

Molecular Characterization of Whole-Genome SARS-CoV-2 from the First Suspected Cases of the XE Variant in the Lazio Region, Italy

 , ,
, ,  , ,
, ,  , , , , ,
, , , , ,
Abstract
:1. Introduction
1.1. SARS-CoV-2 Recombination Events and Evolutionary Pathways
1.2. Recombination Events Involving Omicron Variant
2. Materials and Methods
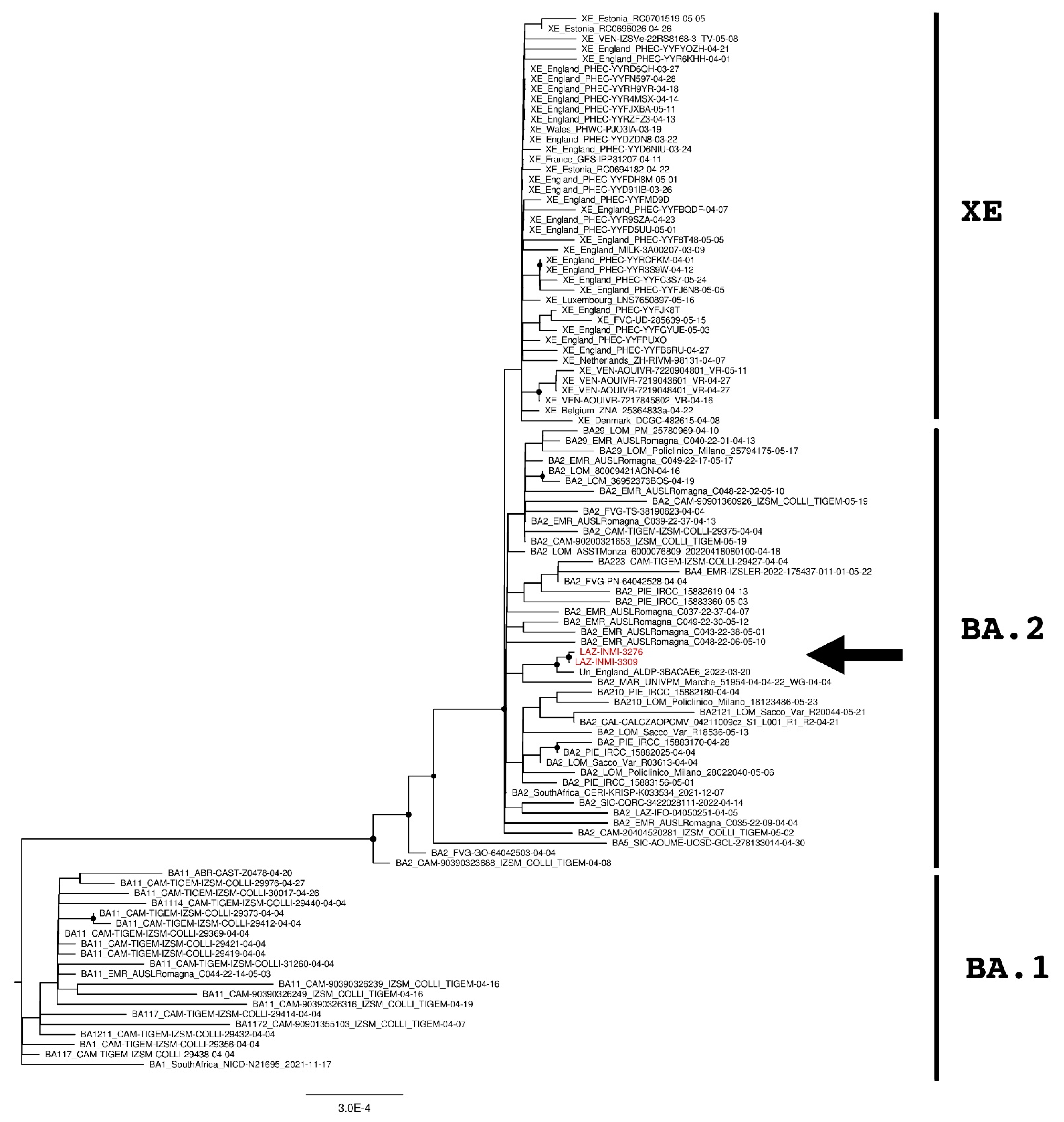
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Palero, F.; González-Candelas, F. Recombination in viruses: Mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 2015, 30, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Bentley, K.; Evans, D.J.J. Mechanisms and consequences of positive-strand RNA virus recombination. J. Gen. Virol. 2018, 99, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cui, X.; Cai, X.; An, T. Recombination in positive-strand RNA viruses. Front. Microbiol. 2022, 13, 870759. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, genetic recombination, and pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef]
- Pollett, S.; Conte, M.A.; Sanborn, M.; Jarman, R.G.; Lidl, G.M.; Modjarrad, K.; Berry, I.M. A comparative recombination analysis of human coronaviruses and implications for the SARS-CoV-2 pandemic. Sci. Rep. 2021, 11, 17365. [Google Scholar] [CrossRef]
- Lytras, S.; Xia, W.; Hughes, J.; Jiang, X.; Robertson, D.L. The animal origin of SARS-CoV-2. Science 2021, 373, 968–970. [Google Scholar] [CrossRef]
- Lytras, S.; Hughes, J.; Martin, D.; Swanepoel, P.; de Klerk, A.; Lourens, R.; Pond, S.L.K.; Xia, W.; Jiang, X.; Robertson, D.L. Exploring the Natural Origins of SARS-CoV-2 in the Light of Recombination. Genome Biol. Evol. 2022, 14, evac018. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, K.R.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Souza, P.F.N.; Mesquita, F.P.; Amaral, J.L.; Landim, P.G.C.; Karollyny Lima, K.R.P.; Costa, M.B.; Farias, I.R.; Belém, M.O.; Pinto, Y.O.; Moreira, H.H.T.; et al. The spike glycoprotein of SARS-CoV-2: A review of how mutations of spike glycoproteins have driven the emergence of variants with high transmissibility and immune escape. Int. J. Biol. Macromol. 2022, 208, 105–125. [Google Scholar] [CrossRef]
- Tatsi, E.B.; Filippatos, F.; Michos, A. SARS-CoV-2 variants and effectiveness of vaccines: A review of current evidence. Epidemiol. Infect. 2021, 149, e237. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Kmiec, D.; Koepke, L.; Zech, F.; Jacob, T.; Sparrer, K.M.J.; Kirchhoff, F. Omicron: What Makes the Latest SARS-CoV-2 Variant of Concern So Concerning? J. Virol. 2022, 96, e0207721. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.F.N.; Mesquita, F.P.; Amaral, J.L.; Landim, P.G.C.; Lima, K.R.P.; Costa, M.B.; Farias, I.R.; Lima, L.B.; Montenegro, R.C. The human pandemic coronaviruses on the show: The spike glycoprotein as the main actor in the coronaviruses play. Int. J. Biol. Macromol. 2021, 179, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Cov-Lineages Lineage List. Available online: https://cov-lineages.org/lineage_list.html (accessed on 27 June 2022).
- Chakraborty, C.; Bhattacharya, M.; Sharma, A.R.; Dhama, K. Recombinant SARS-CoV-2 variants XD, XE, and XF: The emergence of recombinant variants requires an urgent call for research-Correspondence. Int. J. Surg. 2022, 102, 106670. [Google Scholar] [CrossRef]
- Jackson, B.; Boni, M.F.; Bull, M.J.; Colleran, A.; Colquhoun, R.M.; Darby, A.C.; Haldenby, S.; Hill, V.; Lucaci, A.; McCrone, J.T.; et al. Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic. Cell 2021, 184, 5179–5188.e3. [Google Scholar] [CrossRef]
- Novazzi, F.; Focosi, D.; Baj, A.; Ambrosini, A.; Bouthar, S.; Maggi, F. SARS-CoV-2 recombinant XN, Italy. J. Clin. Virol. Plus 2022, 2, 100084. [Google Scholar] [CrossRef]
- Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants (accessed on 27 June 2022).
- UK Health Security Agency. Technical briefing 39. In SARS-CoV-2 Variants of Concern and Variants under Investigation in England; Crown Copyright 2022, England, March 2022; GOV-11753; Department of Health and Social Care: London, UK, 2022. [Google Scholar]
- SARS-CoV-2 Variants of Concern as of 25 August 2022 by ECDC. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 31 August 2022).
- Stalls, V.; Lindenberger, J.; Gobeil, S.M.; Henderson, R.; Parks, R.; Barr, M.; Deyton, M.; Martin, M.; Janowska, K.; Huang, X.; et al. Cryo-EM structures of SARS-CoV-2 Omicron BA.2 spike. Cell Rep. 2022, 39, 111009. [Google Scholar] [CrossRef]
- UK Health Security Agency. SARS-CoV-2 Variants of Concern and Variants under Investigation in England Technical Briefing. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1063424/tech-briefing-39-25march2022_final.pdf (accessed on 12 April 2022).
- UK Health Security Agency. SARS-CoV-2 Variants of Concern and Variants under Investigation in England. Technical Briefing 40. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1067672/Technical-Briefing-40-8April2022.pdf (accessed on 12 April 2022).
- Akash, K.; Sharma, A.; Kumar, D.; Singh, S.K.; Gupta, G.; Chellappan, D.K.; Nagraik, R. Molecular aspects of Omicron, vaccine development, and recombinant strain XE: A review. J. Med. Virol. 2022, 94, 4628–4643. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Rueca, M.; Giombini, E.; Messina, F.; Bartolini, B.; Di Caro, A.; Capobianchi, M.R.; Gruber, C.E.M. The Easy-to-Use SARS-CoV-2 Assembler for Genome Sequencing: Development Study. JMIR Bioinform. Biotech. 2022, 3, e31536. [Google Scholar] [CrossRef]
- Rachiglio, A.M.; De Sabato, L.; Roma, C.; Cennamo, M.; Fiorenza, M.; Terracciano, D.; Pasquale, R.; Bergantino, F.; Cavalcanti, E.; Botti, G.; et al. SARS-CoV-2 complete genome sequencing from the Italian Campania region using a highly automated next generation sequencing system. J. Transl. Med. 2021, 19, 246. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Lo Presti, A.; Di Martino, A.; Faggioni, G.; Giordani, F.; Fillo, S.; Anselmo, A.; Fain, V.V.; Fortunato, A.; Petralito, G.; Molinari, F.; et al. Analysis of Genomic Characteristics of SARS-CoV-2 in Italy, 29 January to 27 March 2020. Viruses 2022, 14, 472. [Google Scholar] [CrossRef]
- Stefanelli, P.; Trentini, F.; Guzzetta, G.; Marziano, V.; Mammone, A.; Sane Schepisi, M.; Poletti, P.; Molina Grané, C.; Manica, M.; Del Manso, M.; et al. Co-circulation of SARS-CoV-2 Alpha and Gamma variants in Italy, February and March 2021. Euro Surveill. 2022, 27, 2100429. [Google Scholar] [CrossRef]
- Focosi, D.; Maggi, F. Recombination in Coronaviruses, with a Focus on SARS-CoV-2. Viruses. 2022, 14, 1239. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gender | Age (Years) | Vaccination | Symptoms Onset (Date) | Clinical Features | Admission (Date) | Discharge (Date) | |
|---|---|---|---|---|---|---|---|
| Pt 1 | Female | 81 | None | 19 April 2022 | Pneumonia | 20 April 2022 | 4 May 2022 |
| Pt 2 | Male | 59 | Pfizer-BioNTech * | 24 May 2022 | Fever, myalgia, arthralgia, rash, asthenia, anorexia, cough, syncope | 25 May 2022 | 1 June 2022 |
| BA.1 | BA.2 | INMI | XE Eu | XE It | |
|---|---|---|---|---|---|
| BA.1 | 2.06 × 10−41 | 1.84 × 10−4 | 1.90 × 10−4 | 1.94 × 10−4 | |
| BA.2 | 1.74 × 10−3 | 1.36 × 10−4 | 1.28 × 10−4 | 1.33 × 10−4 | |
| INMI | 1.22 × 10−3 | 7.76 × 10−4 | 9.68 × 10−4 | 1.03 × 10−4 | |
| XE Eu | 1.34 × 10−3 | 7.95 × 10−4 | 3.57 × 10−4 | 3.68 × 10−5 | |
| XE It | 1.35 × 10−3 | 7.87 × 10−4 | 3.66 × 10−4 | 1.75 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rueca, M.; Giombini, E.; Gramigna, G.; Gruber, C.E.M.; Fabeni, L.; Corpolongo, A.; Mazzotta, V.; Corso, L.; Butera, O.; Valli, M.B.; et al. Molecular Characterization of Whole-Genome SARS-CoV-2 from the First Suspected Cases of the XE Variant in the Lazio Region, Italy. Diagnostics 2022, 12, 2219. https://doi.org/10.3390/diagnostics12092219
Rueca M, Giombini E, Gramigna G, Gruber CEM, Fabeni L, Corpolongo A, Mazzotta V, Corso L, Butera O, Valli MB, et al. Molecular Characterization of Whole-Genome SARS-CoV-2 from the First Suspected Cases of the XE Variant in the Lazio Region, Italy. Diagnostics. 2022; 12(9):2219. https://doi.org/10.3390/diagnostics12092219
Chicago/Turabian StyleRueca, Martina, Emanuela Giombini, Giulia Gramigna, Cesare Ernesto Maria Gruber, Lavinia Fabeni, Angela Corpolongo, Valentina Mazzotta, Luisella Corso, Ornella Butera, Maria Beatrice Valli, and et al. 2022. "Molecular Characterization of Whole-Genome SARS-CoV-2 from the First Suspected Cases of the XE Variant in the Lazio Region, Italy" Diagnostics 12, no. 9: 2219. https://doi.org/10.3390/diagnostics12092219
APA StyleRueca, M., Giombini, E., Gramigna, G., Gruber, C. E. M., Fabeni, L., Corpolongo, A., Mazzotta, V., Corso, L., Butera, O., Valli, M. B., Carletti, F., Pignalosa, S., Vairo, F., Nicastri, E., Antinori, A., Girardi, E., Vaia, F., Maggi, F., & SARS CoV-2 Lazio Surveillance Study Group. (2022). Molecular Characterization of Whole-Genome SARS-CoV-2 from the First Suspected Cases of the XE Variant in the Lazio Region, Italy. Diagnostics, 12(9), 2219. https://doi.org/10.3390/diagnostics12092219