
Kawasaki Disease and Inborn Errors of Immunity: Exploring the Link and Implications


 , , and
, , and
Abstract
:1. Introduction
2. Methods
3. Kawasaki Disease in Inborn Errors of Immunity
3.1. KD and Predominantly Antibody Deficiencies
3.2. KD and Immunodeficiency Disorders Affecting Cellular and Humoral Immunity
3.3. KD and Phagocytic Defects
3.4. KD and Defects in Intrinsic and Innate Immunity
3.5. KD and Autoinflammatory Disorders
4. Discussion
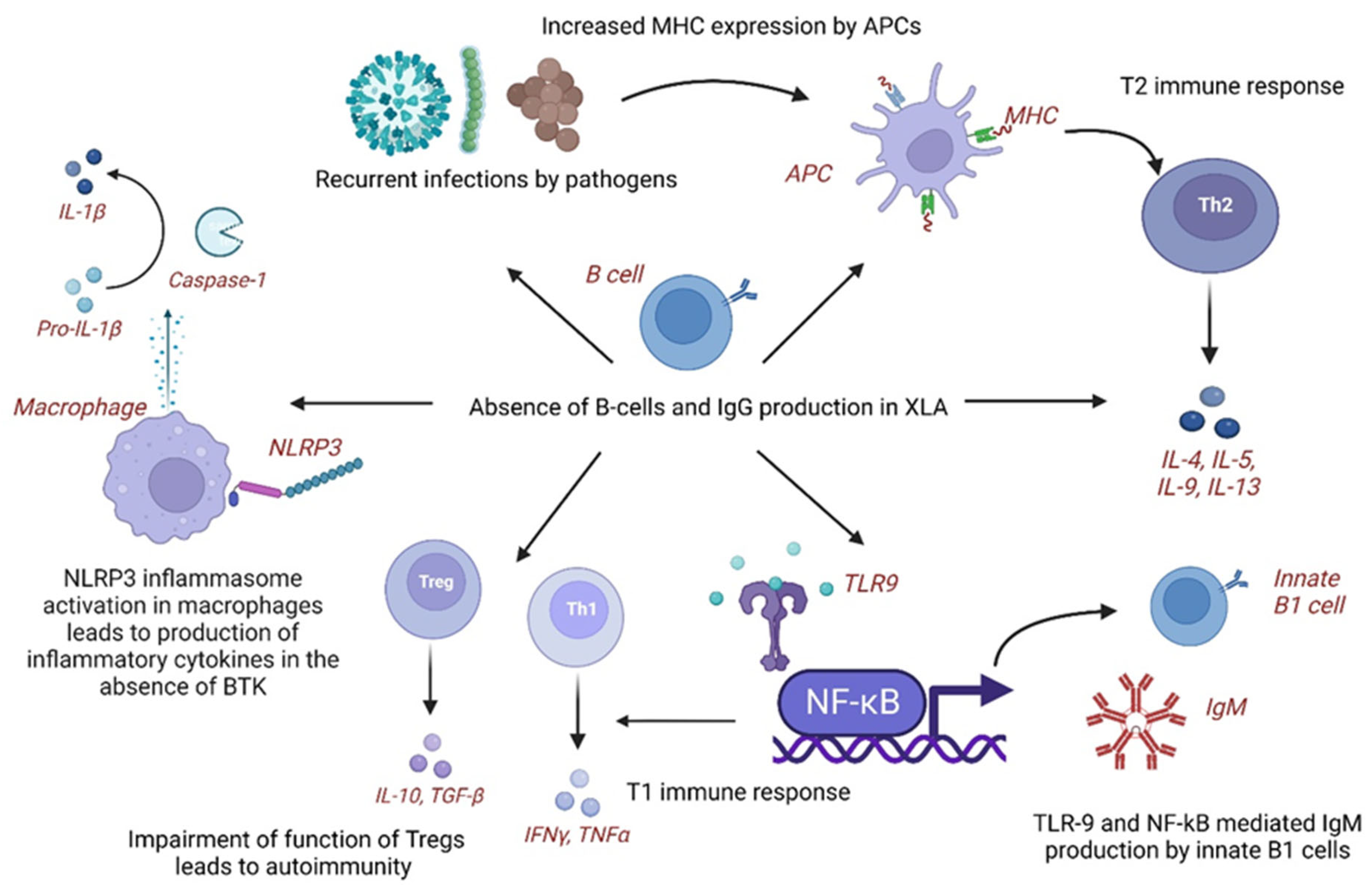
4.1. Pathogenetic Mechanisms of KD in XLA
4.2. Pathogenetic Mechanisms of KD in Selective IgA Deficiency
4.3. Pathogenetic Mechanisms of KD in Transient Hypogammaglobulinemia of Infancy
4.4. Pathogenetic Mechanisms of KD in WAS
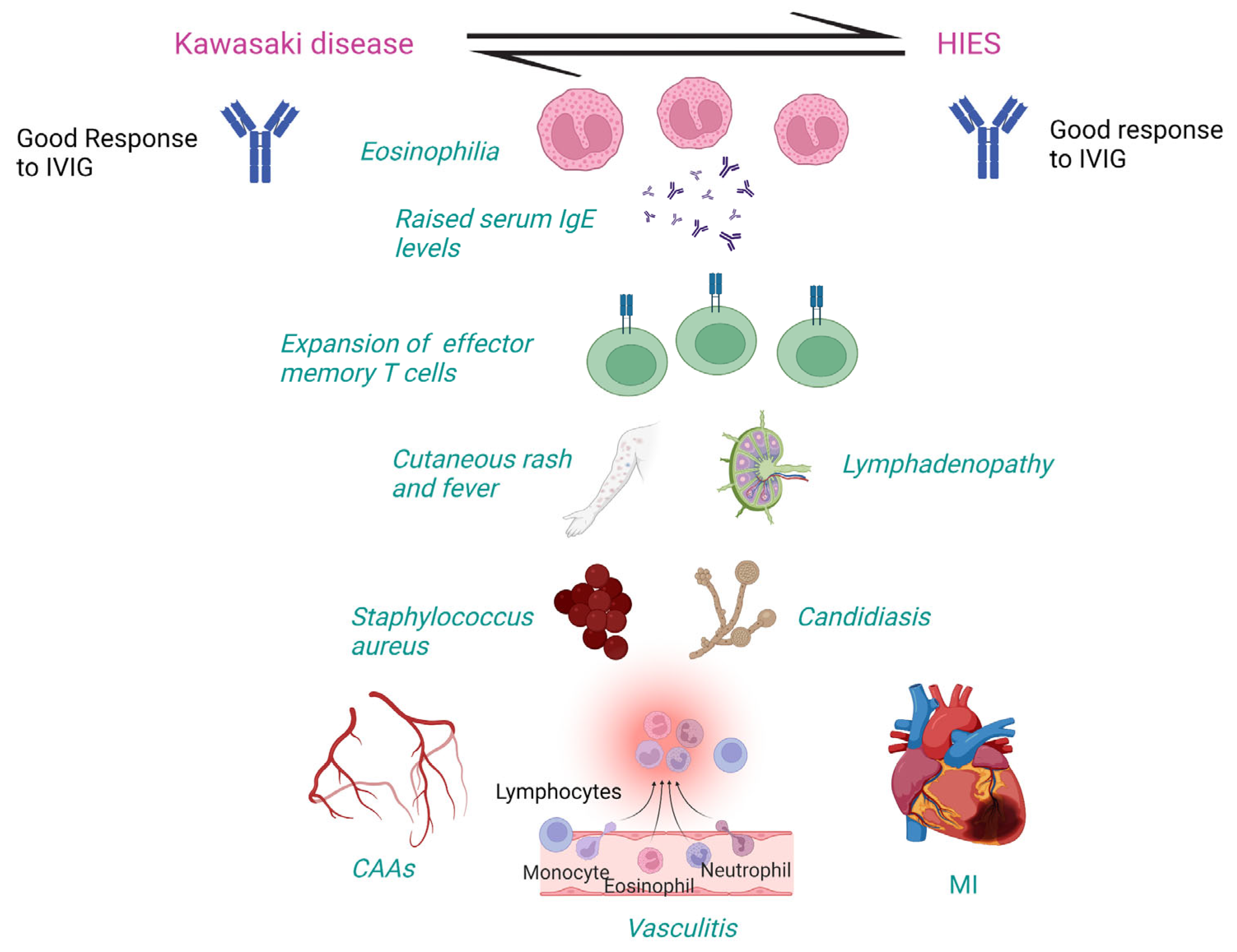
4.5. Pathogenetic Mechanisms of KD in HIES
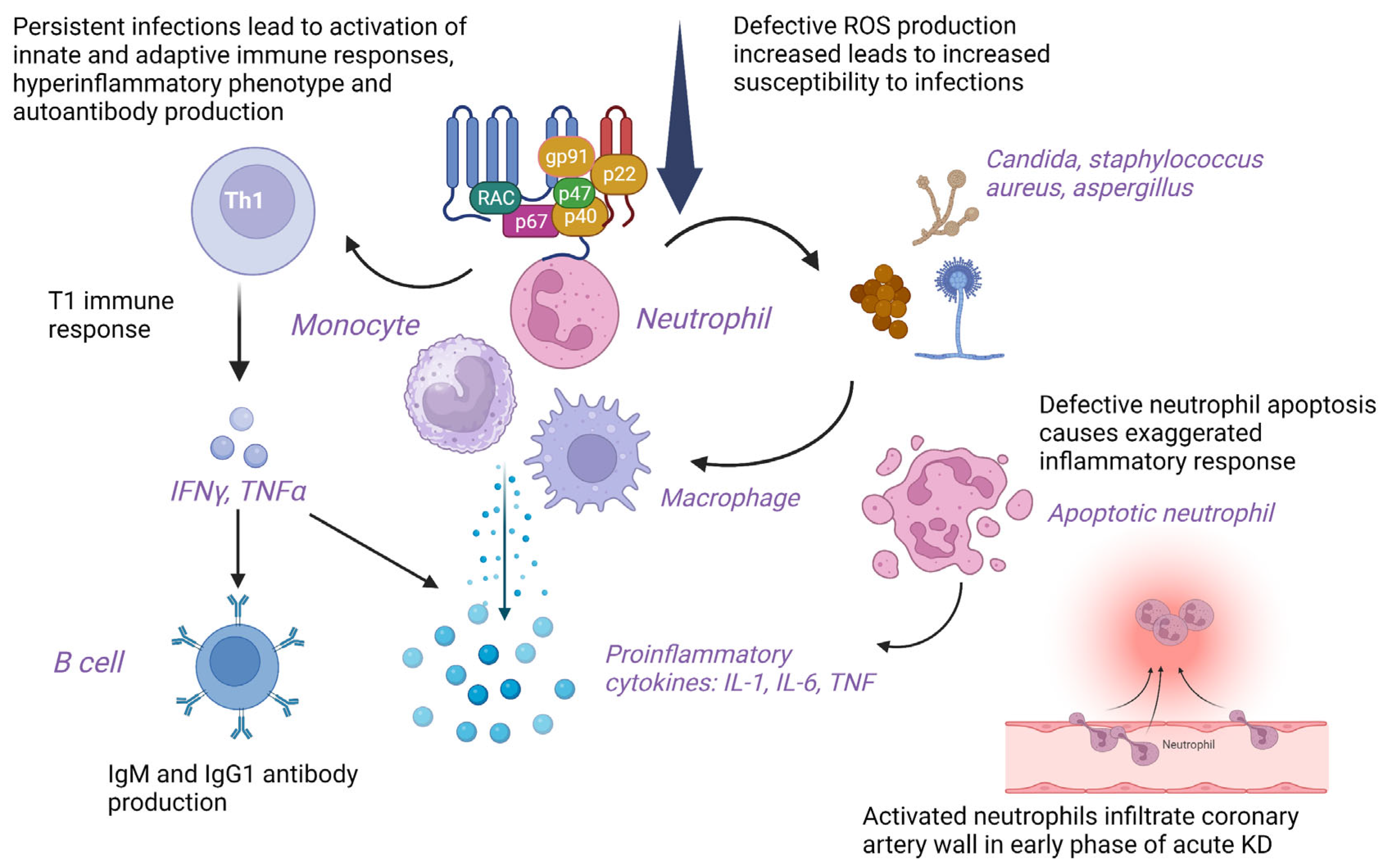
4.6. Pathogenetic Mechanisms of KD in CGD
4.7. Pathogenetic Mechanisms of KD in WHIM Syndrome
4.8. Pathogenetic Mechanisms of KD in PFAPA
4.9. Multisystem Inflammatory Syndrome in Children (MIS-C) and IEIs
5. Future Directions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, S.; Vignesh, P.; Burgner, D. The epidemiology of Kawasaki disease: A global update. Arch. Dis. Child. 2015, 100, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- McCrindle, B.W.; Rowley, A.H.; Newburger, J.W.; Burns, J.C.; Bolger, A.F.; Gewitz, M.; Baker, A.L.; Jackson, M.A.; Takahashi, M.; Shah, P.B.; et al. American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; Council on Cardiovascular Surgery and Anesthesia; and Council on Epidemiology and Prevention. Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals From the American Heart Association. Circulation 2017, 135, e927–e999. [Google Scholar] [PubMed]
- Agarwal, S.; Agrawal, D.K. Kawasaki disease: Etiopathogenesis and novel treatment strategies. Expert Rev. Clin. Immunol. 2017, 13, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Cunningham-Rundles, C.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; Oksenhendler, E.; Picard, C.; et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2022, 42, 1473–1507. [Google Scholar] [CrossRef] [PubMed]
- Marrani, E.; Burns, J.C.; Cimaz, R. How Should We Classify Kawasaki Disease? Front. Immunol. 2018, 9, 2974. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Oharaseki, T.; Yokouchi, Y. Pathogenesis of Kawasaki disease. Clin. Exp. Immunol. 2011, 164 (Suppl. S1), 20–22. [Google Scholar] [CrossRef]
- Rivas-Larrauri, F.; Aguilar-Zanela, L.; Castro-Oteo, P.; Rosales-Hernandez, L.A.; Otero-Mendoza, F.; López-Herrera, G.; Ordoñez-Ortega, J.; Garrido-García, M.; Yamazaki-Nakashimada, M.A. Kawasaki disease and immunodeficiencies in children: Case reports and literature review. Rheumatol. Int. 2019, 39, 1829–1838. [Google Scholar] [CrossRef]
- Behniafard, N.; Aghamohammadi, A.; Abolhassani, H.; Pourjabbar, S.; Sabouni, F.; Rezaei, N. Autoimmunity in X-linked agammaglobulinemia: Kawasaki disease and review of the literature. Expert Rev. Clin. Immunol. 2012, 8, 155–159. [Google Scholar] [CrossRef]
- Malekzadeh, I.; Moradinejad, M.H.; Ziaee, V.; Malek, A.; Khalili, A. Autoimmunity in X-linked agammaglobulinemia; a patient with several episodes of autoimmunity. Iran J. Ped. 2013, 23, 75. [Google Scholar]
- Sharma, D.; Guleria, S.; Suri, D.; Rawat, A.; Garg, R.; Singh, S. A child with X-linked agammaglobulinemia and Kawasaki disease: An unusual association. Rheumatol. Int. 2017, 37, 1401–1403. [Google Scholar] [CrossRef]
- Nishikawa, T.; Nomura, Y.; Kono, Y.; Kawano, Y. Selective IgA deficiency complicated by Kawasaki syndrome. Pediatr. Int. 2008, 50, 816–818. [Google Scholar] [CrossRef] [PubMed]
- Anzai, T.; Minami, T.; Sato, T.; Furui, S.; Yamagata, T. Treatment of a patient with Kawasaki disease associated with selective IgA deficiency by continuous infusion of cyclosporine A without intravenous immunoglobulin. Turk. J. Pediatr. 2016, 58, 666–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Şanlıdağ, B.; Balkan, C.; Bahçeciler, N. A case report: Incomplete Kawasaki disease in a hypogammaglobulinémie child. Arch. Argent Pediatr. 2018, 116, e322–e324. [Google Scholar] [PubMed]
- Kawakami, C.; Miyake, M.; Tamai, H. Kawasaki disease in a patient with Wiskott-Aldrich syndrome: An increase in the platelet count. Int. J. Hematol. 2003, 77, 199–200. [Google Scholar] [CrossRef]
- Kimata, H. High-dose intravenous gamma-globulin treatment for hyperimmunoglobulinemia E syndrome. J. Allergy Clin. Immunol. 1995, 95, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.C.; Freeman, A.F.; Gharib, A.M.; Arai, A.E.; Lederman, R.J.; Rosing, D.R.; Holland, S.M. Coronary artery aneurysms in patients with hyper IgE recurrent infection syndrome. Clin. Immunol. 2007, 122, 255–258. [Google Scholar] [CrossRef]
- Yared, T.; Mohsen, S. Job’s Syndrome with a Family History of Kawasaki Disease: A Case Presentation and Review of Literature. Dis. Diagn. 2021, 10, 19–22. [Google Scholar] [CrossRef]
- Young, T.Y.; Jerome, D.; Gupta, S. Hyperimmunoglobulinemia E syndrome associated with coronary artery aneurysms: Deficiency of central memory CD4+ T cells and expansion of effector memory CD4+ T cells. Ann. Allergy Asthma Immunol. 2007, 98, 389–392. [Google Scholar] [CrossRef]
- Yamazaki-Nakashimada, M.A.; Ramírez-Vargas, N.; De Rubens-Figueroa, J. Chronic granulomatous disease associated with atypical Kawasaki disease. Pediatr. Cardiol. 2008, 29, 169–171. [Google Scholar] [CrossRef]
- Muneuchi, J.; Ishimura, M.; Takada, H.; Hoshina, T.; Utsunomiya, R.; Ikeda, K.; Yamaguchi, K.; Ohga, S.; Kusuhara, K.; Hara, T. Incomplete Kawasaki disease in a patient with chronic granulomatous disease. Pediatr. Int. 2010, 52, e134–e136. [Google Scholar] [CrossRef]
- Tsuge, M.; Shigemitsu, Y.; Yano, Y.; Fujiwara, M.; Miyai, T.; Ueda, K.; Takata, K.; Moriwake, T. Immunoglobulin resistance in Kawasaki disease with chronic granulomatous disease. Pediatr. Int. 2012, 54, e32–e34. [Google Scholar] [CrossRef]
- Hule, G.P.; Kanvinde, P.R.; Kulkarni, M.A.; van Leeuwen, K.; de Boer, M.; Bargir, U.A.; Taur, P.D.; Desai, M.M.; Madkaikar, M.R. p47phox−/− Chronic Granulomatous Disease Patient with Incomplete Kawasaki Disease. J. Clin. Immunol. 2018, 38, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, Y.; Wu, P.; Kang, M.; Hong, Y.; Xue, Y.; Chen, C.; Li, H.; Fang, Y. Case Report: A Novel CXCR4 Mutation in a Chinese Child With Kawasaki Disease Causing WHIM Syndrome. Front. Immunol. 2022, 13, 857527. [Google Scholar] [CrossRef] [PubMed]
- Hambleton, S.; Goodbourn, S.; Young, D.F.; Dickinson, P.; Mohamad, S.M.; Valappil, M.; McGovern, N.; Cant, A.J.; Hackett, S.J.; Ghazal, P.; et al. STAT2 deficiency and susceptibility to viral illness in humans. Proc. Natl. Acad. Sci. USA 2013, 19, 3053–3058. [Google Scholar] [CrossRef] [Green Version]
- Broderick, L.; Tremoulet, A.H.; Burns, J.C.; Bastian, J.F.; Hoffman, H.M. Recurrent fever syndromes in patients after recovery from Kawasaki syndrome. Pediatrics 2011, 127, e489–e493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninomiya, T.; Takada, H.; Nagatomo, Y.; Nanishi, E.; Nagata, H.; Yamamura, K.; Doi, T.; Ikeda, K.; Hara, T. Development of Kawasaki disease in a patient with PFAPA. Pediatr. Int. 2013, 55, 801–802. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, M.; Rikhi, R.; Loganathan, S.K.; Suri, D.; Singh, S. Autoimmunity in Wiskott-Aldrich Syndrome: Updated Perspectives. Appl. Clin. Genet. 2021, 14, 363–388. [Google Scholar] [CrossRef]
- Saikia, B.; Rawat, A.; Minz, R.W.; Suri, D.; Pandiarajan, V.; Jindal, A.; Sahu, S.; Karim, A.; Desai, M.; Taur, P.D.; et al. Clinical Profile of Hyper-IgE Syndrome in India. Front. Immunol. 2021, 12, 626593. [Google Scholar] [CrossRef]
- Goel, R.R.; Nakabo, S.; Dizon, B.L.P.; Urban, A.; Waldman, M.; Howard, L.; Darnell, D.; Buhaya, M.; Carmona-Rivera, C.; Hasni, S.; et al. Lupus-like autoimmunity and increased interferon response in patients with STAT3-deficient hyper-IgE syndrome. J. Allergy Clin. Immunol. 2021, 147, 746–749.e9. [Google Scholar] [CrossRef]
- Yong, P.F.; Freeman, A.F.; Engelhardt, K.R.; Holland, S.; Puck, J.M.; Grimbacher, B. An update on the hyper-IgE syndromes. Arthritis Res. Ther. 2012, 14, 228. [Google Scholar] [CrossRef] [Green Version]
- Henrickson, S.E.; Jongco, A.M.; Thomsen, K.F.; Garabedian, E.K.; Thomsen, I.P. Noninfectious Manifestations and Complications of Chronic Granulomatous Disease. J. Pediatr. Infect. Dis. Soc. 2018, 7, S18–S24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, Y.; Ohashi, Y.; Harada, K.; Asai, T.; Okawa, S.; Nagashima, M.; Katoh, T.; Baba, K.; Frusho, K.; Okuni, M.; et al. Coronary risks after high-dose gamma-globulin in children with Kawasaki disease. Pediatr. Int. 2000, 42, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki-Nakashimada, M.A.; Gámez-González, L.B.; Murata, C.; Honda, T.; Yasukawa, K.; Hamada, H. IgG levels in Kawasaki disease and its association with clinical outcomes. Clin. Rheumatol. 2019, 38, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.C.; Pan, C.T.; Huang, Y.H.; Huang, F.C.; Lin, Y.S.; Li, S.C.; Huang, L.H. Global Investigation of Immune Repertoire Suggests Kawasaki Disease Has Infectious Cause. Circ. J. 2019, 83, 2070–2078. [Google Scholar] [CrossRef] [Green Version]
- Ko, T.M.; Kiyotani, K.; Chang, J.S.; Park, J.H.; Yin Yew, P.; Chen, Y.T.; Wu, J.Y.; Nakamura, Y. Immunoglobulin profiling identifies unique signatures in patients with Kawasaki disease during intravenous immunoglobulin treatment. Hum. Mol. Genet. 2018, 27, 2671–2677. [Google Scholar] [CrossRef]
- Lee, J.K. Hygiene Hypothesis as the Etiology of Kawasaki Disease: Dysregulation of Early B Cell Development. Int. J. Mol. Sci. 2021, 22, 12334. [Google Scholar] [CrossRef]
- Ueno, Y.; Takano, N.; Kanegane, H.; Yokoi, T.; Yachie, A.; Miyawaki, T.; Taniguchi, N. The acute phase nature of interleukin-6: Studies in Kawasaki disease and other febrile illnesses. Clin. Exp. Immunol. 1989, 76, 337–342. [Google Scholar]
- Franco, A.; Shimizu, C.; Tremoulet, A.H.; Burns, J.C. Memory T-cells and characterization of peripheral T-cell clones in acute Kawasaki disease. Autoimmunity 2010, 43, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.H.; Devadas, S.; Kwon, J.; Pinto, L.A.; Williams, M.S. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat. Immunol. 2004, 5, 818–827. [Google Scholar] [CrossRef]
- Sanford, A.N.; Suriano, A.R.; Herche, D.; Dietzmann, K.; Sullivan, K.E. Abnormal apoptosis in chronic granulomatous disease and autoantibody production characteristic of lupus. Rheumatology 2006, 45, 178–181. [Google Scholar] [CrossRef] [Green Version]
- Bucciol, G.; Meyts, I.; COVID Human Genetic Effort. Inherited and acquired errors of type I interferon immunity govern susceptibility to COVID-19 and multisystem inflammatory syndrome in children. J. Allergy Clin. Immunol. 2023, 151, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Gruber, C.N.; Patel, R.S.; Trachtman, R.; Lepow, L.; Amanat, F.; Krammer, F.; Wilson, K.M.; Onel, K.; Geanon, D.; Tuballes, K.; et al. Mapping systemic inflammation and antibody responses in multisystem inflammatory syndrome in children (MIS-C). Cell 2020, 183, 982–995.e14. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, L.R.; Rose, E.B.; Horwitz, S.M.; Collins, J.P.; Newhams, M.M.; Son, M.B.F.; Newburger, J.W.; Kleinman, L.C.; Heidemann, S.M.; Martin, A.A.; et al. Multisystem inflammatory syndrome in U.S. children and adolescents. N. Engl. J. Med. 2020, 383, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Le Pen, J.; Yatim, A.; Dong, B.; Aquino, Y.; Ogishi, M.; Pescarmona, R.; Talouarn, E.; Rinchai, D.; Zhang, P.; et al. Inborn errors of OAS-RNase L in SARS-CoV-2-related multisystem inflammatory syndrome in children. Science 2023, 379, eabo3627. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Author (Year) | IEI | KD | Clinical Phenotype | Laboratory Findings | Treatment |
|---|---|---|---|---|---|
| Predominantly antibody deficiencies | |||||
| Behniafard et al. (2012) [8] | XLA at 10 months | Incomplete KD at 15 months of age | XLA: Absent tonsils, XLA in sibling and maternal uncle, KD: Fever for 5 days, right submandibular lymphadenopathy, peripheral edema, peri-ungual desquamation | XLA: Pan-hypogammaglobulinemia, markedly reduced B cells, BTK mutation confirmed in brother and maternal uncle. No infective organism identified KD: Anemia, thrombocytosis, elevated ESR and CRP, leukocytosis, hypoalbuminemia, gall bladder hydrops, ascites | IVIG (600 mg/kg followed by 2 g/kg) and antibiotics Asymptomatic on follow-up except for periungual desquamation |
| Malekzadeh et al. (2013) [9] | XLA at 6 years | Incomplete KD at 3 years | XLA: recurrent infections, JIA, and MAS, septicemia KD: Prolonged fever > 5 days, rash, cervical adenopathy, conjunctivitis | NA | Methylprednisolone and IVIG for MAS Methotrexate and prednisolone for polyarthritis |
| Sharma et al. (2017) [10] | XLA at 3 years | Incomplete KD at 6 years | XLA: Recurrent infections, CAP unresponsive to antibiotics KD: Prolonged fever, periungual desquamation, irritability | XLA: Hypogammaglobulinemia, absent B cells and BTK expression on flow cytometry, BTK gene exon 9 deletion, CXR showed consolidation, and PE KD: Anemia, thrombocytosis, elevated CRP, and ESR | IVIG (2 g/kg) and aspirin (50 mg/kg/day). Good response |
| Rivas-Larrauri et al. (2019) [7] | XLA at 8 months of age | Incomplete KD at 8 months | XLA: Infections, family H/O XLA in maternal uncles, one died in infancy due to sepsis and other in adulthood due to cancer KD: Fever, skin rash, cough, and hoarseness of voice | XLA: IgG and IgA were low (IgM: WNL), B cells 1%, reduced BTK expression on WB, Pseudomonas aeruginosa on blood culture, and rhinovirus positivity on PCR KD: Anemia with thrombocytosis, pericardial effusion, LCA aneurysm 3.4 mm (z-score > 4). Hypoalbuminemia, increased BNP | Good response to IVIG, corticosteroids, and antibiotics |
| Nishikawa et al. (2008) [11] | Selective IgA deficiency at 2 years | Complete KD at 2 years | Selective IgA deficiency: No specific infections KD: Persistent high-grade fever, conjunctivitis, diffuse skin rash, strawberry tongue, red cracked lips, peripheral edema, anorexia, and tachycardia | Selective IgA deficiency: Normal IgG levels but significantly low IgA levels (2 mg/dL), high anti-IgA antibodies, low IgA levels in sister KD: Leukocytosis, elevated ESR and CRP, hyperbilirubinemia, hyponatremia | Aspirin (30–50/kg per day) and urinastatin (15,000 to 25,000 U/kg per day, i.v. methylprednisolone 30 mg/kg/day for 3 days, followed by oral prednisolone (1 mg/kg/day). Symptoms resolved by 26th day of illness |
| Anzai et al. (2016) [12] | Selective IgA deficiency at 3 years | Complete KD at 5 years of age | IgA deficiency: RTI for 3–5 year and cellulitis post dental procedure KD: Persistent fever for 6 days, cervical LAD, conjunctival congestion, cracked tongue and lips and peripheral edema of hands | IgA deficiency: Low serum IgA level (<5.0 mg/dL) KD: Leukocytosis, elevated ESR and CRP, no CAAs on echo | Oral aspirin and i.v. urinastatin (fever persisted) i.v. CsA (3.0 mg/kg/day), then oral CsA (In remission) |
| Şanlıdağ et al. (2018) [13] | Transient hypogammaglobulinemia of infancy (3.5 years) | Incomplete KD at 4 years | THI: Recurrent tonsillitis and bronchopneumonia KD: Fever, strawberry tongue, conjunctivitis, and periungual desquamation | IgA deficiency: Low serum IgA level (<5.0 mg/dL) KD: Leukocytosis, elevated ESR and CRP, no CAAs on echo | Oral aspirin and i.v. urinastatin (fever persisted) i.v. CsA A (3.0 mg/kg/day), then oral CsA (In remission) |
| Immunodeficiency disorders affecting cellular and humoral immunity | |||||
| Kawakami et al. (2003) [14] | WAS (Early infancy) | Complete KD at 6 months | WAS: eczema, purpura, and recurrent infections KD: prolonged high-grade fever, skin rash, peripheral edema, cervical lymphadenopathy conjunctivitis, and strawberry tongue | WAS: microthrombocytopenia, WASP gene intron 6 597G→A missense mutation, confirmed by Western blotting Platelet count increased transiently in the acute phase of KD and decreased after IVIG | IVIG and oral prednisolone followed by high dose of IVIG (1 g/kg for 2 days and 0.5 g/kg for 2 days) (IVIG therapy may not increase platelet count in WAS) |
| Kimata et al. (1995) [15] | Pt 1: HIES at 1 year (male) Pt 2: HIES at 7 months (female) | Pt 1: KD at 3 years Pt 2: KD at 2.5 years | Pt. 1: HIES: Eczema, chronic otitis media, staphylococcal scalp abscess, pneumonia, pyothorax caused by Staphylococcus aureus KD: Fever | Serum IgE, anti-staphylococcal IgE levels, and in-vitro IgE production were increased pre-IVIG but significantly reduced post-IVIG treatment for 28 days | IVIG (400 mg/day for 28 days, following which fever, eczema, skin infections, and CAAs subsided dramatically in 6 months |
| Ling et al. (2007) [16] | HIES Pt 1: early childhood (male): sporadic Pt 2: 38 years of age (male): AD | Pt 1: CAAs at 43 years of age Pt 2: CAAs at 48 years of age | HIES: Pt 1: eczema, skin boils, recurrent aspergillus pneumonia, pneumatocele formation, coarse facies, scoliosis, and pathologic fractures CAAs: ventricular fibrillation (MI) Pt 2: recurrent pneumonia, recurrent sinusitis, boils, multiple fungal infections (histoplasmosis, cryptococcal meningitis, candidiasis), and coarse facies CAAs: Chest pain and tightness with dyspnea | Pt 1: HIES score: 79 (<40) CAA: fusiform aneurysm of 9 mm in left anterior descending and its first diagonal branch (mid-left anterior descending thrombus), diffuse ectasia in RCA on coronary angiography. Echocardiogram, CEMRI, and CT coronary angiogram: transmural infarction in the left anterior descending territory ECG and cardiac enzymes revealed acute anteroseptal MI Pt 2: HIES score: 79 (<40) ECG: Non-specific T-wave abnormalities Echocardiogram revealed left ventricular hypertrophy Coronary arteriography revealed showed RCA and proximal left anterior descending dilatation CT Coronary angiogram: 6.5 mm ectasia-aneurysm in left anterior descending and long RCA ectasia (6.5 mm) | Pt 1: Aspirin and Clopidogrel Pt 2: Aspirin |
| Yared et al. (2021) [17] | HIES (15 days after birth) | KD in 17-year-old elder brother at 3 years of age | HIES: Eczema, recurrent pneumonia with pneumatoceles, oral candidiasis, recurrent multiple site abscesses, sinusitis, toothache, cold mass in neck, pruritis, cough, left knee pain, coarse facies, joint hyperextensibility, and retained primary teeth | Serum IgE levels were elevated (2500 IU/mL), left submandibular abscess, with multiple lymphadenopathies Pus culture revealed methicillin-resistant Staphylococcus aureus | Antibiotics, antifungal, topical steroids |
| Young et al. (2007) [18] | HIES since childhood | CAAs at 29 years | HIES: Recurrent pneumonia, staphylococcal skin abscesses, pruritis, retained primary teeth, high arched palate, recurrent bacterial infections, hypertension, immune complex–mediated glomerulonephritis CAAs: substernal chest pain, elevated cardiac enzymes | Coronary angiogram revealed RCA aneurysmal dilatation with thrombus, left anterior descending, and circumflex arteries On flow cytometry, CD4+ TCM cells were reduced, whereas, CD4+ effector memory T cells and CD4+ TEMRAs were elevated | Fosinopril sodium, anti-coagulation, recanalization followed by cardiac stenting. Good response |
| Phagocytic defects | |||||
| Yamazaki-Nakashimada et al. (2008) [19] | CGD at 2 months of age (male) | Atypical KD at 1 year of age | CGD: Gastroenteritis leading to hypovolemic shock, pyelonephritis, pneumonia, and empyema. H/O parental consanguinity CGD in elder brother KD: Fever, cough, diffuse maculopapular rash, perineal erythema, BCG site ulcer, conjunctivitis, diffuse crackles, and hypertension post-therapy | CGD: NBT assay showed absence of NBT-positive neutrophils, CXR: Bilateral opacities in lungs KD: Anemia, leukocytosis with neutrophilia, thrombocytosis, elevated CRP, and ALT Echocardiogram: CAAs with dilatation of LCA (4 mm), and RCA (3.7 mm) | IVIG (2 g/kg), aspirin (60 mg/kg/day), Prednisone, Prazocin, and antibiotic On follow-up, asymptomatic with thrombocytosis, low Hb. RCA and LCA reduced in follow-up. Poor response to initial high-dose IVIG |
| Muneuchi et al. (2010) [20] | CGD, X-linked at 4 months of age (male) | Incomplete KD at 2 years | CGD: Disseminated and suppurative BCGosis KD: Fever, painful right cervical lymphadenopathy, conjunctival injection, red lips, BCG site reactivation | CGD: gp91-phox gene mutation <1% Rhodamine-positive neutrophils on DHR assay (70–100%) KD: Anemia, raised CRP, LCA dilatation with a maximum diameter of 4 mm on echo, followed by saccular dilatation with 6 mm diameter. Raised IL-6 | Oral antibiotics and IFN-γ (subcutaneous), i.v. antibiotics, IVIG (2 g/kg and 1 g/kg/day for 2 days), aspirin, and dipyridamole. Poor response to initial high-dose IVIG. On follow-up improved |
| Tsuge et al. (2012) [21] | CGD at 8 months of age (male) | Complete KD at 10 months of age | CGD: perirectal abscess KD: Recurrent fever, maculopapular rash, peripheral edema, reddish lips, pharyngeal erythema, cervical adenopathy, HSM, and BCG site reactivation | CGD: Reduced superoxide production in neutrophils < 2% (70–100%), Frameshift mutation in gp91phox gene (exon 13, c. 1681 del G) KD: Anemia, neutrophilic leukocytosis, thrombocytosis, Raised CRP and AST, echo: WNL Raised IL-8 and sIL-2R levels even after IVIG for a few weeks | Perirectal abscess incision and drainage. High-dose IVIG (2 g/kg for 24 h, (1 g/kg/day for 12 h), flurbiprofen (5 mg/kg daily), antibiotics and antifungal therapy, aspirin (5 mg/kg/day). Poor response to initial high-dose IVIG |
| Hule et al. (2018) [22] | CGD, AR (p47phox−/−) (male) | Incomplete KD at 2 years of age | KD: Fever for 5 days, irritability, cough, erythema of palms and sole, and strawberry tongue CGD: Diagnosed on work-up after elder female sibling (7 years) was diagnosed with CGD | KD: Anemia, raised ESR and CRP, 2D echo: LCA aneurysm (3.5 mm, Z-score + 4.60) CGD: 0% NBT+ cells and 0% rhodamine+ cells (SI: 3.86) and deficient p47phox expression (S/N: 2.0). Homozygous missense mutation in NCF1 gene, c.124C > T, exon 2, p. R42W | IVIG (2 g/kg/day) and aspirin. Good response |
| Defects in Intrinsic and Innate Immunity | |||||
| Ma et al. (2022) [23] | WHIM syndrome (neonatal onset) | Incomplete KD at 1 year of age | WHIM: neonatal jaundice, recurrent infections (H/O leukopenia, neutropenia, lymphopenia, Ig, and circulating B-cells in father) KD: Fever for 6 days, cough, erythematous rash on trunk, red lips, oropharyngeal erythema, tachycardia, conjunctival injection, cervical adenopathy | WHIM: Anemia, leukopenia with neutropenia during neonatal period/early infancy, low IgG and C4 level, CXR: bilateral pneumonia and infiltrates, BME: Hypercellular, mild granulocytic hyperplasia, maturation shift to right, with coarse cytoplasmic granules, and vacuoles, and nuclear multilobation. A novel AD heterozygous frameshift variant in the CXCR4 gene (exon2, c. 1032_1033delTG, p. E345Vfs*12) in patient and father KD: Anemia, leukocytosis, elevated ESR and CRP, thrombocytosis. Echo: Short and thick LCA trunk, dilatation of left anterior descending artery (2.3 mm), and MR | No response to antibiotics. IVIG (2 g/kg), aspirin, and Dipyridamole (improvement in KD symptoms, including coronary artery dilatation but leukopenia and neutropenia continued to persist). IVIG and G-CSF were continued on follow-up |
| Hambleton et al. (2013) [24] | STAT2 deficiency diagnosed at 5 years of age | KD at 18 months of age | STAT2 deficiency: disseminated measles (post-vaccination) KD: fever, rash, conjunctival injection, lymphadenopathy, hepatitis, and pneumonitis. H/O infant sibling death due to an overwhelming viral infection Parental consanguinity was present | Blood and BAL PCR revealed vaccine-strain measles virus. Absent STAT2 expression on immunoblot. DNA sequencing of STAT2 gene revealed homozygous donor splice site mutation (introns 4 and 5, c. 381 + 5 G > C) in affected siblings (parents and other sibling were heterozygous) and 3 homozygotes from extended family. RT-PCR of cDNA revealed longer and less abundant products in patient than in control, indicating aberrant splicing and NMD | Child improved with supplemental oxygen and supportive care |
| Autoinflammatory disorders | |||||
| Broderick et al. (2010) [25] | PFAPA | KD (10, 12, 14, and 36 months of age), M: F-3:1 | KD: Prolonged fever (7–35 days), conjunctival injection, erythematous rash, and lymphadenopathy (4/4), pharyngitis and strawberry tongue (3/4), and periungual desquamation and edema in 2/4, sterile pyuria in 1/4 cases. PFAPA: Recurrent febrile episodes, 2–24 months after IVIG, recurring every 2–6 weeks, lasting for maximum period of 10 days. Pharyngitis, and aphthous ulcers (3/4), lymphadenopathy (4/4), ocular symptoms (2/4), abdominal pain (1/4), and rash (2/4). All 4 patients were apparently well in between febrile episodes with normal CRP levels | Elevated CRP and ESR Echo revealed coronary artery dilatation in ¼ of patients | Responded to primary IVIG therapy (2 g/kg IVIg) followed by recurrent febrile episodes that responded to prednisolone in 3/3 cases. Tonsillectomy, in 1 case, improved the febrile episodes |
| Ninomiya et al. (2013) [26] | PFAPA at 1 year of age | KD at 2 years of age | PFAPA: Febrile episodes, cervical lymphadenopathy, tonsillar enlargement, and pharyngitis Fever persisted despite IVIG, possibly due to PFAPA but declined rapidly after prednisolone therapy KD: Fever, rash, cervical adenopathy, peripheral edema, and conjunctival injection | KD: Neutrophilic leukocytosis, raised CRP. No CAAs | Cimetidine (150 mg/day), IVIG (2 g/kg), Infliximab (high fever, mild tonsillar enlargement, mild cervical adenopathy persisted despite therapy) Prednisolone (2 mg/kg/day), following which fever subsided |
| KD | XLA with KD |
|---|---|
| Maternal IgG (humoral immunity) may be protective as incidence of KD is relatively low below 6 months of age and increases thereafter as the maternal IgG declines | Complete absence of B-cells and IgG in children with XLA may predispose to development of KD |
| Low IgG levels in acute KD have been associated with increased disease severity, CAA, and IVIG resistance | Recurrent or subclinical infections lead to chronic antigen stimulation and immune dysregulation, thereby, inflammatory response of KD. Certain infections like Pseudomonas aeruginosa are common between KD and XLA |
| B cells are increased in proportion compared to T and NK cells in acute KD | Elevated NLRP3 inflammasome activity and proinflammatory cytokines have been reported in XLA as loss of BTK leads to loss of negative regulation of NLRP3 activity |
| In acute KD, BCR repertoire diversity is reduced. B-cell-mediated immune responses are impaired | Increased MHC expression on APCs via molecular mimicry shifts the immune system towards T2 immune response with production of IL-4/5/10. |
| BLK and BCL2L11 genes are downregulated and CD40, FCGR2A, and IGHV genes are upregulated | TLR-9 and NF-kB-mediated immune stimulation leads to IgM production from innate B1 cells and T1 immune response |
| Overall, B-cell function, activation, and development are impaired despite increase in number in acute KD | Leaky antibody production, defective BTK signaling leading to abnormal myeloid maturation and aberrant TLR signaling, autoreactive T cells, and impaired Tregs are other possible mechanisms causing immune dysregulation |
| Functionally impaired B-cells may predispose to KD | B-cell immunodeficiency with activation of non-B-cell-mediated autoimmune and proinflammatory pathways may cause immune dysregulation and predispose to KD in XLA |
| Clinical and Laboratory Features | KD | HIES |
|---|---|---|
| Raised serum IgE and eosinophilia | ✓ | ✓ |
| Fever, rash, and mucocutaneous involvement | ✓ | ✓ |
| Susceptibility to Staphylococcus aureus and Candida albicans | ✓ | ✓ |
| Lymphadenopathy | ✓ | ✓ |
| Vasculitis, CAAs, MI | ✓ | ✓ |
| Response to IVIG with resolution of symptoms | ✓ | ✓ |
| Expansion of CD4+ effector memory T cells | ✓ | ✓ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, S.; Nadig, P.L.; Pilania, R.K.; Sharma, K.; Dhaliwal, M.; Rawat, A.; Singh, S. Kawasaki Disease and Inborn Errors of Immunity: Exploring the Link and Implications. Diagnostics 2023, 13, 2151. https://doi.org/10.3390/diagnostics13132151
Sharma S, Nadig PL, Pilania RK, Sharma K, Dhaliwal M, Rawat A, Singh S. Kawasaki Disease and Inborn Errors of Immunity: Exploring the Link and Implications. Diagnostics. 2023; 13(13):2151. https://doi.org/10.3390/diagnostics13132151
Chicago/Turabian StyleSharma, Saniya, Pallavi L Nadig, Rakesh Kumar Pilania, Kaushal Sharma, Manpreet Dhaliwal, Amit Rawat, and Surjit Singh. 2023. "Kawasaki Disease and Inborn Errors of Immunity: Exploring the Link and Implications" Diagnostics 13, no. 13: 2151. https://doi.org/10.3390/diagnostics13132151
APA StyleSharma, S., Nadig, P. L., Pilania, R. K., Sharma, K., Dhaliwal, M., Rawat, A., & Singh, S. (2023). Kawasaki Disease and Inborn Errors of Immunity: Exploring the Link and Implications. Diagnostics, 13(13), 2151. https://doi.org/10.3390/diagnostics13132151