposing to Congenital Heart Disease

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. Genetic Assay
2.3. Construction of Recombinant Plasmids
2.4. Cell Transfection and Reporter Assay
2.5. Statistics
3. Results
3.1. Phenotypic Data of the Studied Family with CHD
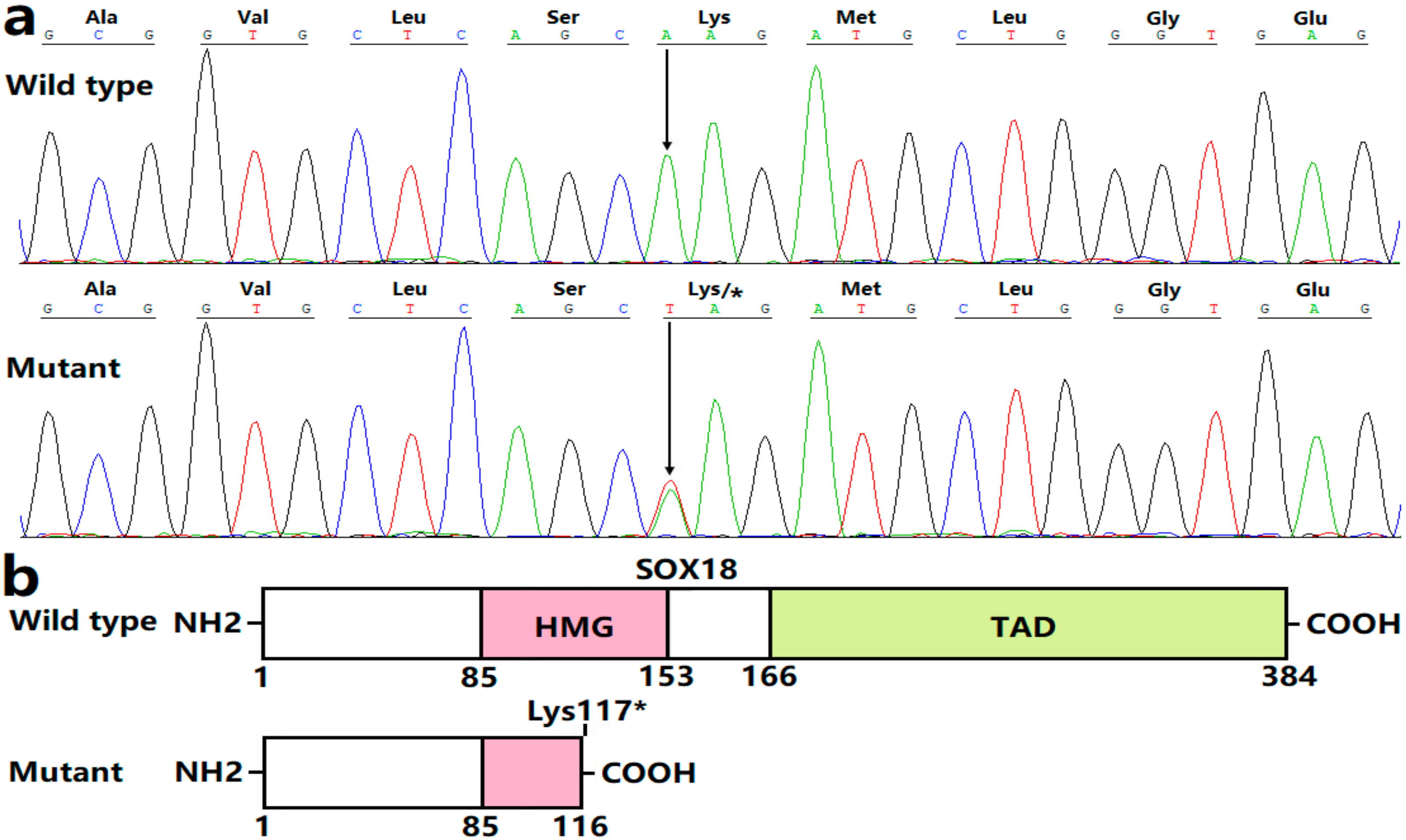
3.2. Discovery of a New CHD-Causative Variation in SOX18
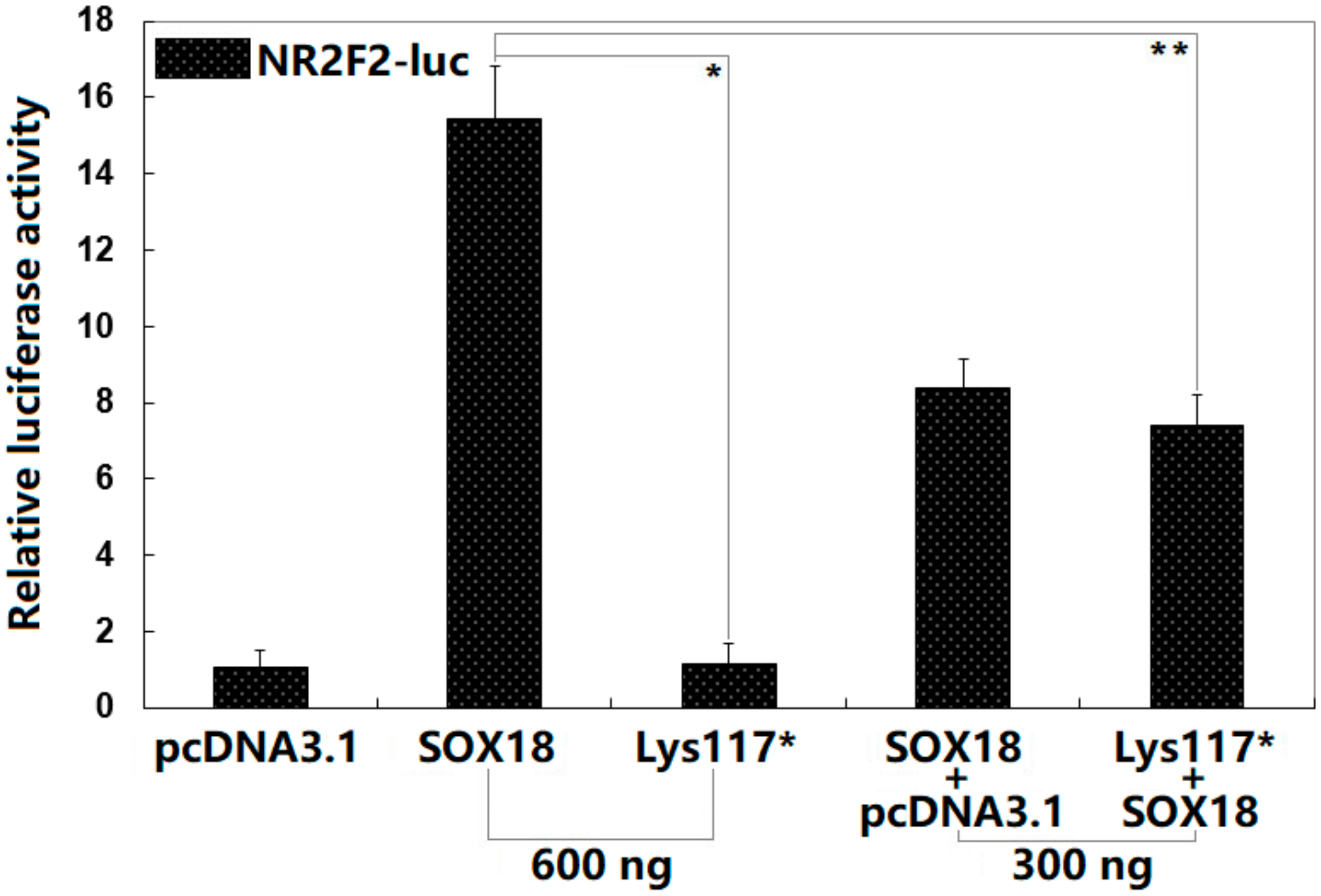
3.3. Functional Failure of Lys117*-Mutant SOX18
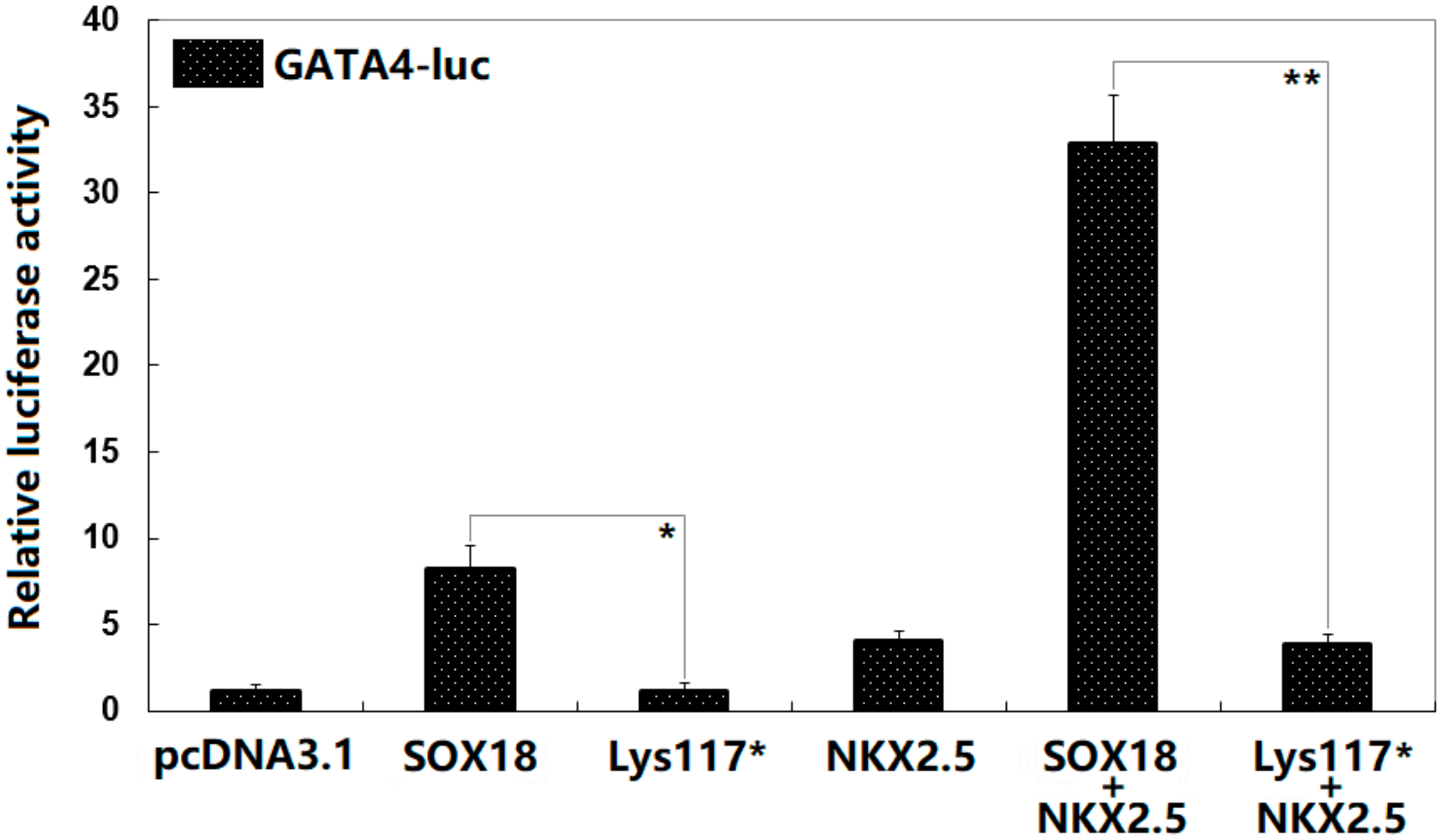
3.4. Synergistic Activation between SOX18 and NKX2.5 Abrogated by the Lys117* Variation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics—2022 Update: A Report From the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Diab, N.S.; Barish, S.; Dong, W.; Zhao, S.; Allington, G.; Yu, X.; Kahle, K.T.; Brueckner, M.; Jin, S.C. Molecular Genetics and Complex Inheritance of Congenital Heart Disease. Genes 2021, 12, 1020. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Benson, D.W. Focused Strategies for Defining the Genetic Architecture of Congenital Heart Defects. Genes 2021, 12, 827. [Google Scholar] [CrossRef] [PubMed]
- Satriano, A.; Varrica, A.; Frigiola, A.; Graziosi, A.; Di Battista, C.; Primavera, A.P.; Centini, G.; Maconi, A.; Strozzi, C.; Gavilanes, A.D.W.; et al. Perioperative GABA Blood Concentrations in Infants with Cyanotic and Non-Cyanotic Congenital Heart Diseases. Diagnostics 2021, 11, 1149. [Google Scholar] [CrossRef]
- Ellmann, S.; Nickel, J.M.; Heiss, R.; El Amrani, N.; Wüst, W.; Rompel, O.; Rueffer, A.; Cesnjevar, R.; Dittrich, S.; Uder, M.; et al. Prognostic Value of CTA-Derived Left Ventricular Mass in Neonates with Congenital Heart Disease. Diagnostics 2021, 11, 1215. [Google Scholar] [CrossRef]
- Kasielska-Trojan, A.; Święchowicz, B.; Antoszewski, B. Coexistence of Thumb Aplasia and Cleft Lip and Alveolus with Aortopulmonary Window-A Tip for Prenatal Diagnostics for Rare Heart Anomalies. Diagnostics 2022, 12, 569. [Google Scholar] [CrossRef]
- Moons, P.; Luyckx, K.; Thomet, C.; Budts, W.; Enomoto, J.; Sluman, M.A.; Lu, C.W.; Jackson, J.L.; Khairy, P.; Cook, S.C.; et al. Physical Functioning, Mental Health, and Quality of Life in Different Congenital Heart Defects: Comparative Analysis in 3538 Patients From 15 Countries. Can. J. Cardiol. 2021, 37, 215–223. [Google Scholar] [CrossRef]
- Brudy, L.; Meyer, M.; Oberhoffer, R.; Ewert, P.; Müller, J. Move more—Be happier? Physical activity and health-related quality of life in children with congenital heart disease. Am. Heart J. 2021, 241, 68–73. [Google Scholar] [CrossRef]
- Liu, H.C.; Chaou, C.H.; Lo, C.W.; Chung, H.T.; Hwang, M.S. Factors Affecting Psychological and Health-Related Quality-of-Life Status in Children and Adolescents with Congenital Heart Diseases. Children 2022, 9, 578. [Google Scholar] [CrossRef]
- Meyer, M.; Brudy, L.; Fuertes-Moure, A.; Hager, A.; Oberhoffer-Fritz, R.; Ewert, P.; Müller, J. E-Health Exercise Intervention for Pediatric Patients with Congenital Heart Disease: A Randomized Controlled Trial. J. Pediatr. 2021, 233, 163–168. [Google Scholar] [CrossRef]
- Masood, I.R.; Detterich, J.; Cerrone, D.; Lewinter, K.; Shah, P.; Kato, R.; Sabati, A. Reduced Forced Vital Capacity and the Number of Chest Wall Surgeries are Associated with Decreased Exercise Capacity in Children with Congenital Heart Disease. Pediatr. Cardiol. 2022, 43, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Saxer, S.; Calendo, L.R.; Lichtblau, M.; Carta, A.; Müller, J.; Gautschi, F.; Berlier, C.; Furian, M.; Schwarz, E.I.; Bloch, K.E.; et al. Effect of oxygen therapy on exercise performance in patients with cyanotic congenital heart disease: Randomized-controlled trial. Int. J. Cardiol. 2022, 348, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Kessler, N.; Feldmann, M.; Schlosser, L.; Rometsch, S.; Brugger, P.; Kottke, R.; Knirsch, W.; Oxenius, A.; Greutmann, M.; Latal, B. Structural brain abnormalities in adults with congenital heart disease: Prevalence and association with estimated intelligence quotient. Int. J. Cardiol. 2020, 306, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Asschenfeldt, B.; Evald, L.; Heiberg, J.; Salvig, C.; Østergaard, L.; Dalby, R.B.; Eskildsen, S.F.; Hjortdal, V.E. Neuropsychological Status and Structural Brain Imaging in Adults with Simple Congenital Heart Defects Closed in Childhood. J. Am. Heart Assoc. 2020, 9, e015843. [Google Scholar] [CrossRef]
- Sadhwani, A.; Wypij, D.; Rofeberg, V.; Gholipour, A.; Mittleman, M.; Rohde, J.; Velasco-Annis, C.; Calderon, J.; Friedman, K.G.; Tworetzky, W.; et al. Fetal Brain Volume Predicts Neurodevelopment in Congenital Heart Disease. Circulation 2022, 145, 1108–1119. [Google Scholar] [CrossRef]
- Parekh, S.A.; Cox, S.M.; Barkovich, A.J.; Chau, V.; Steurer, M.A.; Xu, D.; Miller, S.P.; McQuillen, P.S.; Peyvandi, S. The Effect of Size and Asymmetry at Birth on Brain Injury and Neurodevelopmental Outcomes in Congenital Heart Disease. Pediatr. Cardiol. 2022, 43, 868–877. [Google Scholar] [CrossRef]
- Giang, K.W.; Fedchenko, M.; Dellborg, M.; Eriksson, P.; Mandalenakis, Z. Burden of Ischemic Stroke in Patients with Congenital Heart Disease: A Nationwide, Case-Control Study. J. Am. Heart Assoc. 2021, 10, e020939. [Google Scholar] [CrossRef]
- Karsenty, C.; Waldmann, V.; Mulder, B.; Hascoet, S.; Ladouceur, M. Thromboembolic complications in adult congenital heart disease: The knowns and the unknowns. Clin. Res. Cardiol. 2021, 110, 1380–1391. [Google Scholar] [CrossRef]
- Maser, M.; Freisinger, E.; Bronstein, L.; Köppe, J.; Orwat, S.; Kaleschke, G.; Baumgartner, H.; Diller, G.P.; Lammers, A. Frequency, Mortality, and Predictors of Adverse Outcomes for Endocarditis in Patients with Congenital Heart Disease: Results of a Nationwide Analysis including 2512 Endocarditis Cases. J. Clin. Med. 2021, 10, 5071. [Google Scholar] [CrossRef]
- Snygg-Martin, U.; Giang, K.W.; Dellborg, M.; Robertson, J.; Mandalenakis, Z. Cumulative Incidence of Infective Endocarditis in Patients with Congenital Heart Disease: A Nationwide, Case-Control Study Over Nine Decades. Clin. Infect. Dis. 2021, 73, 1469–1475. [Google Scholar] [CrossRef]
- Meinel, K.; Koestenberger, M.; Sallmon, H.; Hansmann, G.; Pieles, G.E. Echocardiography for the Assessment of Pulmonary Hypertension and Congenital Heart Disease in the Young. Diagnostics 2020, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, E.B.; Krishnan, U. Congenital Heart Disease-Associated Pulmonary Hypertension. Clin. Chest Med. 2021, 42, 9–18. [Google Scholar] [CrossRef]
- Chiu, S.N.; Lu, C.W.; Lin, M.T.; Chen, C.A.; Wu, M.H.; Wang, J.K. Pulmonary Hypertension in Adult Congenital Heart Disease in Asia: A Distinctive Feature of Complex Congenital Heart Disease. J. Am. Heart Assoc. 2022, 11, e022596. [Google Scholar] [CrossRef] [PubMed]
- Fang, N.W.; Chen, Y.C.; Ou, S.H.; Yin, C.H.; Chen, J.S.; Chiou, Y.H. Incidence and risk factors for chronic kidney disease in patients with congenital heart disease. Pediatr. Nephrol. 2021, 36, 3749–3756. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ke, Q.; Weng, G.; Bao, J.; Huang, J.; Yan, L.; Zheng, F. Risk factors of postoperative acute kidney injury in patients with complex congenital heart disease and significance of early detection of serum transcription factor Nkx2.5. Am. J. Transl. Res. 2021, 13, 6468–6477. [Google Scholar]
- Xie, Y.; Jiang, W.; Cao, J.; Xie, H. Dexmedetomidine attenuates acute kidney injury in children undergoing congenital heart surgery with cardiopulmonary bypass by inhibiting the TLR3/NF-κB signaling pathway. Am. J. Transl. Res. 2021, 13, 2763–2773. [Google Scholar]
- Reiter, F.P.; Hadjamu, N.J.; Nagdyman, N.; Zachoval, R.; Mayerle, J.; De Toni, E.N.; Kaemmerer, H.; Denk, G. Congenital heart disease-associated liver disease: A narrative review. Cardiovasc. Diagn. Ther. 2021, 11, 577–590. [Google Scholar] [CrossRef]
- Spiesshoefer, J.; Orwat, S.; Henke, C.; Kabitz, H.J.; Katsianos, S.; Borrelli, C.; Baumgartner, H.; Nofer, J.R.; Spieker, M.; Bengel, P.; et al. Inspiratory muscle dysfunction and restrictive lung function impairment in congenital heart disease: Association with immune inflammatory response and exercise intolerance. Int. J. Cardiol. 2020, 318, 45–51. [Google Scholar] [CrossRef]
- Menachem, J.N.; Schlendorf, K.H.; Mazurek, J.A.; Bichell, D.P.; Brinkley, D.M.; Frischhertz, B.P.; Mettler, B.A.; Shah, A.S.; Zalawadiya, S.; Book, W.; et al. Advanced Heart Failure in Adults with Congenital Heart Disease. JACC Heart Fail. 2020, 8, 87–99. [Google Scholar] [CrossRef]
- Arnaert, S.; De Meester, P.; Troost, E.; Droogne, W.; Van Aelst, L.; Van Cleemput, J.; Voros, G.; Gewillig, M.; Cools, B.; Moons, P.; et al. Heart failure related to adult congenital heart disease: Prevalence, outcome and risk factors. ESC Heart Fail. 2021, 8, 2940–2950. [Google Scholar] [CrossRef]
- Zengin, E.; Sinning, C.; Blaum, C.; Blankenberg, S.; Rickers, C.; von Kodolitsch, Y.; Kirchhof, P.; Drury, N.E.; Stoll, V.M. Heart failure in adults with congenital heart disease: A narrative review. Cardiovasc. Diagn. Ther. 2021, 11, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Brida, M.; Lovrić, D.; Griselli, M.; Riesgo Gil, F.; Gatzoulis, M.A. Heart failure in adults with congenital heart disease. Int. J. Cardiol. 2022, 357, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.W.; Wang, J.K.; Yang, H.L.; Kovacs, A.H.; Luyckx, K.; Ruperti-Repilado, F.J.; Van De Bruaene, A.; Enomoto, J.; Sluman, M.A.; Jackson, J.L.; et al. Heart Failure and Patient-Reported Outcomes in Adults with Congenital Heart Disease from 15 Countries. J. Am. Heart Assoc. 2022, 11, e024993. [Google Scholar] [CrossRef] [PubMed]
- Sakhi, R.; Kauling, R.M.; Theuns, D.A.; Szili-Torok, T.; Bhagwandien, R.E.; van den Bosch, A.E.; Cuypers, J.A.A.E.; Roos-Hesselink, J.W.; Yap, S.C. Early detection of ventricular arrhythmias in adults with congenital heart disease using an insertable cardiac monitor (EDVA-CHD study). Int. J. Cardiol. 2020, 305, 63–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casteigt, B.; Samuel, M.; Laplante, L.; Shohoudi, A.; Apers, S.; Kovacs, A.H.; Luyckx, K.; Thomet, C.; Budts, W.; Enomoto, J.; et al. Atrial arrhythmias and patient-reported outcomes in adults with congenital heart disease: An international study. Heart Rhythm 2021, 18, 793–800. [Google Scholar] [CrossRef]
- Moore, J.P.; Bowman, H.; Gallotti, R.G.; Shannon, K.M. Mechanisms and outcomes of catheter ablation for biatrial tachycardia in adults with congenital heart disease. Heart Rhythm 2021, 18, 1833–1841. [Google Scholar] [CrossRef]
- Fischer, A.J.; Enders, D.; Wasmer, K.; Marschall, U.; Baumgartner, H.; Diller, G.P. Impact of specialized electrophysiological care on the outcome of catheter ablation for supraventricular tachycardias in adults with congenital heart disease: Independent risk factors and gender aspects. Heart Rhythm 2021, 18, 1852–1859. [Google Scholar] [CrossRef]
- Kartas, A.; Papazoglou, A.S.; Kosmidis, D.; Moysidis, D.V.; Baroutidou, A.; Doundoulakis, I.; Despotopoulos, S.; Vrana, E.; Koutsakis, A.; Rampidis, G.P.; et al. The Adult Congenital Heart Disease Anatomic and Physiological Classification: Associations with Clinical Outcomes in Patients with Atrial Arrhythmias. Diagnostics 2022, 12, 466. [Google Scholar] [CrossRef]
- Moore, J.P.; Burrows, A.; Gallotti, R.G.; Shannon, K.M. Electrophysiological characteristics of atrial tachycardia recurrence: Relevance to catheter ablation strategies in adults with congenital heart disease. Heart Rhythm 2022, 19, 272–280. [Google Scholar] [CrossRef]
- Williams, J.L.; Torok, R.D.; D’Ottavio, A.; Spears, T.; Chiswell, K.; Forestieri, N.E.; Sang, C.J.; Paolillo, J.A.; Walsh, M.J.; Hoffman, T.M.; et al. Causes of Death in Infants and Children with Congenital Heart Disease. Pediatr. Cardiol. 2021, 42, 1308–1315. [Google Scholar] [CrossRef]
- Vehmeijer, J.T.; Koyak, Z.; Leerink, J.M.; Zwinderman, A.H.; Harris, L.; Peinado, R.; Oechslin, E.N.; Robbers-Visser, D.; Groenink, M.; Boekholdt, S.M.; et al. Identification of patients at risk of sudden cardiac death in congenital heart disease: The PRospEctiVE study on implaNTable cardIOverter defibrillator therapy and sudden cardiac death in Adults with Congenital Heart Disease (PREVENTION-ACHD). Heart Rhythm 2021, 18, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Constantine, A.; Costola, G.; Bianchi, P.; Chessa, M.; Giamberti, A.; Kempny, A.; Rafiq, I.; Babu-Narayan, S.V.; Gatzoulis, M.A.; Hoschtitzky, A.; et al. Enhanced Assessment of Perioperative Mortality Risk in Adults with Congenital Heart Disease. J. Am. Coll. Cardiol. 2021, 78, 234–242. [Google Scholar] [CrossRef]
- Diller, G.P.; Orwat, S.; Lammers, A.E.; Radke, R.M.; De-Torres-Alba, F.; Schmidt, R.; Marschall, U.; Bauer, U.M.; Enders, D.; Bronstein, L.; et al. Lack of specialist care is associated with increased morbidity and mortality in adult congenital heart disease: A population-based study. Eur. Heart J. 2021, 42, 4241–4248. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.K.; Zmora, R.; Huang, Y.; Oster, M.E.; McCracken, C.; Mahle, W.T.; Kochilas, L.; Kalogeropoulos, A. Long-Term Risk of Heart Failure-Related Death and Heart Transplant After Congenital Heart Surgery in Childhood (from the Pediatric Cardiac Care Consortium). Am. J. Cardiol. 2022, 167, 111–117. [Google Scholar] [CrossRef]
- Triedman, J.K.; Newburger, J.W. Trends in Congenital Heart Disease: The Next Decade. Circulation 2016, 133, 2716–2733. [Google Scholar] [CrossRef] [PubMed]
- Bouma, B.J.; Mulder, B.J. Changing Landscape of Congenital Heart Disease. Circ. Res. 2017, 120, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.; Figueiredo, A.C.V.; Baroneza, J.E.; Afiune, J.Y.; Pic-Taylor, A.; Oliveira, S.F.; Mazzeu, J.F. Genetic and genomics in congenital heart disease: A clinical review. J. Pediatr. 2020, 96, 279–288. [Google Scholar] [CrossRef]
- Moisa, S.M.; Burlacu, A.; Brinza, C.; Țarcă, E.; Butnariu, L.I.; Trandafir, L.M. An Up-to-Date Narrative Review on Congenital Heart Disease Percutaneous Treatment in Children Using Contemporary Devices. Diagnostics 2022, 12, 1189. [Google Scholar] [CrossRef]
- Williams, R.G. Late Causes of Death After Congenital Heart Defects: A Population-Based Study from Finland. J. Am. Coll. Cardiol. 2016, 68, 499–501. [Google Scholar] [CrossRef]
- Baumgartner, H.; De Backer, J.; Babu-Narayan, S.V.; Budts, W.; Chessa, M.; Diller, G.P.; Lung, B.; Kluin, J.; Lang, I.M.; Meijboom, F.; et al. 2020 ESC Guidelines for the management of adult congenital heart disease. Eur. Heart J. 2021, 42, 563–645. [Google Scholar] [CrossRef]
- Niwa, K.; Kaemmerer, H.; von Kodolitsch, Y. Current diagnosis and management of late complications in adult congenital heart disease. Cardiovasc. Diagn. Ther. 2021, 11, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Gurvitz, M.Z.; Beauséjour-Ladouceur, V.; Lawler, P.R.; Therrien, J.; Marelli, A.J. Cancer Risk in Congenital Heart Disease—What Is the Evidence? Can. J. Cardiol. 2019, 35, 1750–1761. [Google Scholar] [CrossRef]
- Wang, G.; Wang, B.; Yang, P. Epigenetics in Congenital Heart Disease. J. Am. Heart Assoc. 2022, 11, e025163. [Google Scholar] [CrossRef] [PubMed]
- Kalisch-Smith, J.I.; Ved, N.; Sparrow, D.B. Environmental Risk Factors for Congenital Heart Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a037234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Digilio, M.C.; Dentici, M.L.; Loddo, S.; Laino, L.; Calcagni, G.; Genovese, S.; Capolino, R.; Bottillo, I.; Calvieri, G.; Dallapiccola, B.; et al. Congenital heart defects in the recurrent 2q13 deletion syndrome. Eur. J. Med. Genet. 2022, 65, 104381. [Google Scholar] [CrossRef] [PubMed]
- Meerschaut, I.; Vergult, S.; Dheedene, A.; Menten, B.; De Groote, K.; De Wilde, H.; Muiño Mosquera, L.; Panzer, J.; Vandekerckhove, K.; Coucke, P.J.; et al. A Reassessment of Copy Number Variations in Congenital Heart Defects: Picturing the Whole Genome. Genes 2021, 12, 1048. [Google Scholar] [CrossRef]
- Richter, F.; Morton, S.U.; Kim, S.W.; Kitaygorodsky, A.; Wasson, L.K.; Chen, K.M.; Zhou, J.; Qi, H.; Patel, N.; DePalma, S.R.; et al. Genomic analyses implicate noncoding de novo variants in congenital heart disease. Nat. Genet. 2020, 52, 769–777. [Google Scholar] [CrossRef]
- Szot, J.O.; Campagnolo, C.; Cao, Y.; Iyer, K.R.; Cuny, H.; Drysdale, T.; Flores-Daboub, J.A.; Bi, W.; Westerfield, L.; Liu, P.; et al. Bi-allelic Mutations in NADSYN1 Cause Multiple Organ Defects and Expand the Genotypic Spectrum of Congenital NAD Deficiency Disorders. Am. J. Hum. Genet. 2020, 106, 129–136. [Google Scholar] [CrossRef]
- Palencia-Campos, A.; Aoto, P.C.; Machal, E.M.F.; Rivera-Barahona, A.; Soto-Bielicka, P.; Bertinetti, D.; Baker, B.; Vu, L.; Piceci-Sparascio, F.; Torrente, I.; et al. Germline and Mosaic Variants in PRKACA and PRKACB Cause a Multiple Congenital Malformation Syndrome. Am. J. Hum. Genet. 2020, 107, 977–988. [Google Scholar] [CrossRef]
- Alankarage, D.; Szot, J.O.; Pachter, N.; Slavotinek, A.; Selleri, L.; Shieh, J.T.; Winlaw, D.; Giannoulatou, E.; Chapman, G.; Dunwoodie, S.L. Functional characterization of a novel PBX1 de novo missense variant identified in a patient with syndromic congenital heart disease. Hum. Mol. Genet. 2020, 29, 1068–1082. [Google Scholar] [CrossRef]
- Chapman, G.; Moreau, J.L.M.; Eddie, I.P.; Szot, J.O.; Iyer, K.R.; Shi, H.; Yam, M.X.; O’Reilly, V.C.; Enriquez, A.; Greasby, J.A.; et al. Functional genomics and gene-environment interaction highlight the complexity of congenital heart disease caused by Notch pathway variants. Hum. Mol. Genet. 2020, 29, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.U.; Agarwal, R.; Madden, J.A.; Genetti, C.A.; Brownstein, C.A.; López-Giráldez, F.; Choi, J.; Seidman, C.E.; Seidman, J.G.; Lyon, G.J.; et al. Congenital Heart Defects Due to TAF1 Missense Variants. Circ. Genom. Precis. Med. 2020, 13, e002843. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.A.; Crutcher, E.; Gill, H.; Nelson, T.N.; Robak, L.A.; Jongmans, M.C.J.; Pfundt, R.; Prasad, C.; Berard, R.A.; Fannemel, M.; et al. The expanding clinical phenotype of germline ABL1-associated congenital heart defects and skeletal malformations syndrome. Hum. Mutat. 2020, 41, 1738–1744. [Google Scholar] [CrossRef]
- Wang, S.S.; Wang, T.M.; Qiao, X.H.; Huang, R.T.; Xue, S.; Dong, B.B.; Xu, Y.J.; Liu, X.Y.; Yang, Y.Q. KLF13 loss-of-function variation contributes to familial congenital heart defects. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 11273–11285. [Google Scholar] [PubMed]
- Zhang, Y.; Sun, Y.M.; Xu, Y.J.; Zhao, C.M.; Yuan, F.; Guo, X.J.; Guo, Y.H.; Yang, C.X.; Gu, J.N.; Qiao, Q.; et al. A New TBX5 Loss-of-Function Mutation Contributes to Congenital Heart Defect and Atrioventricular Block. Int. Heart J. 2020, 61, 761–768. [Google Scholar] [CrossRef]
- Wang, C.; Lv, H.; Ling, X.; Li, H.; Diao, F.; Dai, J.; Du, J.; Chen, T.; Xi, Q.; Zhao, Y.; et al. Association of assisted reproductive technology, germline de novo mutations and congenital heart defects in a prospective birth cohort study. Cell Res. 2021, 31, 919–928. [Google Scholar] [CrossRef]
- Lahrouchi, N.; Postma, A.V.; Salazar, C.M.; De Laughter, D.M.; Tjong, F.; Piherová, L.; Bowling, F.Z.; Zimmerman, D.; Lodder, E.M.; Ta-Shma, A.; et al. Biallelic loss-of-function variants in PLD1 cause congenital right-sided cardiac valve defects and neonatal cardiomyopathy. J. Clin. Investig. 2021, 131, e142148. [Google Scholar] [CrossRef]
- Fu, F.; Li, R.; Lei, T.Y.; Wang, D.; Yang, X.; Han, J.; Pan, M.; Zhen, L.; Li, J.; Li, F.T.; et al. Compound heterozygous mutation of the ASXL3 gene causes autosomal recessive congenital heart disease. Hum. Genet. 2021, 140, 333–348. [Google Scholar] [CrossRef]
- Zheng, S.Q.; Chen, H.X.; Liu, X.C.; Yang, Q.; He, G.W. Genetic analysis of the CITED2 gene promoter in isolated and sporadic congenital ventricular septal defects. J. Cell. Mol. Med. 2021, 25, 2254–2261. [Google Scholar] [CrossRef]
- Gentile, M.; Ranieri, C.; Loconte, D.C.; Ponzi, E.; Ficarella, R.; Volpe, P.; Scalzo, G.; Lepore Signorile, M.; Grossi, V.; Cordella, A.; et al. Functional evidence of mTORβ splice variant involvement in the pathogenesis of congenital heart defects. Clin. Genet. 2021, 99, 425–429. [Google Scholar] [CrossRef]
- Roifman, M.; Chung, B.H.Y.; Reid, D.M.; Teitelbaum, R.; Martin, N.; Nield, L.E.; Thompson, M.; Shannon, P.; Chitayat, D. Heterozygous NOTCH1 deletion associated with variable congenital heart defects. Clin. Genet. 2021, 99, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Massadeh, S.; Albeladi, M.; Albesher, N.; Alhabshan, F.; Kampe, K.D.; Chaikhouni, F.; Kabbani, M.S.; Beetz, C.; Alaamery, M. Novel Autosomal Recessive Splice-Altering Variant in PRKD1 Is Associated with Congenital Heart Disease. Genes 2021, 12, 612. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Jiang, W.F.; Yang, C.X.; Qiao, Q.; Xu, Y.J.; Shi, H.Y.; Qiu, X.B.; Wu, S.H.; Yang, Y.Q. SOX17 loss-of-function variation underlying familial congenital heart disease. Eur. J. Med. Genet. 2021, 64, 104211. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Li, B.; Chen, S.; Shi, Z.; Yu, L.; Gao, Y.; Yang, X.; Lu, L.; Wang, H. Deleterious Rare Mutations of GLI1 Dysregulate Sonic Hedgehog Signaling in Human Congenital Heart Disease. Front. Cardiovasc. Med. 2022, 9, 798033. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahman, H.A.; Akawi, N.; Al-Shamsi, A.M.; Ali, A.; Al-Jasmi, F.; John, A.; Hertecant, J.; Al-Gazali, L.; Ali, B.R. Bi-allelic null variant in matrix metalloproteinase-15, causes congenital cardiac defect, cholestasis jaundice, and failure to thrive. Clin. Genet. 2022, 101, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.Q.; Chen, W.C.; Li, Y.J.; Suo, M.J.; Tian, G.X.; Sheng, W.; Huang, G.Y. PTPN11 Gene Mutations and Its Association with the Risk of Congenital Heart Disease. Dis. Markers 2022, 2022, 8290779. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Qiao, X.H.; Xu, Y.J.; Liu, X.Y.; Huang, R.T.; Xue, S.; Qiu, H.Y.; Yang, Y.Q. SMAD1 Loss-of-Function Variant Responsible for Congenital Heart Disease. BioMed Res. Int. 2022, 2022, 9916325. [Google Scholar] [CrossRef]
- Huang, R.T.; Guo, Y.H.; Yang, C.X.; Gu, J.N.; Qiu, X.B.; Shi, H.Y.; Xu, Y.J.; Xue, S.; Yang, Y.Q. SOX7 loss-of-function variation as a cause of familial congenital heart disease. Am. J. Transl. Res. 2022, 14, 1672–1684. [Google Scholar]
- Abhinav, P.; Zhang, G.F.; Zhao, C.M.; Xu, Y.J.; Wang, J.; Yang, Y.Q. A novel KLF13 mutation underlying congenital patent ductus arteriosus and ventricular septal defect, as well as bicuspid aortic valve. Exp. Ther. Med. 2022, 23, 311. [Google Scholar] [CrossRef]
- Ke, Z.P.; Zhang, G.F.; Guo, Y.H.; Sun, Y.M.; Wang, J.; Li, N.; Qiu, X.B.; Xu, Y.J.; Yang, Y.Q. A novel PRRX1 loss-of-function variation contributing to familial atrial fibrillation and congenital patent ductus arteriosus. Genet. Mol. Biol. 2022, 45, e20210378. [Google Scholar] [CrossRef]
- Brancaccio, M.; Mennitti, C.; Cesaro, A.; Monda, E.; D’Argenio, V.; Casaburi, G.; Mazzaccara, C.; Ranieri, A.; Fimiani, F.; Barretta, F.; et al. Multidisciplinary In-Depth Investigation in a Young Athlete Suffering from Syncope Caused by Myocardial Bridge. Diagnostics 2021, 11, 2144. [Google Scholar] [CrossRef] [PubMed]
- Cenni, C.; Mansard, L.; Blanchet, C.; Baux, D.; Vaché, C.; Baudoin, C.; Moclyn, M.; Faugère, V.; Mondain, M.; Jeziorski, E.; et al. When Familial Hearing Loss Means Genetic Heterogeneity: A Model Case Report. Diagnostics 2021, 11, 1636. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, A.; Farina, A.; Seidenari, A.; Azzaroli, F.; Serra, C.; Della Gatta, A.; Zuffardi, O.; Giglio, S.R. Prenatal Noninvasive Trio-WES in a Case of Pregnancy-Related Liver Disorder. Diagnostics 2021, 11, 1904. [Google Scholar] [CrossRef] [PubMed]
- Traisrisilp, K.; Luewan, S.; Sirilert, S.; Jatavan, P.; Tongsong, T. Prenatal Sonographic and Molecular Genetic Diagnosis of Popliteal Pterygium Syndrome. Diagnostics 2021, 11, 1819. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Yang, Y.Q. An update on the molecular diagnosis of congenital heart disease: Focus on loss-of-function mutations. Expert Rev. Mol. Diagn. 2017, 17, 393–401. [Google Scholar] [CrossRef]
- Lilly, A.J.; Lacaud, G.; Kouskoff, V. SOXF transcription factors in cardiovascular development. Semin. Cell Dev. Biol. 2017, 63, 50–57. [Google Scholar] [CrossRef]
- Azuma, T.; Seki, N.; Yoshikawa, T.; Saito, T.; Masuho, Y.; Muramatsu, M. cDNA cloning, tissue expression, and chromosome mapping of human homolog of SOX18. J. Hum. Genet. 2000, 45, 192–195. [Google Scholar] [CrossRef]
- Wangberg, H.; Wigby, K.; Jones, M.C. A novel autosomal dominant mutation in SOX18 resulting in a fatal case of hypotrichosis-lymphedema-telangiectasia syndrome. Am. J. Med. Genet. Part A 2018, 176, 2824–2828. [Google Scholar] [CrossRef]
- Swift, M.R.; Pham, V.N.; Castranova, D.; Bell, K.; Poole, R.J.; Weinstein, B.M. SoxF factors and Notch regulate nr2f2 gene expression during venous differentiation in zebrafish. Dev. Biol. 2014, 390, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Hosking, B.M.; Wang, S.C.; Chen, S.L.; Penning, S.; Koopman, P.; Muscat, G.E. SOX18 directly interacts with MEF2C in endothelial cells. Biochem. Biophys. Res. Commun. 2001, 287, 493–500. [Google Scholar] [CrossRef]
- Al Turki, S.; Manickaraj, A.K.; Mercer, C.L.; Gerety, S.S.; Hitz, M.P.; Lindsay, S.; D’Alessandro, L.C.; Swaminathan, G.J.; Bentham, J.; Arndt, A.K.; et al. Rare variants in NR2F2 cause congenital heart defects in humans. Am. J. Hum. Genet. 2014, 94, 574–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, X.H.; Wang, F.; Zhang, X.L.; Huang, R.T.; Xue, S.; Wang, J.; Qiu, X.B.; Liu, X.Y.; Yang, Y.Q. MEF2C loss-of-function mutation contributes to congenital heart defects. Int. J. Med. Sci. 2017, 14, 1143–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, Y.; Hara, K.; Kanai-Azuma, M.; Matsui, T.; Miura, Y.; Tsunekawa, N.; Kurohmaru, M.; Saijoh, Y.; Koopman, P.; Kanai, Y. Redundant roles of Sox17 and Sox18 in early cardiovascular development of mouse embryos. Biochem. Biophys. Res. Commun. 2007, 360, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Basta, T.; Klymkowsky, M.W. SOX7 and SOX18 are essential for cardiogenesis in Xenopus. Dev. Dyn. 2005, 234, 878–891. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Chen, L.; Yang, L.; Ye, Z.; Huang, W.; Li, X.; Liu, Q.; Qiu, J.; Ding, X. KCTD1 mutants in scalp-ear-nipple syndrome and AP-2α P59A in Char syndrome reciprocally abrogate their interactions, but can regulate Wnt/β-catenin signaling. Mol. Med. Rep. 2020, 22, 3895–3903. [Google Scholar] [CrossRef]
- Lewis, T.R.; Shelton, E.L.; Van Driest, S.L.; Kannankeril, P.J.; Reese, J. Genetics of the patent ductus arteriosus (PDA) and pharmacogenetics of PDA treatment. Semin. Fetal Neonatal Med. 2018, 23, 232–238. [Google Scholar] [CrossRef]
- Gong, Q.; Zhou, Z. Nonsense-mediated mRNA decay of hERG mutations in long QT syndrome. Methods Mol. Biol. 2018, 1684, 37–49. [Google Scholar]
- Inácio, A.; Silva, A.L.; Pinto, J.; Ji, X.; Morgado, A.; Almeida, F.; Faustino, P.; Lavinha, J.; Liebhaber, S.A.; Romão, L. Nonsense mutations in close proximity to the initiation codon fail to trigger full nonsense-mediated mRNA decay. J. Biol. Chem. 2004, 279, 32170–32180. [Google Scholar] [CrossRef] [Green Version]
- Coulie, R.; Niyazov, D.M.; Gambello, M.J.; Fastré, E.; Brouillard, P.; Vikkula, M. Hypotrichosis-lymphedema-telangiectasia syndrome: Report of ileal atresia associated with a SOX18 de novo pathogenic variant and review of the phenotypic spectrum. Am. J. Med. Genet. Part A 2021, 185, 2153–2159. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Individual (Family 1) | Gender | Age (Years) | Cardiac Structural Deformities | SOX18 Variation (Lys117*) |
|---|---|---|---|---|
| II-1 | Male | 62 | PDA, PS | +/− |
| II-3 | Male | 60 | PDA | +/− |
| II-8 | Female | 55 | PDA | +/− |
| III-3 | Male | 37 | PDA, PS | +/− |
| III-8 | Female | 34 | PDA | +/− |
| III-13 | Male | 31 | PDA | +/− |
| IV-2 | Female | 13 | PDA, PS | +/− |
| IV-8 | Male | 5 | PDA | +/− |
| Chr | Position (GRCh37) | Ref | Alt | Gene | Variation |
|---|---|---|---|---|---|
| 1 | 91,403,874 | T | A | ZNF644 | NM_201269.3: c.3037T>A; p.(Phe1013Ile) |
| 1 | 196,434,495 | A | C | KCNT2 | NM_198503.5: c.566A>C; p.(Gln189Pro) |
| 2 | 207,953,267 | T | A | KLF7 | NM_003709.4: c.772T>A; p.(Trp258Arg) |
| 2 | 25,982,422 | C | G | ASXL2 | NM_018263.6: c.868C>G; p.(His290Asp) |
| 3 | 178,745,488 | G | A | ZMAT3 | NM_022470.4: c.503G>A; p.(Gly168Glu) |
| 4 | 114,239,688 | A | T | ANK2 | NM_001148.6: c.2812A>T; p.(Lys938*) |
| 6 | 152,762,351 | T | C | SYNE1 | NM_182961.4: c.4063T>C; p.(Tyr1355His) |
| 9 | 101,907,169 | A | T | TGFBR1 | NM_004612.4: c.1129A>T; p.(Arg377Trp) |
| 10 | 112,590,865 | T | G | RBM20 | NM_001134363.3: c.3498T>G; p.(Cys1166Trp) |
| 10 | 64,573,251 | G | A | EGR2 | NM_000399.5: c.1147G>A; p.(Asp383Asn) |
| 14 | 64,990,070 | A | T | ZBTB1 | NM_001123329.2: c.1848A>T; p.(Leu616Phe) |
| 20 | 62,680,521 | A | T | SOX18 | NM_018419.3: c.349A>T; p.(Lys117*) |
| Coding Exons | Forward Primers (5′→3′) | Backward Primers (5′→3′) | Amplicons (bp) |
|---|---|---|---|
| 1 (a) | GGCCCTGAGCCGCTATATCT | CTTTGCCCACACCATGAAGG | 457 |
| 1 (b) | CAGCTGGAATGCAGAGATCG | TCAGCTCCTTCCACGCTTTG | 583 |
| 2 (a) | CAGCTGGAATGCAGAGATCG | CGGCCGGTACTTGTAGTTGG | 672 |
| 2 (b) | AAGCGTGGAAGGAGCTGAAC | GGCTGCAGTTGAGGTACTGG | 642 |
| 2 (c) | GCTCGCTGGCCTGTACTACG | TGTAACCCTGGCAACTCTGC | 622 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, H.-Y.; Xie, M.-S.; Yang, C.-X.; Huang, R.-T.; Xue, S.; Liu, X.-Y.; Xu, Y.-J.; Yang, Y.-Q. Identification of SOX18 as a New Gene Predisposing to Congenital Heart Disease. Diagnostics 2022, 12, 1917. https://doi.org/10.3390/diagnostics12081917
Shi H-Y, Xie M-S, Yang C-X, Huang R-T, Xue S, Liu X-Y, Xu Y-J, Yang Y-Q. Identification of SOX18 as a New Gene Predisposing to Congenital Heart Disease. Diagnostics. 2022; 12(8):1917. https://doi.org/10.3390/diagnostics12081917
Chicago/Turabian StyleShi, Hong-Yu, Meng-Shi Xie, Chen-Xi Yang, Ri-Tai Huang, Song Xue, Xing-Yuan Liu, Ying-Jia Xu, and Yi-Qing Yang. 2022. "Identification of SOX18 as a New Gene Predisposing to Congenital Heart Disease" Diagnostics 12, no. 8: 1917. https://doi.org/10.3390/diagnostics12081917