
Interstitial Lung Disease in Immunocompromised Children


Abstract
1. Introduction
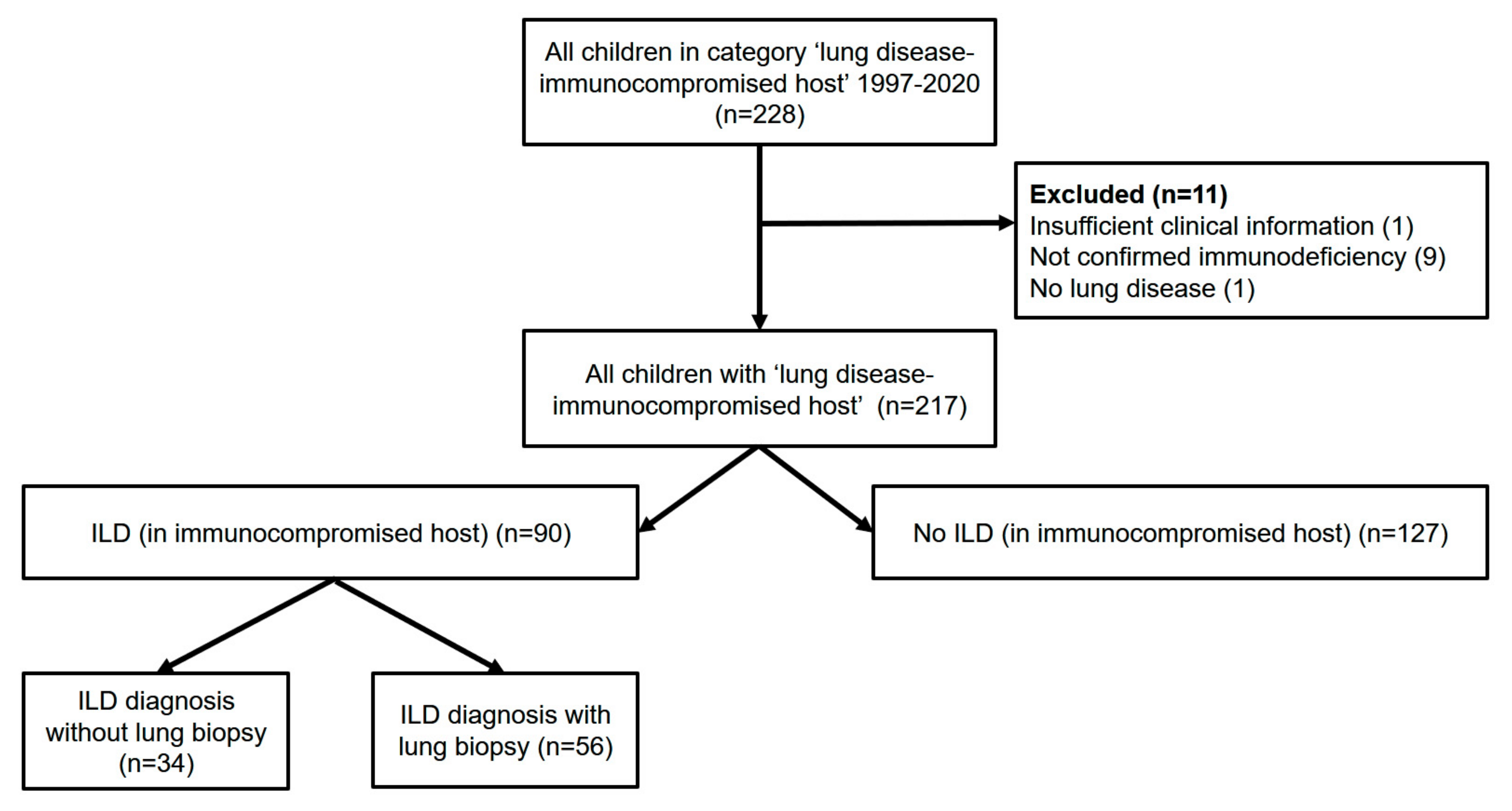
2. Materials and Methods
3. Results
3.1. Characteristics of the Immunodeficiency Population and Spectrum of Associated Lung Diseases
3.2. Comparison of Immunodeficient Children with and without ILD
3.3. Features of ILDs in Immunodeficient Children
3.4. ILD in Genetically Defined Primary Immunodeficiency: Experience from a Single Pediatric Pneumology Center and Review of Literature
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Autosomal dominant transmission |
| ADA | Adenosine deaminase |
| AGS7 | Aicardi–Goutières syndrome 7 |
| ALL | Acute lymphocytic leukemia |
| AML | Acute myeloid leukemia |
| APLAID | Auto-inflammation and phospholipase Cγ2 (PLCγ2)-associated antibody deficiency and immune dysregulation |
| AR | Autosomal recessive transmission |
| ATM | Ataxia Telangiectasia, Mutated |
| BO | Bronchiolitis obliterans |
| CD | Cluster of differentiation |
| CD40LG | CD40 Ligand |
| CGD | Chronic granulomatous disease |
| CID | Combined immunodeficiencies |
| CLL | Chronic lymphocytic leukemia |
| CML | Chronic myelogenous leukemia |
| CVID | Common variable immunodeficiency |
| CYBA | Cytochrome B-245 Alpha Chain |
| CYBB | Cytochrome b-245 beta chain |
| def | deficiency |
| DKC | Dyskeratosis congenita |
| DNMT3B | DNA methyltransferase 3b |
| EDA | Anhidrotic ectodermodysplasia |
| FOXP3 | Forkhead box protein P3 |
| GOF | Gain-of-function |
| HIV | Human immunodeficiency virus |
| ICF | Immunodeficiency, Centromeric region instability, Facial anomalies syndrome |
| ID | Immunodeficiency |
| Ig | Immunoglobulin |
| IKBA | Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha |
| IL | Interleukin |
| IL2RG | Interleukin 2 Receptor Subunit Gamma |
| ILD | Interstitial lung disease |
| IPEX | Immune dysregulation, polyendocrinopathy, enteropathy X-linked |
| JMML | Juvenile myelomonocytic leukemia |
| LOF | Loss-of-function |
| MCM4 | Minichromosome maintenance |
| MDA | Melanoma differentiation-associated protein |
| MHC | Major histocompatibility complex |
| MIRAGE | Myelodysplasia, infection, restriction of growth, adrenal hypoplasia, genital phenotypes, enteropathy |
| NBS | Nijmegen breakage syndrome |
| NCF | Neutrophil cytosolic factor |
| NFKB1 | Nuclear factor-kappaB1 |
| PAP | Pulmonary alveolar proteinosis |
| PAS | Periodic acid–Schiff |
| PHT | Pulmonary hypertension |
| PIK3CD | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Delta |
| PLAID | PLCγ2 associated antibody deficiency and immune dysregulation |
| PLCG2 | phospholipase C gamma 2 |
| RF | Respiratory failure |
| RFXAP | Regulatory Factor X-Associated Protein |
| SAMD9 | Sterile Alpha Motif Domain Containing 9 |
| SCID | Severe combined immunodeficiency |
| SID | Secondary Immunodeficiency |
| STAT | Signal transducer and activator of transcription |
| TACI | Transmembrane activator calcium modulator and cyclophilin ligand interactor |
| TERC | Telomerase RNA Component |
| TERT | Telomerase Reverse Transcriptase |
| TNFRSF13B | Tumor Necrosis Factor Receptor Superfamily Member 13B |
| TNFRSF1A | TNF Receptor Superfamily Member 1A |
| TRAPS | TNF receptor-associated periodic syndrome |
| TTC7A | Tetratricopeptide repeat domain 7A gene |
| UNC13D | Protein unc-13 homolog D |
| ZNFX-1 | NFX1-type zinc finger-containing 1 |
References
- Andrews, J.T.; Voth, D.E.; Huang, S.C.; Huang, L. Breathe in, breathe out: Metabolic regulation of lung macrophages in host defense against bacterial infection. Front. Cell. Infect. Microbiol. 2022, 12, 934460. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Burgel, P.R.; Lepage, P.; Andrejak, C.; de Blic, J.; Bourdin, A.; Brouard, J.; Chanez, P.; Dalphin, J.C.; Deslee, G.; et al. Host-microbe interactions in distal airways: Relevance to chronic airway diseases. Eur. Respir. Rev. 2015, 24, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Ptasinski, V.A.; Stegmayr, J.; Belvisi, M.G.; Wagner, D.E.; Murray, L.A. Targeting alveolar repair in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 65, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Soler-Palacin, P.; de Gracia, J.; Gonzalez-Granado, L.I.; Martin, C.; Rodriguez-Gallego, C.; Sanchez-Ramon, S.; Lung, I.D.S.G. Primary immunodeficiency diseases in lung disease: Warning signs, diagnosis and management. Respir. Res. 2018, 19, 219. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Geng, B.; Cameron, D.W.; Murphy, L.M.; Schulman, E.S. Primary immune deficiency diseases as unrecognized causes of chronic respiratory disease. Respir. Med. 2017, 132, 181–188. [Google Scholar] [CrossRef]
- Neumann, M.; von Bredow, C.; Ratjen, F.; Griese, M. Bronchoalveolar lavage protein patterns in children with malignancies, immunosuppression, fever and pulmonary infiltrates. Proteomics 2002, 2, 683–689. [Google Scholar] [CrossRef]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Cunningham-Rundles, C.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; Oksenhendler, E.; Picard, C.; et al. The ever-increasing array of novel inborn errors of immunity: An interim update by the IUIS committee. J. Clin. Immunol. 2021, 41, 666–679. [Google Scholar] [CrossRef]
- Baumann, U.; Routes, J.M.; Soler-Palacin, P.; Jolles, S. The lung in primary immunodeficiencies: New concepts in infection and inflammation. Front. Immunol. 2018, 9, 1837. [Google Scholar] [CrossRef]
- Raje, N.; Dinakar, C. Overview of immunodeficiency disorders. Immunol. Allergy Clin. N. Am. 2015, 35, 599–623. [Google Scholar] [CrossRef]
- Griese, M.; Rampf, U.; Hofmann, D.; Fuhrer, M.; Reinhardt, D.; Bender-Gotze, C. Pulmonary complications after bone marrow transplantation in children: Twenty-four years of experience in a single pediatric center. Pediatr. Pulmonol. 2000, 30, 393–401. [Google Scholar] [CrossRef]
- Kaul, B.; Cottin, V.; Collard, H.R.; Valenzuela, C. Variability in global prevalence of interstitial lung disease. Front. Med. 2021, 8, 751181. [Google Scholar] [CrossRef] [PubMed]
- Griese, M.; Felber, J.; Reiter, K.; Strong, P.; Reid, K.; Belohradsky, B.H.; Jager, G.; Nicolai, T. Airway inflammation in children with tracheostomy. Pediatr. Pulmonol. 2004, 37, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Griese, M.; Neumann, M.; von Bredow, T.; Schmidt, R.; Ratjen, F. Surfactant in children with malignancies, immunosuppression, fever and pulmonary infiltrates. Eur. Respir. J. 2002, 20, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Griese, M. Etiologic classification of diffuse parenchymal (interstitial) lung diseases. J. Clin. Med. 2022, 11, 1747. [Google Scholar] [CrossRef]
- Bryan Corrin, A.G.N.D. Chapter 10—Pulmonary manifestations of systemic disease. In Pathology of the Lungs, 3rd ed.; Churchill Livingstone: London, UK, 2011. [Google Scholar]
- Grunebaum, E.; Cutz, E.; Roifman, C.M. Pulmonary alveolar proteinosis in patients with adenosine deaminase deficiency. J. Allergy Clin. Immunol. 2012, 129, 1588–1593. [Google Scholar] [CrossRef] [PubMed]
- Al-Saud, B.K.; Al-Sum, Z.; Alassiri, H.; Al-Ghonaium, A.; Al-Muhsen, S.; Al-Dhekri, H.; Arnaout, R.; Alsmadi, O.; Borrero, E.; Abu-Staiteh, A.; et al. Clinical, immunological, and molecular characterization of hyper-IgM syndrome due to CD40 deficiency in eleven patients. J. Clin. Immunol. 2013, 33, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Karaca, N.E.; Forveille, M.; Aksu, G.; Durandy, A.; Kutukculer, N. Hyper-immunoglobulin M syndrome type 3 with normal CD40 cell surface expression. Scand. J. Immunol. 2012, 76, 21–25. [Google Scholar] [CrossRef]
- Du, X.; Tang, W.; Chen, X.; Zeng, T.; Wang, Y.; Chen, Z.; Xu, T.; Zhou, L.; Tang, X.; An, Y.; et al. Clinical, genetic and immunological characteristics of 40 Chinese patients with CD40 ligand deficiency. Scand. J. Immunol. 2019, 90, e12798. [Google Scholar] [CrossRef]
- Winkelstein, J.A.; Marino, M.C.; Ochs, H.; Fuleihan, R.; Scholl, P.R.; Geha, R.; Stiehm, E.R.; Conley, M.E. The X-linked hyper-IgM syndrome: Clinical and immunologic features of 79 patients. Medicine 2003, 82, 373–384. [Google Scholar] [CrossRef]
- Tuovinen, E.A.; Gronholm, J.; Ohman, T.; Poysti, S.; Toivonen, R.; Kreutzman, A.; Heiskanen, K.; Trotta, L.; Toiviainen-Salo, S.; Routes, J.M.; et al. Novel hemizygous IL2RG p.(Pro58Ser) mutation impairs IL-2 receptor complex expression on lymphocytes causing X-linked combined immunodeficiency. J. Clin. Immunol. 2020, 40, 503–514. [Google Scholar] [CrossRef]
- Aydin, S.E.; Kilic, S.S.; Aytekin, C.; Kumar, A.; Porras, O.; Kainulainen, L.; Kostyuchenko, L.; Genel, F.; Kutukculer, N.; Karaca, N.; et al. DOCK8 deficiency: Clinical and immunological phenotype and treatment options—A review of 136 patients. J. Clin. Immunol. 2015, 35, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.Y.; Freeman, A.F.; Jing, H.; Cowen, E.W.; Davis, J.; Su, H.C.; Holland, S.M.; Turner, M.L. Cutaneous manifestations of DOCK8 deficiency syndrome. Arch. Dermatol. 2012, 148, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Matheux, F.; Ikinciogullari, A.; Zapata, D.A.; Barras, E.; Zufferey, M.; Dogu, F.; Regueiro, J.R.; Reith, W.; Villard, J. Direct genetic correction as a new method for diagnosis and molecular characterization of MHC class II deficiency. Mol. Ther. 2002, 6, 824–829. [Google Scholar] [CrossRef] [PubMed]
- Dunn, H.G.; Meuwissen, H.; Livingstone, C.S.; Pump, K.K. Ataxia-telangiectasia. Can. Med. Assoc. J. 1964, 91, 1106–1118. [Google Scholar] [PubMed]
- Schroeder, S.A.; Swift, M.; Sandoval, C.; Langston, C. Interstitial lung disease in patients with ataxia-telangiectasia. Pediatr. Pulmonol. 2005, 39, 537–543. [Google Scholar] [CrossRef]
- Cirillo, E.; Polizzi, A.; Soresina, A.; Prencipe, R.; Giardino, G.; Cancrini, C.; Finocchi, A.; Rivalta, B.; Dellepiane, R.M.; Baselli, L.A.; et al. Progressive depletion of b and t lymphocytes in patients with ataxia telangiectasia: Results of the italian primary immunodeficiency network. J. Clin. Immunol. 2022, 42, 783–797. [Google Scholar] [CrossRef]
- Voigt, R.; Maier-Weidmann, M.; Lange, P.E.; Haaf, T. Chromosome 10p13-14 and 22q11 deletion screening in 100 patients with isolated and syndromic conotruncal heart defects. J. Med. Genet. 2002, 39, e16. [Google Scholar] [CrossRef][Green Version]
- Lichtner, P.; Konig, R.; Hasegawa, T.; Van Esch, H.; Meitinger, T.; Schuffenhauer, S. An HDR (hypoparathyroidism, deafness, renal dysplasia) syndrome locus maps distal to the DiGeorge syndrome region on 10p13/14. J. Med. Genet. 2000, 37, 33–37. [Google Scholar] [CrossRef]
- Gineau, L.; Cognet, C.; Kara, N.; Lach, F.P.; Dunne, J.; Veturi, U.; Picard, C.; Trouillet, C.; Eidenschenk, C.; Aoufouchi, S.; et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J. Clin. Investig. 2012, 122, 821–832. [Google Scholar] [CrossRef]
- Hughes, C.R.; Guasti, L.; Meimaridou, E.; Chuang, C.H.; Schimenti, J.C.; King, P.J.; Costigan, C.; Clark, A.J.; Metherell, L.A. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J. Clin. Investig. 2012, 122, 814–820. [Google Scholar] [CrossRef]
- Weemaes, C.M.; van Tol, M.J.; Wang, J.; van Ostaijen-ten Dam, M.M.; van Eggermond, M.C.; Thijssen, P.E.; Aytekin, C.; Brunetti-Pierri, N.; van der Burg, M.; Graham Davies, E.; et al. Heterogeneous clinical presentation in ICF syndrome: Correlation with underlying gene defects. Eur. J. Hum. Genet. 2013, 21, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Granados, E.; Keenan, J.E.; Kinney, M.C.; Leo, H.; Jain, N.; Ma, C.A.; Quinones, R.; Gelfand, E.W.; Jain, A. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency. Hum. Mutat. 2008, 29, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Boisson, B.; Puel, A.; Picard, C.; Casanova, J.L. Human ikappabalpha gain of function: A severe and syndromic immunodeficiency. J. Clin. Immunol. 2017, 37, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Nijmegen Breakage Syndrome. The International Nijmegen Breakage Syndrome Study Group. Arch. Dis. Child. 2000, 82, 400–406. [Google Scholar] [CrossRef]
- Marczak, H.; Heropolitanska-Pliszka, E.; Langfort, R.; Roik, D.; Grzela, K. Nijmegen breakage syndrome complicated with primary pulmonary granulomas. Pediatrics 2018, 142, e20180122. [Google Scholar] [CrossRef]
- Wang, L.; Cheng, J.; Gao, J.; Wang, J.; Liu, X.; Xiong, L. Association between the NBS1 Glu185Gln polymorphism and lung cancer risk: A systemic review and meta-analysis. Mol. Biol. Rep. 2013, 40, 2711–2715. [Google Scholar] [CrossRef]
- Samuels, M.E.; Majewski, J.; Alirezaie, N.; Fernandez, I.; Casals, F.; Patey, N.; Decaluwe, H.; Gosselin, I.; Haddad, E.; Hodgkinson, A.; et al. Exome sequencing identifies mutations in the gene TTC7A in French-Canadian cases with hereditary multiple intestinal atresia. J. Med. Genet. 2013, 50, 324–329. [Google Scholar] [CrossRef]
- Fang, Y.; Luo, Y.; Yu, J.; Chen, J. A case of severe malnutrition infant with neonatal onset intractable diarrhea. BMC Pediatr. 2020, 20, 133. [Google Scholar] [CrossRef]
- Nambu, R.; Warner, N.; Mulder, D.J.; Kotlarz, D.; McGovern, D.P.B.; Cho, J.; Klein, C.; Snapper, S.B.; Griffiths, A.M.; Iwama, I.; et al. A systematic review of monogenic inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 2022, 20, e653–e663. [Google Scholar] [CrossRef]
- McDonald-McGinn, D.M.; Sullivan, K.E. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine 2011, 90, 1–18. [Google Scholar] [CrossRef]
- Sood, A.K.; Funkhouser, W.; Handly, B.; Weston, B.; Wu, E.Y. Granulomatous-lymphocytic interstitial lung disease in 22q11.2 deletion syndrome: A case report and literature review. Curr. Allergy Asthma Rep. 2018, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Kiaee, F.; Zaki-Dizaji, M.; Hafezi, N.; Almasi-Hashiani, A.; Hamedifar, H.; Sabzevari, A.; Shirkani, A.; Zian, Z.; Jadidi-Niaragh, F.; Aghamahdi, F.; et al. Clinical, immunologic and molecular spectrum of patients with immunodeficiency, centromeric instability, and facial anomalies (ICF) syndrome: A systematic review. Endocr. Metab. Immune Disord. Drug Targ. 2021, 21, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, H.A.; Tashkandi, S.A.; Alidrissi, E.M.; Aledielah, R.D.; AlSaidi, K.A.; Alharbi, E.S.; Habazi, M.K.; Alzahrani, M.S. Three types of immunodeficiency, centromeric instability, and facial anomalies (ICF) syndrome identified by whole-exome sequencing in saudi hypogammaglobulinemia patients: Clinical, molecular, and cytogenetic features. J. Clin. Immunol. 2018, 38, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Erdem, S.B.; Gulez, N.; Genel, F.; Karaman, S.; Nacaroglu, H.T. Characteristics of the patients followed with the diagnosis of common variable immunodeficiency and the complications. Cent. Eur. J. Immunol. 2019, 44, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Shinya, Y.; Hiraide, T.; Momoi, M.; Goto, S.; Suzuki, H.; Katsumata, Y.; Kurebayashi, Y.; Endo, J.; Sano, M.; Fukuda, K.; et al. TNFRSF13B c.226G>A (p.Gly76Ser) as a novel causative mutation for pulmonary arterial hypertension. J. Am. Heart Assoc. 2021, 10, e019245. [Google Scholar] [CrossRef]
- Han, S.P.; Lin, Y.F.; Weng, H.Y.; Tsai, S.F.; Fu, L.S. A novel BTK gene mutation in a child with atypical x-linked agammaglobulinemia and recurrent hemophagocytosis: A case report. Front. Immunol. 2019, 10, 1953. [Google Scholar] [CrossRef]
- Quartier, P.; Debre, M.; De Blic, J.; de Sauverzac, R.; Sayegh, N.; Jabado, N.; Haddad, E.; Blanche, S.; Casanova, J.L.; Smith, C.I.; et al. Early and prolonged intravenous immunoglobulin replacement therapy in childhood agammaglobulinemia: A retrospective survey of 31 patients. J. Pediatr. 1999, 134, 589–596. [Google Scholar] [CrossRef]
- Tuijnenburg, P.; Lango Allen, H.; Burns, S.O.; Greene, D.; Jansen, M.H.; Staples, E.; Stephens, J.; Carss, K.J.; Biasci, D.; Baxendale, H.; et al. Loss-of-function nuclear factor kappaB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J. Allergy Clin. Immunol. 2018, 142, 1285–1296. [Google Scholar] [CrossRef]
- Schroder, C.; Sogkas, G.; Fliegauf, M.; Dork, T.; Liu, D.; Hanitsch, L.G.; Steiner, S.; Scheibenbogen, C.; Jacobs, R.; Grimbacher, B.; et al. Late-onset antibody deficiency due to monoallelic alterations in NFKB1. Front. Immunol. 2019, 10, 2618. [Google Scholar] [CrossRef]
- Coulter, T.I.; Chandra, A.; Bacon, C.M.; Babar, J.; Curtis, J.; Screaton, N.; Goodlad, J.R.; Farmer, G.; Steele, C.L.; Leahy, T.R.; et al. Clinical spectrum and features of activated phosphoinositide 3-kinase delta syndrome: A large patient cohort study. J. Allergy Clin. Immunol. 2017, 139, 597–606.e4. [Google Scholar] [CrossRef]
- Fekrvand, S.; Delavari, S.; Chavoshzadeh, Z.; Sherkat, R.; Mahdaviani, S.A.; Sadeghi Shabestari, M.; Azizi, G.; Arzanian, M.T.; Shahin Shamsian, B.; Eskandarzadeh, S.; et al. The first iranian cohort of pediatric patients with activated phosphoinositide 3-kinase-delta (PI3Kdelta) syndrome (APDS). Immunol. Investig. 2022, 51, 644–659. [Google Scholar] [CrossRef] [PubMed]
- Gambineri, E.; Ciullini Mannurita, S.; Hagin, D.; Vignoli, M.; Anover-Sombke, S.; DeBoer, S.; Segundo, G.R.S.; Allenspach, E.J.; Favre, C.; Ochs, H.D.; et al. Clinical, immunological, and molecular heterogeneity of 173 patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Front. Immunol. 2018, 9, 2411. [Google Scholar] [CrossRef] [PubMed]
- Fabre, A.; Marchal, S.; Barlogis, V.; Mari, B.; Barbry, P.; Rohrlich, P.S.; Forbes, L.R.; Vogel, T.P.; Giovannini-Chami, L. Clinical aspects of STAT3 gain-of-function germline mutations: A systematic review. J. Allergy Clin. Immunol. Pract. 2019, 7, 1958–1969. [Google Scholar] [CrossRef] [PubMed]
- Gothe, F.; Gehrig, J.; Rapp, C.K.; Knoflach, K.; Reu-Hofer, S.; Langer, F.; Schramm, D.; Ley-Zaporozhan, J.; Ehl, S.; Schwerk, N.; et al. Early-onset, fatal interstitial lung disease in STAT3 gain-of-function patients. Pediatr. Pulmonol. 2021, 56, 3934–3941. [Google Scholar] [CrossRef] [PubMed]
- Werner, L.; Lee, Y.N.; Rechavi, E.; Lev, A.; Yerushalmi, B.; Ling, G.; Shah, N.; Uhlig, H.H.; Weiss, B.; Somech, R.; et al. Alterations in T and B cell receptor repertoires patterns in patients with IL10 signaling defects and history of infantile-onset IBD. Front. Immunol. 2020, 11, 109. [Google Scholar] [CrossRef]
- Zheng, C.; Huang, Y.; Hu, W.; Shi, J.; Ye, Z.; Qian, X.; Huang, Z.; Xue, A.; Wang, Y.; Lu, J.; et al. Phenotypic characterization of very early-onset inflammatory bowel disease with interleukin-10 signaling deficiency: Based on a large cohort study. Inflamm. Bowel Dis. 2019, 25, 756–766. [Google Scholar] [CrossRef]
- Meeths, M.; Chiang, S.C.; Wood, S.M.; Entesarian, M.; Schlums, H.; Bang, B.; Nordenskjold, E.; Bjorklund, C.; Jakovljevic, G.; Jazbec, J.; et al. Familial hemophagocytic lymphohistiocytosis type 3 (FHL3) caused by deep intronic mutation and inversion in UNC13D. Blood 2011, 118, 5783–5793. [Google Scholar] [CrossRef]
- Hu, X.; Liu, D.; Jiang, X.; Gao, B.; Chen, C. Identification of a novel nonsense mutation in the UNC13D gene from a patient with hemophagocytic lymphohistiocytosis: A case report. BMC Med. Genet. 2018, 19, 82. [Google Scholar] [CrossRef]
- Hildebrandt, J.; Yalcin, E.; Bresser, H.G.; Cinel, G.; Gappa, M.; Haghighi, A.; Kiper, N.; Khalilzadeh, S.; Reiter, K.; Sayer, J.; et al. Characterization of CSF2RA mutation related juvenile pulmonary alveolar proteinosis. Orph. J. Rare Dis. 2014, 9, 171. [Google Scholar] [CrossRef]
- Whitsett, J.A.; Wert, S.E.; Weaver, T.E. Diseases of pulmonary surfactant homeostasis. Annu. Rev. Pathol. 2015, 10, 371–393. [Google Scholar] [CrossRef]
- Rawat, A.; Vignesh, P.; Sudhakar, M.; Sharma, M.; Suri, D.; Jindal, A.; Gupta, A.; Shandilya, J.K.; Loganathan, S.K.; Kaur, G.; et al. Clinical, immunological, and molecular profile of chronic granulomatous disease: A multi-centric study of 236 patients from India. Front. Immunol. 2021, 12, 625320. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, W.F.; Zhang, Y.D.; Chen, T.X. Clinical features and genetic analysis of 48 patients with chronic granulomatous disease in a single center study from Shanghai, China (2005–2015): New studies and a literature review. J. Immunol. Res. 2017, 2017, 8745254. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, J.M.; van Koppen, E.; Ahlin, A.; Belohradsky, B.H.; Bernatowska, E.; Corbeel, L.; Espanol, T.; Fischer, A.; Kurenko-Deptuch, M.; Mouy, R.; et al. Chronic granulomatous disease: The European experience. PLoS ONE 2009, 4, e5234. [Google Scholar] [CrossRef] [PubMed]
- Roos, D.; Kuhns, D.B.; Maddalena, A.; Bustamante, J.; Kannengiesser, C.; de Boer, M.; van Leeuwen, K.; Koker, M.Y.; Wolach, B.; Roesler, J.; et al. Hematologically important mutations: The autosomal recessive forms of chronic granulomatous disease (second update). Blood Cells Mol. Dis. 2010, 44, 291–299. [Google Scholar] [CrossRef]
- Lamborn, I.T.; Jing, H.; Zhang, Y.; Drutman, S.B.; Abbott, J.K.; Munir, S.; Bade, S.; Murdock, H.M.; Santos, C.P.; Brock, L.G.; et al. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J. Exp. Med. 2017, 214, 1949–1972. [Google Scholar] [CrossRef]
- Zheng, S.; Lee, P.Y.; Wang, J.; Wang, S.; Huang, Q.; Huang, Y.; Liu, Y.; Zhou, Q.; Li, T. Interstitial lung disease and psoriasis in a child with Aicardi-Goutieres syndrome. Front. Immunol. 2020, 11, 985. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, X.; Gao, G.; Xing, S.; Zhou, L.; Tang, X.; Zhao, X.; An, Y. Clinical relevance of gain- and loss-of-function germline mutations in STAT1: A systematic review. Front. Immunol. 2021, 12, 654406. [Google Scholar] [CrossRef]
- Capo, V.; Penna, S.; Merelli, I.; Barcella, M.; Scala, S.; Basso-Ricci, L.; Draghici, E.; Palagano, E.; Zonari, E.; Desantis, G.; et al. Expanded circulating hematopoietic stem/progenitor cells as novel cell source for the treatment of TCIRG1 osteopetrosis. Haematologica 2021, 106, 74–86. [Google Scholar] [CrossRef]
- Vavassori, S.; Chou, J.; Faletti, L.E.; Haunerdinger, V.; Opitz, L.; Joset, P.; Fraser, C.J.; Prader, S.; Gao, X.; Schuch, L.A.; et al. Multisystem inflammation and susceptibility to viral infections in human ZNFX1 deficiency. J. Allergy Clin. Immunol. 2021, 148, 381–393. [Google Scholar] [CrossRef]
- Le Voyer, T.; Neehus, A.L.; Yang, R.; Ogishi, M.; Rosain, J.; Alroqi, F.; Alshalan, M.; Blumental, S.; Al Ali, F.; Khan, T.; et al. Inherited deficiency of stress granule ZNFX1 in patients with monocytosis and mycobacterial disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2102804118. [Google Scholar] [CrossRef]
- Vece, T.J.; Watkin, L.B.; Nicholas, S.; Canter, D.; Braun, M.C.; Guillerman, R.P.; Eldin, K.W.; Bertolet, G.; McKinley, S.; de Guzman, M.; et al. Copa syndrome: A novel autosomal dominant immune dysregulatory disease. J. Clin. Immunol. 2016, 36, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Watkin, L.B.; Jessen, B.; Wiszniewski, W.; Vece, T.J.; Jan, M.; Sha, Y.; Thamsen, M.; Santos-Cortez, R.L.; Lee, K.; Gambin, T.; et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat. Genet. 2015, 47, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Magg, T.; Okano, T.; Koenig, L.M.; Boehmer, D.F.R.; Schwartz, S.L.; Inoue, K.; Heimall, J.; Licciardi, F.; Ley-Zaporozhan, J.; Ferdman, R.M.; et al. Heterozygous OAS1 gain-of-function variants cause an autoinflammatory immunodeficiency. Sci. Immunol. 2021, 6, eabf9564. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Yamada, M.; Agematsu, K.; Kanegane, H.; Miyake, N.; Ueki, M.; Akimoto, T.; Kobayashi, N.; Ikemoto, S.; Tanino, M.; et al. Heterozygous mutations in OAS1 cause infantile-onset pulmonary alveolar proteinosis with hypogammaglobulinemia. Am. J. Hum. Genet. 2018, 102, 480–486. [Google Scholar] [CrossRef]
- Aderibigbe, O.M.; Priel, D.L.; Lee, C.C.; Ombrello, M.J.; Prajapati, V.H.; Liang, M.G.; Lyons, J.J.; Kuhns, D.B.; Cowen, E.W.; Milner, J.D. Distinct cutaneous manifestations and cold-induced leukocyte activation associated with PLCG2 mutations. JAMA Dermatol. 2015, 151, 627–634. [Google Scholar] [CrossRef]
- Kutukculer, N.; Topyildiz, E.; Berdeli, A.; Guven Bilgin, B.; Aykut, A.; Durmaz, A.; Cogulu, O.; Aksu, G.; Edeer Karaca, N. Four diseases, PLAID, APLAID, FCAS3 and CVID and one gene (PHOSPHOLIPASE C, GAMMA-2; PLCG2): Striking clinical phenotypic overlap and difference. Clin. Case Rep. 2021, 9, 2023–2031. [Google Scholar] [CrossRef]
- Ombrello, M.J.; Remmers, E.F.; Sun, G.; Freeman, A.F.; Datta, S.; Torabi-Parizi, P.; Subramanian, N.; Bunney, T.D.; Baxendale, R.W.; Martins, M.S.; et al. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N. Engl. J. Med. 2012, 366, 330–338. [Google Scholar] [CrossRef]
- Fremond, M.L.; Hadchouel, A.; Berteloot, L.; Melki, I.; Bresson, V.; Barnabei, L.; Jeremiah, N.; Belot, A.; Bondet, V.; Brocq, O.; et al. Overview of STING-associated vasculopathy with onset in infancy (SAVI) among 21 patients. J. Allergy Clin. Immunol. Pract. 2021, 9, 803–818.e11. [Google Scholar] [CrossRef]
- Liu, Y.; Jesus, A.A.; Marrero, B.; Yang, D.; Ramsey, S.E.; Sanchez, G.A.M.; Tenbrock, K.; Wittkowski, H.; Jones, O.Y.; Kuehn, H.S.; et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 2014, 371, 507–518. [Google Scholar] [CrossRef]
- Amari, S.; Tsukamoto, K.; Ishiguro, A.; Yanagi, K.; Kaname, T.; Ito, Y. An extremely severe case of Aicardi-Goutieres syndrome 7 with a novel variant in IFIH1. Eur. J. Med. Genet. 2020, 63, 103646. [Google Scholar] [CrossRef]
- Adang, L.A.; Frank, D.B.; Gilani, A.; Takanohashi, A.; Ulrick, N.; Collins, A.; Cross, Z.; Galambos, C.; Helman, G.; Kanaan, U.; et al. Aicardi goutieres syndrome is associated with pulmonary hypertension. Mol. Genet. Metab. 2018, 125, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Adang, L.; Gavazzi, F.; De Simone, M.; Fazzi, E.; Galli, J.; Koh, J.; Kramer-Golinkoff, J.; De Giorgis, V.; Orcesi, S.; Peer, K.; et al. Developmental outcomes of aicardi goutieres syndrome. J. Child Neurol. 2020, 35, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Cekin, N.; Akyurek, M.E.; Pinarbasi, E.; Ozen, F. MEFV mutations and their relation to major clinical symptoms of familial mediterranean fever. Gene 2017, 626, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Makay, B.; Gulez, N. Long-term follow-up of paediatric MEFV carriers. Clin. Rheumatol. 2018, 37, 1683–1687. [Google Scholar] [CrossRef] [PubMed]
- Caglayan, A.O.; Demiryilmaz, F.; Ozyazgan, I.; Gumus, H. MEFV gene compound heterozygous mutations in familial Mediterranean fever phenotype: A retrospective clinical and molecular study. Nephrol. Dial. Transpl. 2010, 25, 2520–2523. [Google Scholar] [CrossRef][Green Version]
- Cudrici, C.; Deuitch, N.; Aksentijevich, I. Revisiting TNF receptor-associated periodic syndrome (TRAPS): Current perspectives. Int. J. Mol. Sci. 2020, 21, 3263. [Google Scholar] [CrossRef]
- Davidsson, J.; Puschmann, A.; Tedgard, U.; Bryder, D.; Nilsson, L.; Cammenga, J. SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 2018, 32, 1106–1115. [Google Scholar] [CrossRef]
- Nagata, Y.; Narumi, S.; Guan, Y.; Przychodzen, B.P.; Hirsch, C.M.; Makishima, H.; Shima, H.; Aly, M.; Pastor, V.; Kuzmanovic, T.; et al. Germline loss-of-function SAMD9 and SAMD9L alterations in adult myelodysplastic syndromes. Blood 2018, 132, 2309–2313. [Google Scholar] [CrossRef]
- Mushiroda, T.; Wattanapokayakit, S.; Takahashi, A.; Nukiwa, T.; Kudoh, S.; Ogura, T.; Taniguchi, H.; Kubo, M.; Kamatani, N.; Nakamura, Y.; et al. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J. Med. Genet. 2008, 45, 654–656. [Google Scholar] [CrossRef]
- Armanios, M.Y.; Chen, J.J.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A.; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef]
- Doubkova, M.; Speldova, J.; Chovancova, Z. Immunodeficiency in the differential diagnosis of interstitial lung diseases. Vnitr. Lek. 2019, 65, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Pac, M.; Bielecka, T.; Grzela, K.; Komarnicka, J.; Langfort, R.; Koltan, S.; Dabrowska-Leonik, N.; Bernat-Sitarz, K.; Pronicki, M.; Dmenska, H.; et al. Interstitial lung disease in children with selected primary immunodeficiency disorders—A multicenter observational study. Front. Immunol. 2020, 11, 1950. [Google Scholar] [CrossRef] [PubMed]
- Maglione, P.J.; Overbey, J.R.; Radigan, L.; Bagiella, E.; Cunningham-Rundles, C. Pulmonary radiologic findings in common variable immunodeficiency: Clinical and immunological correlations. Ann. Allergy Asthma Immunol. 2014, 113, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Grimbacher, B.; Hurst, J.R. Lung disease in primary antibody deficiency. Lancet Respir. Med. 2015, 3, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Maarschalk-Ellerbroek, L.J.; de Jong, P.A.; van Montfrans, J.M.; Lammers, J.W.; Bloem, A.C.; Hoepelman, A.I.; Ellerbroek, P.M. CT screening for pulmonary pathology in common variable immunodeficiency disorders and the correlation with clinical and immunological parameters. J. Clin. Immunol. 2014, 34, 642–654. [Google Scholar] [CrossRef]
- Bates, C.A.; Ellison, M.C.; Lynch, D.A.; Cool, C.D.; Brown, K.K.; Routes, J.M. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J. Allergy Clin. Immunol. 2004, 114, 415–421. [Google Scholar] [CrossRef]
- Lopes, J.P.; Ho, H.E.; Cunningham-Rundles, C. Interstitial lung disease in common variable immunodeficiency. Front. Immunol. 2021, 12, 605945. [Google Scholar] [CrossRef]
- Van De Ven, A.A.J.M.; De Jong, P.A.; Van Konijnenburg, D.P.H.; Kessels, O.A.M.; Boes, M.; Sanders, E.A.M.; Terheggen-Lagro, S.W.J.; Van Montfrans, J.M. Airway and interstitial lung disease are distinct entities in paediatric common variable immunodeficiency. Clin. Exp. Immunol. 2011, 165, 235–242. [Google Scholar] [CrossRef]
- Ho, H.E.; Cunningham-Rundles, C. Non-infectious complications of common variable immunodeficiency: Updated clinical spectrum, sequelae, and insights to pathogenesis. Front. Immunol. 2020, 11, 149. [Google Scholar] [CrossRef]
- Lopez, A.L.; Paolini, M.V.; Fernandez Romero, D.S. Lung disease in patients with common variable immunodeficiency. Allergol. Immunopathol. 2020, 48, 720–728. [Google Scholar] [CrossRef]
- Shin, J.J.; Liauw, D.; Siddiqui, S.; Lee, J.; Chung, E.J.; Steele, R.; Hsu, F.I.; Price, C.; Kang, I. Immunological and clinical phenotyping in primary antibody deficiencies: A growing disease spectrum. J. Clin. Immunol. 2020, 40, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Berbers, R.M.; Mohamed Hoesein, F.A.A.; Ellerbroek, P.M.; van Montfrans, J.M.; Dalm, V.; van Hagen, P.M.; Paganelli, F.L.; Viveen, M.C.; Rogers, M.R.C.; de Jong, P.A.; et al. Low IgA associated with oropharyngeal microbiota changes and lung disease in primary antibody deficiency. Front. Immunol. 2020, 11, 1245. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, T.; Fuleihan, R.; Cunningham-Rundles, C.; Maglione, P.J. Factors beyond lack of antibody govern pulmonary complications in primary antibody deficiency. J. Clin. Immunol. 2019, 39, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Bruns, L.; Panagiota, V.; von Hardenberg, S.; Schmidt, G.; Adriawan, I.R.; Sogka, E.; Hirsch, S.; Ahrenstorf, G.; Witte, T.; Schmidt, R.E.; et al. Common variable immunodeficiency-associated cancers: The role of clinical phenotypes, immunological and genetic factors. Front. Immunol. 2022, 13, 742530. [Google Scholar] [CrossRef] [PubMed]
- Maglione, P.J.; Gyimesi, G.; Cols, M.; Radigan, L.; Ko, H.M.; Weinberger, T.; Lee, B.H.; Grasset, E.K.; Rahman, A.H.; Cerutti, A.; et al. BAFF-driven B cell hyperplasia underlies lung disease in common variable immunodeficiency. J. Clin. Investig. 2019, 4, e122728. [Google Scholar] [CrossRef]
- Cabanero-Navalon, M.D.; Garcia-Bustos, V.; Forero-Naranjo, L.F.; Baettig-Arriagada, E.J.; Nunez-Beltran, M.; Canada-Martinez, A.J.; Forner Giner, M.J.; Catalan-Caceres, N.; Martinez Frances, M.; Moral Moral, P. Integrating clinics, laboratory, and imaging for the diagnosis of common variable immunodeficiency-related granulomatous-lymphocytic interstitial lung disease. Front. Immunol. 2022, 13, 813491. [Google Scholar] [CrossRef]
- Kellner, E.S.; Fuleihan, R.; Cunningham-Rundles, C.; Wechsler, J.B. Cellular defects in CVID patients with chronic lung disease in the USIDNET registry. J. Clin. Immunol. 2019, 39, 569–576. [Google Scholar] [CrossRef]
- Fraz, M.S.A.; Michelsen, A.E.; Moe, N.; Aaløkken, T.M.; Macpherson, M.E.; Nordøy, I.; Aukrust, P.; Taraldsrud, E.; Holm, A.M.; Ueland, T.; et al. Raised serum markers of t cell activation and exhaustion in granulomatous-lymphocytic interstitial lung disease in common variable immunodeficiency. J. Clin. Immunol. 2022, 42, 1553–1563. [Google Scholar] [CrossRef]
- Popa, V.; Colby, T.V.; Reich, S.B. Pulmonary interstitial disease in Ig deficiency. Chest 2002, 122, 1594–1603. [Google Scholar] [CrossRef][Green Version]
- Kawai, T.; Watanabe, N.; Yokoyama, M.; Nakazawa, Y.; Goto, F.; Uchiyama, T.; Higuchi, M.; Maekawa, T.; Tamura, E.; Nagasaka, S.; et al. Interstitial lung disease with multiple microgranulomas in chronic granulomatous disease. J. Clin. Immunol. 2014, 34, 933–940. [Google Scholar] [CrossRef]
- Hurst, J.R.; Verma, N.; Lowe, D.; Baxendale, H.E.; Jolles, S.; Kelleher, P.; Longhurst, H.J.; Patel, S.Y.; Renzoni, E.A.; Sander, C.R.; et al. British lung foundation/united kingdom primary immunodeficiency network consensus statement on the definition, diagnosis, and management of granulomatous-lymphocytic interstitial lung disease in common variable immunodeficiency disorders. J. Allergy Clin. Immunol. Pract. 2017, 5, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Hurst, J.R.; Warnatz, K.; ERS eGLILDnet Clinical Research Collaboration. Interstitial lung disease in primary immunodeficiency: Towards a brighter future. Eur. Respir. J. 2020, 55, 2000089. [Google Scholar] [CrossRef] [PubMed]
- Larsen, B.T. GLILD revisited pulmonary pathology of common variable and selective IgA immunodeficiency. Am. J. Surg. Pathol. 2020, 44, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics data integration, interpretation, and its application. Bioinform. Biol. Insights 2020, 14, 117793221989905. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| All Immunodeficiency Patients | Immunodeficiency with ILD | Immunodeficiency without ILD | Comparison between with/without ILD P | |
|---|---|---|---|---|
| Total number | 217 | 90 (41%) | 127 (59%) | |
| Sex (male/female) | 129 (59%)/88 (41%) | 53 (59%)/37 (41%) | 76 (60%)/51 (40%) | 0.888 * |
| Age at onset of lung disease in years (range) | 2.0 (0.0–20.1) | 2.9 (0.0–15.2) | 1.5 (0.0–20.1) | 0.116 ** |
| Follow-up duration in years (range) | 4.9 (0.0–30.2) | 4.9 (0.1–19.4) | 4.9 (0.0–30.2) | 0.893 ** |
| ILD family history (yes/no) | 10 (9%)/97 (91%) | 8 (12%)/59 (88%) | 2 (5%)/38 (95%) | 0.233 * |
| Consanguinity (yes/no) | 27 (25%)/79 (75%) | 20 (30%)/46 (70%) | 7 (18%)/33 (82%) | 0.143 * |
| Gestational age (range) | 40 (29–42) | 40 (31–41) | 40 (29–42) | 0.696 ** |
| O2 supplement in neonatal period (yes/no) | 11/132 | 6/58 | 5/74 | 0.497 * |
| Mechanical ventilation in neonatal period (yes/no) | 8/135 | 4/60 | 4/75 | 0.759 * |
| Outcome of lung disease at the end of follow-up | ||||
| Sick-better | 96 | 39 | 57 | 0.853 * |
| Sick-same | 46 | 23 | 23 | 0.177 * |
| Sick-worse | 32 | 7 | 25 | 0.015 * |
| Dead | 39 | 19 | 20 | 0.299 * |
| Lung Disease; n (% of Immunodeficiency Group) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Immunodeficiency Type (n) | Interstitial Lung Disease | Pulmonary Hypertension | Infections (Opportunistic/Recurrent) | Bronchiolitis Obliterans | Bronchiectasis | PTLD | Respiratory Failure | ARDS | Diffuse Alveolar Damage | Pneumothorax | Asthma | Pleural Disease |
| All immunodeficiencies (217) | 90 (41) | 11 (5) | 142 (65) | 32 (15) | 23 (11) | 3 (1) | 58 (27) | 15 (7) | 3 (1) | 9 (4) | 21 (10) | 12 (6) |
| Primary immunodeficiencies (120) | 52 (44) | 6 (5) | 88 (73) | 6 (5) | 19 (16) | - | 31 (26) | 9 (8) | - | 1 (1) | 13 (11) | 4 (3) |
| Combined immunodeficiencies (22) | 9 (41) | - | 18 (82) | - | 2 (9) | - | 8 (36) | 3(14) | - | 1 (5) | 1 (5) | - |
| Well-defined syndromes (15) | 4 (27) | 2 (13) | 11 (73) | 2 (13) | 4 (27) | - | 1 (7) | 2 (13) | - | - | 1 (7) | - |
| Antibody deficiencies (21) | 4 (19) | 1 (5) | 17 (81) | 1 (5) | 8 (38) | - | 2 (10) | 1 (5) | - | - | 6 (29) | - |
| Immune dysregulation (5) | 2 (40) | - | 3 (60) | 1 (20) | - | - | 1 (20) | - | - | - | - | - |
| Defects of phagocytes (30) | 18 (60) | 1 (3) | 20 (67) | 2 (7) | 3 (10) | - | 10 (33) | - | - | - | 3 (10) | 2 (7) |
| Defects of innate immunity (7) | 3 (43) | - | 6 (86) | - | 1 (14) | - | 4 (57) | 2 (29) | - | - | - | 1 (14) |
| Autoinflammatory syndromes (17) | 10 (59) | 2 (12) | 12 (71) | - | 1 (6) | - | 4 (24) | 1 (6) | - | - | 2 (12) | 1 (6) |
| Bone marrow failure (3) | 2 (67) | - | 1 (33) | - | - | - | 1 (33) | - | - | - | - | - |
| Secondary immunodeficiencies (97) | 38 (39) | 5 (5) | 54 (56) | 26 (27) | 4 (4) | 3 (3) | 27 (28) | 6 (6) | 3 (3) | 8 (8) | 8 (8) | 8 (8) |
| ALL (15) | 4 (27) | - | 11 (73) | 1 (7) | 1 (7) | 10(7) | 5 (33) | 1 (7) | - | - | 1 (7) | 1 (7) |
| AML (10) | 3 (30) | - | 6 (60) | 1 (10) | - | - | 3 (30) | - | 1 (10) | - | - | 1 (10) |
| Cancer, other (10) | 4 (40) | - | 4 (40) | 1 (10) | - | - | - | 1 (10) | - | 1 (10) | 1 (10) | - |
| CLL (2) | 1 (50) | - | 1 (50) | - | - | - | - | - | - | - | 1 (50) | 1 (50) |
| CML (1) | - | - | - | - | - | - | 1 (100) | - | - | - | - | 1 (100) |
| HIV (2) | 1 (50) | - | 1 (50) | - | - | - | - | - | - | - | - | - |
| Hodgkin lymphoma (3) | - | - | 2 (67) | - | - | - | - | - | - | - | 1 (33) | - |
| JMML (3) | 2 (66) | 1 (33) | 2 (67) | 1 (33) | - | - | 1 (33) | 1 (33) | - | - | - | - |
| MDS (5) | 4 (80) | 1 (20) | 2 (40) | 1 (20) | - | - | 2 (40) | - | - | 1 (20) | 2 (40) | 1 (20) |
| Non-Hodgkin lymphoma (1) | - | - | 1 (100) | - | - | - | - | - | - | - | - | - |
| Other therap. intervention (1) | - | - | 1 (100) | - | - | - | - | - | - | - | - | - |
| Transplant-heart (3) | 2 (67) | - | 2 (67) | - | - | 1 (33) | - | - | - | - | - | 1 (33) |
| Transplant-heart and lung (6) | 3 (50) | 3 (50) | 2 (33) | 2 (33) | - | - | 2 (33) | - | 1 (17) | - | - | - |
| Transplant-kidney (1) | - | - | 1 (100) | - | - | - | 1 (100) | - | - | - | - | - |
| Transplant-lung (4) | 1 (25) | - | 3 (75) | 1 (25) | 1 (25) | 1 (25) | 2 (50) | - | - | 1 (25) | - | - |
| Transplant-stem cell (30) | 14 (47) | - | 15 (50) | 18 (60) | 2 (7) | 1(3) | 10 (33) | 3 (10) | 1 (3) | 5 (17) | 2 (7) | 2 (7) |
| All Immunodeficiency Patients | Immunodeficiency with ILD | Immunodeficiency without ILD | Comparison between with/without ILD P * | |
|---|---|---|---|---|
| Cell concentration (/µL) | 405.3 ± 1209.4 (114) | 343.5 ± 308.1 (28) | 425.4 ± 1383.0 (86) | 0.216 |
| Macrophages (%) | 60.1 ± 27.9 (138) | 64.7 ± 23.6 (46) | 57.8 ± 29.6 (92) | 0.299 |
| PMN (%) | 19.4 ± 25.9 (138) | 16.3 ± 19.2 (46) | 21.0 ± 28.7 (92) | 0.626 |
| Lymphocytes (%) | 16.2 ± 17.0 (138) | 13.5 ± 14.0 (46) | 17.5 ± 18.2 (92) | 0.437 |
| Eosinophils (%) | 2.1 ± 6.6 (138) | 4.0 ± 10.5 (46) | 1.2 ± 2.9 (92) | 0.101 |
| Mast cells (%) | 0.2 ± 1.0 (138) | 0.2 ± 0.8 (46) | 0.3 ± 1.2 (92) | 0.995 |
| Plasma cells (%) | 0.04 ± 0.3 (138) | 0.03 ± 0.1 (46) | 0.05 ± 0.4 (92) | 0.388 |
| Cell viability (%) | 79.0 ± 23.0 (105) | 80.4 ± 25.9 (23) | 78.5 ± 22.2 (82) | 0.283 |
| Cell recovery (/µL) | 53.0 ± 24.2 (107) | 50.7 ± 28.5 (30) | 53.9 ± 22.5 (77) | 0.539 ** |
| Sex (m/f) | ILD Family History (y/n) | Consanguinity (y/n) | Age at Onset of Lung Disease (Years) | Gestational Age (Week) | O2 Supplement in Neonatal Period (y/n) | Mechanical Ventilation in Neonatal Period (y/n) | Follow-Up Duration (Years) | Outcome of Lung Disease | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sick-Better | Sick-Same | Sick-Worse | Dead | |||||||||
| All types of immunodeficiency | 53/37 | 8/59 | 20/46 | 4.4 ± 4.4 | 31–41 (40) | 6/58 | 4/60 | 6.0 ± 5.2 | 39 | 23 | 7 | 19 |
| Primary immunodeficiency | 28/24 | 8/38 | 18/28 | 3.5 ± 4.2 | 31–41 (40) | 4/38 | 2/40 | 6.8 ± 5.8 | 22 | 15 | 4 | 10 |
| Combined immunodeficiencies | 8/1 | 0/6 | 3/4 | 1.4 ± 3.2 | 34–41 (40) | 0/8 | 0/8 | 2.4 ± 1.7 | 6 | 2 | 0 | 1 |
| Well-defined syndromes | 3/1 | 0/3 | 1/2 | 6.1 ± 5.5 | 37–40 (40) | 1/1 | 1/1 | 3.6 ± 4.0 | 1 | 1 | 1 | 1 |
| Antibody deficiencies | 2/2 | 0/4 | 0/4 | 5.2 ± 6.9 | 37–40 (38) | 0/4 | 0/4 | 6.3 ± 4.2 | 3 | 1 | 0 | 0 |
| Immune dysregulation | 2/0 | 0/2 | 0/2 | 2.7 ± 2.8 | 39–40 (40) | 1/1 | 0/2 | 4.4 ± 3.8 | 1 | 0 | 1 | 0 |
| Defects of phagocytes | 4/14 | 3/13 | 9/6 | 4.5 ± 3.9 | 31–41 (40) | 0/12 | 0/12 | 8.9 ± 5.2 | 8 | 5 | 2 | 2 |
| Defects of innate immunity | 2/1 | 2/1 | 3/0 | 0.6 ± 0.2 | 40 (40) | 1/2 | 1/2 | 5.2 ± 8.1 | 0 | 0 | 0 | 3 |
| Autoinflammatory syndromes | 5/5 | 3/7 | 2/8 | 2.5 ± 2.8 | 34–40 (40) | 1/8 | 0/9 | 9.4 ± 7.3 | 3 | 5 | 0 | 2 |
| Bone marrow failure | 2/0 | 0/2 | 0/2 | 7.7 ± 9.5 | 40 (40) | 0/2 | 0/2 | 3.6 ± 4.0 | 0 | 1 | .0 | 1 |
| Secondary immunodeficiency | 25/13 | 0/21 | 2/18 | 5.5 ± 4.4 | 32–40 (40) | 2/20 | 2/20 | 5.0 ± 4.3 | 17 | 8 | 3 | 9 |
| Comparisons between primary and secondary immunodeficiency | 0.255 * | 0.042 * | 0.018 * | 0.612 ** | 0.901 ** | 1.0 * | 0.603 * | 0.445 ** | 0.217 * | 0.412 * | 1.000 * | 0.596 |
| Numbers; % | |
|---|---|
| Chest CT completed (yes/nk; % yes of all patients) | 80/10; 89 |
| ILD consistent with CT diagnosis (yes/no; % yes of patients with this test) | 64/16; 80 |
| Lung biopsy completed (yes/nk; % yes of all patients) | 59/31; 66 |
| Lung biopsy diagnosis proving ILD (yes/no; % yes of patients with this test) | 56/3; 95 |
| Genetic testing completed (yes/nk; % yes of all patients) | 44/46; 49 |
| Gene identified known to be associated with ILD (yes/no; % yes of patients with this test) | 34/10; 76 |
| ILD supported by genetics and lung biopsy (yes/no; % yes of patients with these tests) | 21/28; 75 |
| ILD supported by genetics and CT (yes/no; % yes of patients with these tests) | 26/38; 68 |
| ILD supported by lung biopsy and CT (yes/no; % yes of patients with these tests) | 43/56; 77 |
| ILD supported by genetics, biopsy and CT (yes/no; % yes of patients with these tests) | 18/26; 69 |
| ILD diagnosis only according to clinical records | 3/87; 3 |
| Immunodeficiency (n, Percentage of Histopathological ILD Diagnosis in Immunodeficiency Subcategories) | Gender (Male/Female) | Histopathological Diagnosis and Pattern (n) |
|---|---|---|
| Primary immunodeficiency (32, 27%) | ||
| Combined deficiencies (5) | 4/1 | NSIP (1), GLILD (1), Interstitial pneumonia (1), Intra-alveolar haemorrhage (1), Follicular bronchiolitis (1) |
| Well-defined syndromes (2) | 2/0 | Interstitial pneumonia (1), CPI (1) |
| Antibody deficiencies (2) | 2/0 | GLILD (1), Interstitial pneumonia (1) |
| Immune dysregulation (2) | 2/0 | LIP (2) |
| Defects of phagocytes (11) | 1/10 | Cholesterol pneumonia (1), DIP (2), PAP (7), Interstitial pneumonia (1) |
| Autoinflammatory syndromes (8) | 4/4 | LIP (1), Follicular bronchiolitis (1), NSIP (2), Interstitial pneumonia (1), DIP (1), PAP (1), Lung hypoplasia (1) |
| Bone marrow failure (2) | 2/0 | NSIP (1), Lung fibrosis (1) |
| Secondary immunodeficiency (24, 25%) | ||
| ALL (3) | 2/1 | GLILD (1), NSIP (1), Follicular bronchiolitis (1) |
| AML (1) | 1/0 | PAP (1) |
| Cancer (2) | 2/0 | BPD (1), DIP (1) |
| JMML (2) | 2/0 | Follicular bronchiolitis (1), Pulmonary hemosiderosis (1) |
| MDS (2) | 2/0 | Lung fibrosis (1), NSIP (1) |
| Transplanted (14) | 9/5 | LIP (1), DIP + NSIP (1), DAD (1), Lung fibrosis (4), Cholesterol pneumonia (2), NSIP (5) |
| Immunodeficiency Subcategories (Number of Patients with ILD/Number of Patients with Genetically Defined Immunodeficiency) | Disease Genetically Defined in Our Cohort (No.) | No. of Cases with ILD in Our Cohort (ILD Percentage) | Pulmonary Diseases Other than ILD (n) | Prevalence of ILD (%) in Primary Immunodeficiency Genetic Defect, as Reported in the Literature (May 1999 to May 2022) | Gene Identified, Known to Be Associated with a Condition Presenting with an ILD |
|---|---|---|---|---|---|
| Combined deficiencies (4/8) | ADA (2) | 1 (50%) | ARDS, Respiratory failure, Opportunistic/recurrent infections (1) | 44% [16] ** | Y |
| CD40 (2) | 2 (100%) | 0% [17] **, [18] * | N | ||
| CD40LG (1) | 0 (0%) | Opportunistic/recurrent infections (1) | 20% [19] ***, 0% [20] *** | N | |
| IL2RG (1) | 1 (100%) | 7% ILD [21] *** | Y | ||
| DOCK8 (1) | 0 (0%) | Bronchiolitis obliterans, Bronchiectasis (1) | 0% [22] ***, [23] *** | N | |
| RFXAP (1) | 0 (0%) | Opportunistic/recurrent infections (1) | 50% [24] * | Y | |
| Well-defined syndromes (4/14) | ATM (3) | 1 (33%) | Bronchiectasis, Pulmonary hypertension (1), Bronchiectasis, Opportunistic/recurrent infections (1) | 50% [25] *, 26% [26] ***, 14% [27] *** | Y |
| 10p13-p14DS (1) | 1 (100%) | 0% [28] ***, [29] * | N | ||
| MCM4 (1) | 1 (100%) | 50% [30] **, 0% [31] ** | Y | ||
| DNMT3B (1) | 0 (0%) | Opportunistic/recurrent infections (1) | 0% [32] *** | N | |
| IKBA (1) | 0 (0%) | Opportunistic/recurrent infections (1) | 0% [33] *, [34] ** | N | |
| NBS1 (1) | 0 (0%) | Bronchiectasis, Opportunistic/recurrent infections (1) | 0% [35] *. [36] *, [37] *** | N | |
| TTC7A (1) | 0 (0%) | Opportunistic/recurrent infections (1) | 0% [38] *, [39] *, [40] *** | N | |
| DiGeorge (4) | 0 (0%) | Opportunistic/recurrent infections (2), Asthma, Pulmonary hypertension (1), Opportunistic/recurrent infections, ADRS (1) | 0% [41] ***, 100% [42] * | Y | |
| HELLS (1) | 1 (100%) | 0% [43] **, [44] * | N | ||
| Antibody deficiencies (1/4) | TNFRSF13B (1) | 1 (100%) | 0% [45] **, [46] * | N | |
| BTK (1) | 0 (0%) | Opportunistic/recurrent infections, Respiratory failure (1) | 0% [47] *, [48] *** | N | |
| NFKB1 (1) | 0 (0%) | Opportunistic/recurrent infections, ARDS (1) | 12% [49] ***, 0% [50] ** | Y | |
| PIK3CD (1) | 0 (0%) | Bronchiectasis, Opportunistic/recurrent infections (1) | 0% [51] **, [52] ** | N | |
| Immune dysregulation (2/5) | FOXP3 (2) | 1 (50%) | Bronchiolitis obliterans, Respiratory failure (1) | 23% [53] ***, | Y |
| STAT3 GOF (1) | 1 (100%) | 36% [54] ***, 100% [55] * | Y | ||
| IL10 (1) | 0 (0%) | Opportunistic/recurrent infections (1) | 0% [56] **, [57] *** | N | |
| UNC13D (1) | 0 (0%) | Opportunistic/recurrent infections (1) | 0% [58] **, [59] * | N | |
| Defects of phagocytes (18/21) | CSF2RA (15) | 15 (100%) | 100% [60] **, [61] *** | Y | |
| CYBA (3) | 2 (67%) | Bronchiolitis obliterans, Opportunistic/recurrent infections, Asthma (1) | 0% [62] **, [63] *, [64] ***, [65] *** | N | |
| CYBB (1) | 0 (0%) | Opportunistic/recurrent infections (1) | 0% [62] ***, [63] ***, [64] ***, [65] *** | N | |
| NCF4 (1) | 0 (0%) | Bronchiectasis (1) | 0% [65]* | N | |
| NCF2 (1) | 1 (100%) | 0% [62] **, [63] *, [64] ***, [65] *** | N | ||
| Defects of innate immunity (3/7) | MDA5 def (LOF). IFIH1 (1) | 0 (0%) | Respiratory failure (1) | 0% [66] *, 100% [67] * | Y |
| STAT1 (AD LOF) (1) | 0 (0%) | Bronchiectasis, Opportunistic/recurrent infections (1) | 5% [68] *** | Y | |
| TCIRG1 (1) | 0 (0%) | Opportunistic/recurrent infections (1) | 0% [69]*** | N | |
| ZNFX-1 (4) | 3 (75%) | Opportunistic/recurrent infections (1) | 13% [70] **, 50% [71] * | Y | |
| Autoinflammatory syndromes (10/16) | COPA (7) | 7 (100%) | 100% [72] ***, [73] *** | Y | |
| OAS1 (1) | 1 (100%) | 100% [74] **, [75] * | Y | ||
| PLCG2 (1) | 1 (100%) | 0% [76] **, [77] *, [78] *** | N | ||
| STING (1) | 1 (100%) | 100% [79] ***, 85% [80] ** | Y | ||
| AGS7.IFIH1 (1) | 0 (0%) | Opportunistic/recurrent infections, Respiratory failure, ARDS (1) | 0% [81] *, [82] *, [83] ** | N | |
| MEFV (2) | 0 (0%) | Opportunistic/recurrent infections (2) | 0% [84] ***, [85] ***, [86] *** | N | |
| TMEM173 (1) | 0 (0%) | Opportunistic/recurrent infections (1) | 100% [79] ***, 85% [80] ** | Y | |
| TNFRSF1A (2) | 0 (0%) | Asthma (1), Opportunistic/recurrent infections (1) | 0% [87] *** | N | |
| Bone marrow failure (2/3) | SAMD9 (1) | 0 (0%) | Opportunistic/recurrent infections, Respiratory failure (1) | 0% [88] **, [89] ** | N |
| TERC (1) | 1 (100%) | 50% [90] * | Y | ||
| TERT (1) | 1 (100%) | 16% [90] ***, 56% [91] ** | Y |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, X.; Michel, K.; Griese, M. Interstitial Lung Disease in Immunocompromised Children. Diagnostics 2023, 13, 64. https://doi.org/10.3390/diagnostics13010064
Gao X, Michel K, Griese M. Interstitial Lung Disease in Immunocompromised Children. Diagnostics. 2023; 13(1):64. https://doi.org/10.3390/diagnostics13010064
Chicago/Turabian StyleGao, Xianfei, Katarzyna Michel, and Matthias Griese. 2023. "Interstitial Lung Disease in Immunocompromised Children" Diagnostics 13, no. 1: 64. https://doi.org/10.3390/diagnostics13010064
APA StyleGao, X., Michel, K., & Griese, M. (2023). Interstitial Lung Disease in Immunocompromised Children. Diagnostics, 13(1), 64. https://doi.org/10.3390/diagnostics13010064