
Exploring the Molecular Complexity of Medulloblastoma: Implications for Diagnosis and Treatment

 , , , and
, , , and
Abstract
1. Introduction
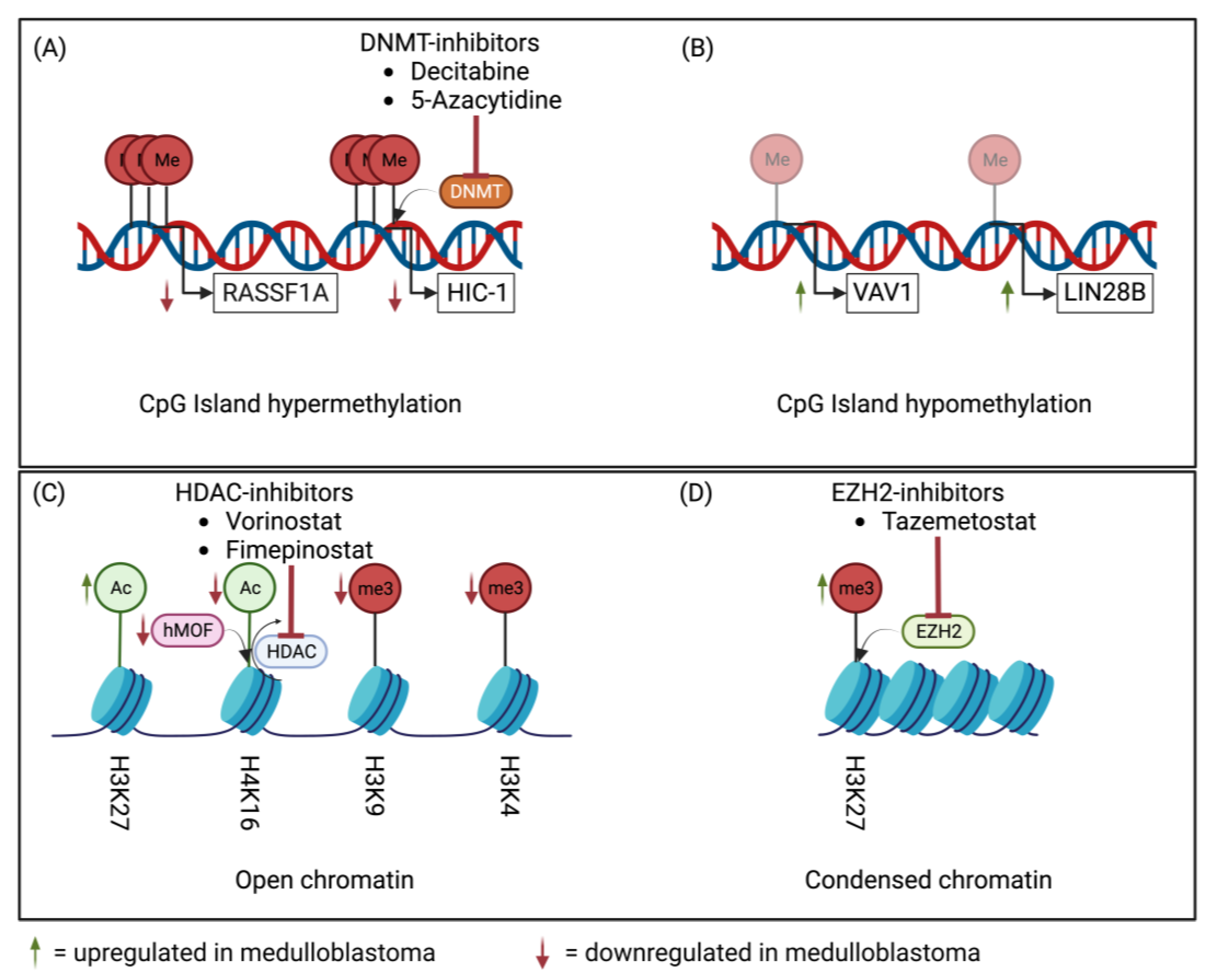
2. Epigenetic Machinery in Medulloblastoma
3. Current Treatment Paradigms and Novel Therapies
3.1. Standard-of-Care Therapy
3.2. Targeted Therapies
3.2.1. Hedgehog Signaling Pathway
3.2.2. PI3K/AKT/mTOR Pathway
3.2.3. CDK Signaling Pathway
3.2.4. Epigenetic Deregulation
3.3. Tumor Microenvironment and Immunotherapies
3.3.1. Adoptive Cellular Therapy/Cellular Immunotherapy
3.3.2. Immune Checkpoint Inhibition
3.3.3. Cancer Vaccination
3.3.4. Oncolytic Virotherapy
4. Mechanisms of Resistance
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Cotter, J.A.; Hawkins, C. Medulloblastoma: WHO 2021 and beyond. Pediatr. Dev. Pathol. 2022, 25, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Schwalbe, E.C.; Lindsey, J.C.; Nakjang, S.; Crosier, S.; Smith, A.J.; Hicks, D.; Rafiee, G.; Hill, R.M.; Iliasova, A.; Stone, T.; et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: A cohort study. Lancet Oncol. 2017, 18, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef]
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754.E6. [Google Scholar] [CrossRef]
- Gorini, F.; Miceli, M.; de Antonellis, P.; Amente, S.; Zollo, M.; Ferrucci, V. Epigenetics and immune cells in medulloblastoma. Front. Genet. 2023, 14, 1135404. [Google Scholar] [CrossRef]
- Pugh, T.J.; Weeraratne, S.D.; Archer, T.C.; Pomeranz Krummel, D.A.; Auclair, D.; Bochicchio, J.; Carneiro, M.O.; Carter, S.L.; Cibulskis, K.; Erlich, R.L.; et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 2012, 488, 106–110. [Google Scholar] [CrossRef]
- Yi, J.; Wu, J. Epigenetic regulation in medulloblastoma. Mol. Cell. Neurosci. 2018, 87, 65–76. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef]
- Haltom, A.R.; Toll, S.A.; Cheng, D.; Maegawa, S.; Gopalakrishnan, V.; Khatua, S. Medulloblastoma epigenetics and the path to clinical innovation. J. Neurooncol. 2020, 150, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Schwalbe, E.C.; Williamson, D.; Lindsey, J.C.; Hamilton, D.; Ryan, S.L.; Megahed, H.; Garami, M.; Hauser, P.; Dembowska-Baginska, B.; Perek, D.; et al. DNA methylation profiling of medulloblastoma allows robust subclassification and improved outcome prediction using formalin-fixed biopsies. Acta Neuropathol. 2013, 125, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Lusher, M.E.; Lindsey, J.C.; Latif, F.; Pearson, A.D.; Ellison, D.W.; Clifford, S.C. Biallelic epigenetic inactivation of the RASSF1A tumor suppressor gene in medulloblastoma development. Cancer Res. 2002, 62, 5906–5911. [Google Scholar] [PubMed]
- Zuzak, T.J.; Steinhoff, D.F.; Sutton, L.N.; Phillips, P.C.; Eggert, A.; Grotzer, M.A. Loss of caspase-8 mRNA expression is common in childhood primitive neuroectodermal brain tumour/medulloblastoma. Eur. J. Cancer. 2002, 38, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Rood, B.R.; Zhang, H.; Weitman, D.M.; Cogen, P.H. Hypermethylation of HIC-1 and 17p allelic loss in medulloblastoma. Cancer Res. 2002, 62, 3794–3797. [Google Scholar]
- Pfister, S.; Schlaeger, C.; Mendrzyk, F.; Wittmann, A.; Benner, A.; Kulozik, A.; Scheurlen, W.; Radlwimmer, B.; Lichter, P. Array-based profiling of reference-independent methylation status (aPRIMES) identifies frequent promoter methylation and consecutive downregulation of ZIC2 in pediatric medulloblastoma. Nucleic Acids Res. 2007, 35, e51. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, J.C.; Kawauchi, D.; Schwalbe, E.C.; Solecki, D.J.; Selby, M.P.; McKinnon, P.J.; Olson, J.M.; Hayden, J.T.; Grundy, R.G.; Ellison, D.W.; et al. Cross-species epigenetics identifies a critical role for VAV1 in SHH subgroup medulloblastoma maintenance. Oncogene 2015, 34, 4746–4757. [Google Scholar] [CrossRef]
- Lindsey, J.C.; Lusher, M.E.; Anderton, J.A.; Gilbertson, R.J.; Ellison, D.W.; Clifford, S.C. Epigenetic deregulation of multiple S100 gene family members by differential hypomethylation and hypermethylation events in medulloblastoma. Br. J. Cancer 2007, 97, 267–274. [Google Scholar] [CrossRef]
- Hovestadt, V.; Jones, D.T.; Picelli, S.; Wang, W.; Kool, M.; Northcott, P.A.; Sultan, M.; Stachurski, K.; Ryzhova, M.; Warnatz, H.J.; et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature 2014, 510, 537–541. [Google Scholar] [CrossRef]
- Northcott, P.A.; Nakahara, Y.; Wu, X.; Feuk, L.; Ellison, D.W.; Croul, S.; Mack, S.; Kongkham, P.N.; Peacock, J.; Dubuc, A.; et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 2009, 41, 465–472. [Google Scholar] [CrossRef]
- Batora, N.V.; Sturm, D.; Jones, D.T.; Kool, M.; Pfister, S.M.; Northcott, P.A. Transitioning from genotypes to epigenotypes: Why the time has come for medulloblastoma epigenomics. Neuroscience 2014, 264, 171–185. [Google Scholar] [CrossRef]
- Dubuc, A.M.; Remke, M.; Korshunov, A.; Northcott, P.A.; Zhan, S.H.; Mendez-Lago, M.; Kool, M.; Jones, D.T.; Unterberger, A.; Morrissy, A.S.; et al. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 2013, 125, 373–384. [Google Scholar] [CrossRef]
- Pfister, S.; Rea, S.; Taipale, M.; Mendrzyk, F.; Straub, B.; Ittrich, C.; Thuerigen, O.; Sinn, H.P.; Akhtar, A.; Lichter, P. The histone acetyltransferase hMOF is frequently downregulated in primary breast carcinoma and medulloblastoma and constitutes a biomarker for clinical outcome in medulloblastoma. Int. J. Cancer 2008, 122, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Grobner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Genovesi, L.A.; Carter, K.W.; Gottardo, N.G.; Giles, K.M.; Dallas, P.B. Integrated analysis of miRNA and mRNA expression in childhood medulloblastoma compared with neural stem cells. PLoS ONE 2011, 6, e23935. [Google Scholar] [CrossRef] [PubMed]
- Mollashahi, B.; Aghamaleki, F.S.; Movafagh, A. The Roles of miRNAs in Medulloblastoma: A Systematic Review. J. Cancer Prev. 2019, 24, 79–90. [Google Scholar] [CrossRef]
- Ferretti, E.; De Smaele, E.; Po, A.; Di Marcotullio, L.; Tosi, E.; Espinola, M.S.; Di Rocco, C.; Riccardi, R.; Giangaspero, F.; Farcomeni, A.; et al. MicroRNA profiling in human medulloblastoma. Int. J. Cancer 2009, 124, 568–577. [Google Scholar] [CrossRef]
- Leary, S.E.S.; Packer, R.J.; Li, Y.; Billups, C.A.; Smith, K.S.; Jaju, A.; Heier, L.; Burger, P.; Walsh, K.; Han, Y.; et al. Efficacy of Carboplatin and Isotretinoin in Children with High-risk Medulloblastoma: A Randomized Clinical Trial from the Children’s Oncology Group. JAMA Oncol. 2021, 7, 1313–1321. [Google Scholar] [CrossRef]
- Northcott, P.A.; Jones, D.T.; Kool, M.; Robinson, G.W.; Gilbertson, R.J.; Cho, Y.J.; Pomeroy, S.L.; Korshunov, A.; Lichter, P.; Taylor, M.D.; et al. Medulloblastomics: The end of the beginning. Nat. Rev. Cancer 2012, 12, 818–834. [Google Scholar] [CrossRef]
- Clifford, S.C.; Lusher, M.E.; Lindsey, J.C.; Langdon, J.A.; Gilbertson, R.J.; Straughton, D.; Ellison, D.W. Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle 2006, 5, 2666–2670. [Google Scholar] [CrossRef]
- Ellison, D.W.; Kocak, M.; Dalton, J.; Megahed, H.; Lusher, M.E.; Ryan, S.L.; Zhao, W.; Nicholson, S.L.; Taylor, R.E.; Bailey, S.; et al. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J. Clin. Oncol. 2011, 29, 1400–1407. [Google Scholar] [CrossRef]
- Ellison, D.W.; Onilude, O.E.; Lindsey, J.C.; Lusher, M.E.; Weston, C.L.; Taylor, R.E.; Pearson, A.D.; Clifford, S.C. beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: The United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J. Clin. Oncol. 2005, 23, 7951–7957. [Google Scholar] [CrossRef]
- Maier, H.; Dalianis, T.; Kostopoulou, O.N. New Approaches in Targeted Therapy for Medulloblastoma in Children. Anticancer Res. 2021, 41, 1715–1726. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.A.; Ness, K.K.; Whitton, J.; Recklitis, C.; Zebrack, B.; Robison, L.L.; Zeltzer, L.; Mertens, A.C. Behavioral and social outcomes in adolescent survivors of childhood cancer: A report from the childhood cancer survivor study. J. Clin. Oncol. 2007, 25, 3649–3656. [Google Scholar] [CrossRef] [PubMed]
- Ness, K.K.; Gurney, J.G.; Zeltzer, L.K.; Leisenring, W.; Mulrooney, D.A.; Nathan, P.C.; Robison, L.L.; Mertens, A.C. The impact of limitations in physical, executive, and emotional function on health-related quality of life among adult survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. Arch. Phys. Med. Rehabil. 2008, 89, 128–136. [Google Scholar] [CrossRef]
- Thompson, E.M.; Ashley, D.; Landi, D. Current medulloblastoma subgroup specific clinical trials. Transl. Pediatr. 2020, 9, 157–162. [Google Scholar] [CrossRef]
- Liu, X.; Ding, C.; Tan, W.; Zhang, A. Medulloblastoma: Molecular understanding, treatment evolution, and new developments. Pharmacol. Ther. 2020, 210, 107516. [Google Scholar] [CrossRef]
- Khatua, S.; Song, A.; Citla Sridhar, D.; Mack, S.C. Childhood Medulloblastoma: Current Therapies, Emerging Molecular Landscape and Newer Therapeutic Insights. Curr. Neuropharmacol. 2018, 16, 1045–1058. [Google Scholar] [CrossRef]
- Pak, E.; Segal, R.A. Hedgehog Signal Transduction: Key Players, Oncogenic Drivers, and Cancer Therapy. Dev. Cell 2016, 38, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Petrirena, G.J.; Masliah-Planchon, J.; Sala, Q.; Pourroy, B.; Frappaz, D.; Tabouret, E.; Graillon, T.; Gentet, J.-C.; Delattre, O.; Chinot, O.; et al. Recurrent extraneural sonic hedgehog medulloblastoma exhibiting sustained response to vismodegib and temozolomide monotherapies and inter-metastatic molecular heterogeneity at progression. Oncotarget 2018, 9, 10175–10183. [Google Scholar] [CrossRef]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results from Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Jones, D.T.; Jäger, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Kaste, S.C.; Chemaitilly, W.; Bowers, D.C.; Laughton, S.; Smith, A.; Gottardo, N.G.; Partap, S.; Bendel, A.; Wright, K.D.; et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 2017, 8, 69295–69302. [Google Scholar] [CrossRef] [PubMed]
- Kieran, M.W.; Chisholm, J.; Casanova, M.; Brandes, A.A.; Aerts, I.; Bouffet, E.; Bailey, S.; Leary, S.; MacDonald, T.J.; Mechinaud, F.; et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro Oncol. 2017, 19, 1542–1552. [Google Scholar] [CrossRef]
- Yuile, A.K.; Kastelan, M.; Lee, A.P.; Back, M.; Drummond, J.; Wheeler, H.R. The use of Sonidegib in the adjuvant and advanced phases of Sonic Hedge Hog Mutant Medulloblastomas. Oxf. Med. Case Rep. 2022, 2022, omac019. [Google Scholar] [CrossRef]
- Infante, P.; Alfonsi, R.; Botta, B.; Mori, M.; Di Marcotullio, L. Targeting GLI factors to inhibit the Hedgehog pathway. Trends Pharmacol. Sci. 2015, 36, 547–558. [Google Scholar] [CrossRef]
- Coni, S.; Antonucci, L.; D’Amico, D.; Di Magno, L.; Infante, P.; De Smaele, E.; Giannini, G.; Di Marcotullio, L.; Screpanti, I.; Gulino, A.; et al. Gli2 acetylation at lysine 757 regulates hedgehog-dependent transcriptional output by preventing its promoter occupancy. PLoS ONE 2013, 8, e65718. [Google Scholar] [CrossRef] [PubMed]
- De Braganca, K.C.; Packer, R.J. Treatment Options for Medulloblastoma and CNS Primitive Neuroectodermal Tumor (PNET). Curr. Treat. Options Neurol. 2013, 15, 593–606. [Google Scholar] [CrossRef]
- Holzhauser, S.; Lukoseviciute, M.; Andonova, T.; Ursu, R.G.; Dalianis, T.; Wickström, M.; Kostopoulou, O.N. Targeting Fibroblast Growth Factor Receptor (FGFR) and Phosphoinositide 3-kinase (PI3K) Signaling Pathways in Medulloblastoma Cell Lines. Anticancer Res. 2020, 40, 53–66. [Google Scholar] [CrossRef]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef]
- Holzhauser, S.; Lukoseviciute, M.; Papachristofi, C.; Vasilopoulou, C.; Herold, N.; Wickström, M.; Kostopoulou, O.N.; Dalianis, T. Effects of PI3K and FGFR inhibitors alone and in combination, and with/without cytostatics in childhood neuroblastoma cell lines. Int. J. Oncol. 2021, 58, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Becher, O.J.; Rosenblum, M.K.; Pandolfi, P.P.; Manova-Todorova, K.; Holland, E.C. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008, 22, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Eckerdt, F.; Clymer, J.; Bell, J.B.; Beauchamp, E.M.; Blyth, G.T.; Goldman, S.; Platanias, L.C. Pharmacological mTOR targeting enhances the antineoplastic effects of selective PI3Kα inhibition in medulloblastoma. Sci. Rep. 2019, 9, 12822. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Janeway, K.A.; Patton, D.R.; Winter, C.L.; Coffey, B.; Williams, P.M.; Roy-Chowdhuri, S.; Tsongalis, G.J.; Routbort, M.; Ramirez, N.C.; et al. Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients with Refractory Cancers in the National Cancer Institute-Children’s Oncology Group Pediatric MATCH Trial. J. Clin. Oncol. 2022, 40, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, N.K.; Kling, M.J.; Coulter, D.W.; McGuire, T.R.; Ray, S.; Kesherwani, V.; Joshi, S.S.; Sharp, J.G. Improved therapy for medulloblastoma: Targeting hedgehog and PI3K-mTOR signaling pathways in combination with chemotherapy. Oncotarget 2018, 9, 16619–16633. [Google Scholar] [CrossRef]
- Northcott, P.A.; Shih, D.J.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stütz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef]
- Shapiro, G.I. Cyclin-dependent kinase pathways as targets for cancer treatment. J. Clin. Oncol. 2006, 24, 1770–1783. [Google Scholar] [CrossRef]
- Roberts, P.J.; Bisi, J.E.; Strum, J.C.; Combest, A.J.; Darr, D.B.; Usary, J.E.; Zamboni, W.C.; Wong, K.K.; Perou, C.M.; Sharpless, N.E. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J. Natl. Cancer Inst. 2012, 104, 476–487. [Google Scholar] [CrossRef]
- Cook Sangar, M.L.; Genovesi, L.A.; Nakamoto, M.W.; Davis, M.J.; Knobluagh, S.E.; Ji, P.; Millar, A.; Wainwright, B.J.; Olson, J.M. Inhibition of CDK4/6 by Palbociclib Significantly Extends Survival in Medulloblastoma Patient-Derived Xenograft Mouse Models. Clin. Cancer Res. 2017, 23, 5802–5813. [Google Scholar] [CrossRef]
- Roberts, P.J.; Kumarasamy, V.; Witkiewicz, A.K.; Knudsen, E.S. Chemotherapy and CDK4/6 Inhibitors: Unexpected Bedfellows. Mol. Cancer Ther. 2020, 19, 1575–1588. [Google Scholar] [CrossRef]
- Whiteway, S.L.; Harris, P.S.; Venkataraman, S.; Alimova, I.; Birks, D.K.; Donson, A.M.; Foreman, N.K.; Vibhakar, R. Inhibition of cyclin-dependent kinase 6 suppresses cell proliferation and enhances radiation sensitivity in medulloblastoma cells. J. Neurooncol. 2013, 111, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Roussel, M.F.; Stripay, J.L. Epigenetic Drivers in Pediatric Medulloblastoma. Cerebellum 2018, 17, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Fouladi, M.; Park, J.R.; Stewart, C.F.; Gilbertson, R.J.; Schaiquevich, P.; Sun, J.; Reid, J.M.; Ames, M.M.; Speights, R.; Ingle, A.M.; et al. Pediatric phase I trial and pharmacokinetic study of vorinostat: A Children’s Oncology Group phase I consortium report. J. Clin. Oncol. 2010, 28, 3623–3629. [Google Scholar] [CrossRef] [PubMed]
- Hummel, T.R.; Wagner, L.; Ahern, C.; Fouladi, M.; Reid, J.M.; McGovern, R.M.; Ames, M.M.; Gilbertson, R.J.; Horton, T.; Ingle, A.M.; et al. A pediatric phase 1 trial of vorinostat and temozolomide in relapsed or refractory primary brain or spinal cord tumors: A Children’s Oncology Group phase 1 consortium study. Pediatr. Blood Cancer 2013, 60, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Griesinger, A.M.; Birks, D.K.; Donson, A.M.; Amani, V.; Hoffman, L.M.; Waziri, A.; Wang, M.; Handler, M.H.; Foreman, N.K. Characterization of distinct immunophenotypes across pediatric brain tumor types. J. Immunol. 2013, 191, 4880–4888. [Google Scholar] [CrossRef]
- Bockmayr, M.; Mohme, M.; Klauschen, F.; Winkler, B.; Budczies, J.; Rutkowski, S.; Schüller, U. Subgroup-specific immune and stromal microenvironment in medulloblastoma. Oncoimmunology 2018, 7, e1462430. [Google Scholar] [CrossRef]
- Margol, A.S.; Robison, N.J.; Gnanachandran, J.; Hung, L.T.; Kennedy, R.J.; Vali, M.; Dhall, G.; Finlay, J.L.; Erdreich-Epstein, A.; Krieger, M.D.; et al. Tumor-associated macrophages in SHH subgroup of medulloblastomas. Clin. Cancer Res. 2015, 21, 1457–1465. [Google Scholar] [CrossRef]
- Zhang, J.; Yuan, X.; Wang, Y.; Liu, J.; Li, Z.; Li, S.; Liu, Y.; Gong, X.; Sun, Y.; Wu, W.; et al. Tumor-Associated Macrophages Correlate with Prognosis in Medulloblastoma. Front. Oncol. 2022, 12, 893132. [Google Scholar] [CrossRef]
- O’Connor, T.; Heikenwalder, M. CCL2 in the Tumor Microenvironment. In Tumor Microenvironment; Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2021; Volume 1302, pp. 1–14. [Google Scholar] [CrossRef]
- Maximov, V.; Chen, Z.; Wei, Y.; Robinson, M.H.; Herting, C.J.; Shanmugam, N.S.; Rudneva, V.A.; Goldsmith, K.C.; MacDonald, T.J.; Northcott, P.A.; et al. Tumour-associated macrophages exhibit anti-tumoural properties in Sonic Hedgehog medulloblastoma. Nat. Commun. 2019, 10, 2410. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Crotty, E.E.; Smith, S.M.C.; Brasel, K.; Pakiam, F.; Girard, E.J.; Connor, Y.D.; Zindy, F.; Mhyre, A.J.; Roussel, M.F.; Olson, J.M. Medulloblastoma recurrence and metastatic spread are independent of colony-stimulating factor 1 receptor signaling and macrophage survival. J. Neurooncol. 2021, 153, 225–237. [Google Scholar] [CrossRef]
- Lee, C.; Lee, J.; Choi, S.A.; Kim, S.K.; Wang, K.C.; Park, S.H.; Kim, S.H.; Lee, J.Y.; Phi, J.H. M1 macrophage recruitment correlates with worse outcome in SHH Medulloblastomas. BMC Cancer 2018, 18, 535. [Google Scholar] [CrossRef] [PubMed]
- Patt, S.; Zimmer, C. Age-related immunoreactivity pattern in medulloblastoma. Child’s Nerv. Syst. 1992, 8, 326–331. [Google Scholar] [CrossRef]
- Haberthur, K.; Brennan, K.; Hoglund, V.; Balcaitis, S.; Chinn, H.; Davis, A.; Kreuser, S.; Winter, C.; Leary, S.E.; Deutsch, G.H.; et al. NKG2D ligand expression in pediatric brain tumors. Cancer Biol. Ther. 2016, 17, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, M.; Mulla, N.; Malibary, H.; Bamaga, A.K.; Fadul, M.M.; Faizo, E.; Hakamy, S.; Baeesa, S. Immune microenvironment of medulloblastoma: The association between its molecular subgroups and potential targeted immunotherapeutic receptors. World J. Clin. Oncol. 2023, 14, 117–130. [Google Scholar] [CrossRef]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef]
- Gururangan, S.; Reap, E.; Schmittling, R.; Kocak, M.; Reynolds, R.; Grant, G.; Onar-Thomas, A.; Baxter, P.; Pollack, I.F.; Phillips, P.; et al. Regulatory T cell subsets in patients with medulloblastoma at diagnosis and during standard irradiation and chemotherapy (PBTC N-11). Cancer Immunol. Immunother. 2017, 66, 1589–1595. [Google Scholar] [CrossRef]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H., Jr. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, S87–S97. [Google Scholar] [CrossRef] [PubMed]
- Schakelaar, M.Y.; Monnikhof, M.; Crnko, S.; Pijnappel, E.W.; Meeldijk, J.; Ten Broeke, T.; Bovenschen, N. Cellular immunotherapy for medulloblastoma. Neuro Oncol. 2023, 25, 617–627. [Google Scholar] [CrossRef]
- Voskamp, M.J.; Li, S.; van Daalen, K.R.; Crnko, S.; Ten Broeke, T.; Bovenschen, N. Immunotherapy in Medulloblastoma: Current State of Research, Challenges, and Future Perspectives. Cancers 2021, 13, 5387. [Google Scholar] [CrossRef] [PubMed]
- Audi, Z.F.; Saker, Z.; Rizk, M.; Harati, H.; Fares, Y.; Bahmad, H.F.; Nabha, S.M. Immunosuppression in Medulloblastoma: Insights into Cancer Immunity and Immunotherapy. Curr. Treat. Options Oncol. 2021, 22, 83. [Google Scholar] [CrossRef]
- Ward, S.A.; Warrington, N.M.; Taylor, S.; Kfoury, N.; Luo, J.; Rubin, J.B. Reprogramming Medulloblastoma-Propagating Cells by a Combined Antagonism of Sonic Hedgehog and CXCR4. Cancer Res. 2017, 77, 1416–1426. [Google Scholar] [CrossRef]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, D.W.; Spanos, W.C.; Vermeer, P.D.; Bruns, A.M.; Lee, K.M.; Lee, J.H. Radiation-induced loss of cell surface CD47 enhances immune-mediated clearance of human papillomavirus-positive cancer. Int. J. Cancer 2013, 133, 120–129. [Google Scholar] [CrossRef]
- Das, A.; McDonald, D.; Lowe, S.; Bredlau, A.L.; Vanek, K.; Patel, S.J.; Cheshier, S.; Eskandari, R. Immunological low-dose radiation modulates the pediatric medulloblastoma antigens and enhances antibody-dependent cellular cytotoxicity. Child’s Nerv. Syst. 2017, 33, 429–436. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, Y.J. Adoptive Cell Therapy Targeting Neoantigens: A Frontier for Cancer Research. Front. Immunol. 2020, 11, 176. [Google Scholar] [CrossRef]
- Ligon, J.A.; Wessel, K.M.; Shah, N.N.; Glod, J. Adoptive Cell Therapy in Pediatric and Young Adult Solid Tumors: Current Status and Future Directions. Front. Immunol. 2022, 13, 846346. [Google Scholar] [CrossRef]
- Kabir, T.F.; Kunos, C.A.; Villano, J.L.; Chauhan, A. Immunotherapy for Medulloblastoma: Current Perspectives. ImmunoTargets Ther. 2020, 9, 57–77. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Rα2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef]
- Gajjar, A.; Hernan, R.; Kocak, M.; Fuller, C.; Lee, Y.; Mckinnon, P.J.; Wallace, D.; Lau, C.; Chintagumpala, M.; Ashley, D.M.; et al. Clinical, Histopathologic, and Molecular Markers of Prognosis: Toward a New Disease Risk Stratification System for Medulloblastoma. J. Clin. Oncol. 2004, 22, 984–993. [Google Scholar] [CrossRef]
- Patereli, A.; Alexiou, G.A.; Stefanaki, K.; Moschovi, M.; Doussis-Anagnostopoulou, I.; Prodromou, N.; Karentzou, O. Expression of epidermal growth factor receptor and HER-2 in pediatric embryonal brain tumors. Pediatr. Neurosurg. 2010, 46, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.Y.K.; Hui, A.B.Y.; Yin, X.-L.; Pang, J.C.S.; Zhu, X.-L.; Poon, W.-S.; Ng, H.-K. Detection of oncogene amplifications in medulloblastomas by comparative genomic hybridization and array-based comparative genomic hybridization. J. Neurosurg. Pediatr. 2004, 100, 187–193. [Google Scholar] [CrossRef]
- Ahmed, N.; Ratnayake, M.; Savoldo, B.; Perlaky, L.; Dotti, G.; Wels, W.S.; Bhattacharjee, M.B.; Gilbertson, R.J.; Shine, H.D.; Weiss, H.L.; et al. Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Res. 2007, 67, 5957–5964. [Google Scholar] [CrossRef]
- Nellan, A.; Rota, C.; Majzner, R.; Lester-McCully, C.M.; Griesinger, A.M.; Mulcahy Levy, J.M.; Foreman, N.K.; Warren, K.E.; Lee, D.W. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J. Immunother. Cancer 2018, 6, 30. [Google Scholar] [CrossRef]
- Dai, H.-j.; Sun, B.; Yang, D.; Xu, H.; Zhu, J.; Wei, J.; Zhao, X. Eradication of medulloblastoma by NKG2D-specific CAR T-cells. J. Clin. Oncol. 2020, 38, 2522. [Google Scholar] [CrossRef]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; Van Ommeren, R.; et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med. 2020, 26, 720–731. [Google Scholar] [CrossRef]
- Fares, J.; Davis, Z.B.; Rechberger, J.S.; Toll, S.A.; Schwartz, J.D.; Daniels, D.J.; Miller, J.S.; Khatua, S. Advances in NK cell therapy for brain tumors. NPJ Precis. Oncol. 2023, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.B.; Yadavilli, S.; Saunders, D.; Van Pelt, S.; Chorvinsky, E.; Burga, R.A.; Albihani, S.; Hanley, P.J.; Xu, Z.; Pei, Y.; et al. Medulloblastoma rendered susceptible to NK-cell attack by TGFβ neutralization. J. Transl. Med. 2019, 17, 321. [Google Scholar] [CrossRef] [PubMed]
- Khatua, S.; Cooper, L.J.N.; Sandberg, D.I.; Ketonen, L.; Johnson, J.M.; Rytting, M.E.; Liu, D.D.; Meador, H.; Trikha, P.; Nakkula, R.J.; et al. Phase I study of intraventricular infusions of autologous ex vivo expanded NK cells in children with recurrent medulloblastoma and ependymoma. Neuro Oncol. 2020, 22, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- May, K.F., Jr.; Roychowdhury, S.; Bhatt, D.; Kocak, E.; Bai, X.F.; Liu, J.Q.; Ferketich, A.K.; Martin, E.W., Jr.; Caligiuri, M.A.; Zheng, P.; et al. Anti-human CTLA-4 monoclonal antibody promotes T-cell expansion and immunity in a hu-PBL-SCID model: A new method for preclinical screening of costimulatory monoclonal antibodies. Blood 2005, 105, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bondarenko, I.; Luft, A.; Serwatowski, P.; Barlesi, F.; Chacko, R.; Sebastian, M.; Neal, J.; Lu, H.; Cuillerot, J.M.; et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: Results from a randomized, double-blind, multicenter phase II study. J. Clin. Oncol. 2012, 30, 2046–2054. [Google Scholar] [CrossRef]
- Small, E.J.; Tchekmedyian, N.S.; Rini, B.I.; Fong, L.; Lowy, I.; Allison, J.P. A pilot trial of CTLA-4 blockade with human anti-CTLA-4 in patients with hormone-refractory prostate cancer. Clin. Cancer Res. 2007, 13, 1810–1815. [Google Scholar] [CrossRef]
- Ansell, S.M.; Hurvitz, S.A.; Koenig, P.A.; LaPlant, B.R.; Kabat, B.F.; Fernando, D.; Habermann, T.M.; Inwards, D.J.; Verma, M.; Yamada, R.; et al. Phase I study of ipilimumab, an anti-CTLA-4 monoclonal antibody, in patients with relapsed and refractory B-cell non-Hodgkin lymphoma. Clin. Cancer Res. 2009, 15, 6446–6453. [Google Scholar] [CrossRef]
- Kvistborg, P.; Philips, D.; Kelderman, S.; Hageman, L.; Ottensmeier, C.; Joseph-Pietras, D.; Welters, M.J.; van der Burg, S.; Kapiteijn, E.; Michielin, O.; et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci. Transl. Med. 2014, 6, 254ra128. [Google Scholar] [CrossRef] [PubMed]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [PubMed]
- Friedlaender, A.; Addeo, A.; Banna, G. New emerging targets in cancer immunotherapy: The role of TIM3. ESMO Open 2019, 4, e000497. [Google Scholar] [CrossRef]
- Moon, Y.W.; Hajjar, J.; Hwu, P.; Naing, A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J. Immunother. Cancer 2015, 3, 51. [Google Scholar] [CrossRef]
- Vonderheide, R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020, 71, 47–58. [Google Scholar] [CrossRef]
- Vermeulen, J.F.; Van Hecke, W.; Adriaansen, E.J.M.; Jansen, M.K.; Bouma, R.G.; Villacorta Hidalgo, J.; Fisch, P.; Broekhuizen, R.; Spliet, W.G.M.; Kool, M.; et al. Prognostic relevance of tumor-infiltrating lymphocytes and immune checkpoints in pediatric medulloblastoma. Oncoimmunology 2018, 7, e1398877. [Google Scholar] [CrossRef]
- Martin, A.M.; Nirschl, C.J.; Polanczyk, M.J.; Bell, W.R.; Nirschl, T.R.; Harris-Bookman, S.; Phallen, J.; Hicks, J.; Martinez, D.; Ogurtsova, A.; et al. PD-L1 expression in medulloblastoma: An evaluation by subgroup. Oncotarget 2018, 9, 19177–19191. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.D.; Flores, C.; Yang, C.; Pinheiro, E.M.; Yearley, J.H.; Sayour, E.J.; Pei, Y.; Moore, C.; McLendon, R.E.; Huang, J.; et al. Differential Immune Microenvironments and Response to Immune Checkpoint Blockade among Molecular Subtypes of Murine Medulloblastoma. Clin. Cancer Res. 2016, 22, 582–595. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Penas-Prado, M.; Zhou, S.; Wei, J.; Khatua, S.; Hodges, T.R.; Sanai, N.; Xiu, J.; Gatalica, Z.; Kim, L.; et al. Rethinking medulloblastoma from a targeted therapeutics perspective. J. Neurooncol. 2018, 139, 713–720. [Google Scholar] [CrossRef]
- Sayour, E.J.; Mitchell, D.A. Immunotherapy for Pediatric Brain Tumors. Brain Sci. 2017, 7, 137. [Google Scholar] [CrossRef]
- Jähnisch, H.; Füssel, S.; Kiessling, A.; Wehner, R.; Zastrow, S.; Bachmann, M.; Rieber, E.P.; Wirth, M.P.; Schmitz, M. Dendritic cell-based immunotherapy for prostate cancer. Clin. Dev. Immunol. 2010, 2010, 517493. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Bandopadhayay, P.; Jenkins, M.R. Towards Immunotherapy for Pediatric Brain Tumors. Trends Immunol. 2019, 40, 748–761. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination with Extension of Survival Among Patients with Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121. [Google Scholar] [CrossRef]
- Ardon, H.; De Vleeschouwer, S.; Van Calenbergh, F.; Claes, L.; Kramm, C.M.; Rutkowski, S.; Wolff, J.E.; Van Gool, S.W. Adjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours. Pediatr. Blood Cancer 2010, 54, 519–525. [Google Scholar] [CrossRef]
- McNamara, M.A.; Nair, S.K.; Holl, E.K. RNA-Based Vaccines in Cancer Immunotherapy. J. Immunol. Res. 2015, 2015, 794528. [Google Scholar] [CrossRef]
- Jahanafrooz, Z.; Baradaran, B.; Mosafer, J.; Hashemzaei, M.; Rezaei, T.; Mokhtarzadeh, A.; Hamblin, M.R. Comparison of DNA and mRNA vaccines against cancer. Drug Discov. Today 2020, 25, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, O.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Kreiter, S.; Selmi, A.; Diken, M.; Koslowski, M.; Britten, C.M.; Huber, C.; Türeci, O.; Sahin, U. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 2010, 70, 9031–9040. [Google Scholar] [CrossRef] [PubMed]
- Coban, C.; Kobiyama, K.; Aoshi, T.; Takeshita, F.; Horii, T.; Akira, S.; Ishii, K.J. Novel strategies to improve DNA vaccine immunogenicity. Curr. Gene Ther. 2011, 11, 479–484. [Google Scholar] [CrossRef]
- Nair, S.K.; Driscoll, T.; Boczkowski, D.; Schmittling, R.; Reynolds, R.; Johnson, L.A.; Grant, G.; Fuchs, H.; Bigner, D.D.; Sampson, J.H.; et al. Ex vivo generation of dendritic cells from cryopreserved, post-induction chemotherapy, mobilized leukapheresis from pediatric patients with medulloblastoma. J. Neurooncol. 2015, 125, 65–74. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Aurelian, L. Oncolytic viruses as immunotherapy: Progress and remaining challenges. OncoTargets Ther. 2016, 9, 2627–2637. [Google Scholar] [CrossRef]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef]
- Liu, T.C.; Hwang, T.; Park, B.H.; Bell, J.; Kirn, D.H. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol. Ther. 2008, 16, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Kellish, P.; Shabashvili, D.; Rahman, M.M.; Nawab, A.; Guijarro, M.V.; Zhang, M.; Cao, C.; Moussatche, N.; Boyle, T.; Antonia, S.; et al. Oncolytic virotherapy for small-cell lung cancer induces immune infiltration and prolongs survival. J. Clin. Investig. 2019, 129, 2279–2292. [Google Scholar] [CrossRef]
- Stanford, M.M.; Breitbach, C.J.; Bell, J.C.; McFadden, G. Innate immunity, tumor microenvironment and oncolytic virus therapy: Friends or foes? Curr. Opin. Mol. Ther. 2008, 10, 32–37. [Google Scholar] [PubMed]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267.e255. [Google Scholar] [CrossRef]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, F. Advances and potential pitfalls of oncolytic viruses expressing immunomodulatory transgene therapy for malignant gliomas. Cell Death Dis. 2020, 11, 485. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Ino, Y.; Ohtsu, H.; Shibahara, J.; Tanaka, M. A phase I/II study of triple-mutated oncolytic herpes virus G47∆ in patients with progressive glioblastoma. Nat. Commun. 2022, 13, 4119. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef]
- Lun, X.Q.; Zhou, H.; Alain, T.; Sun, B.; Wang, L.; Barrett, J.W.; Stanford, M.M.; McFadden, G.; Bell, J.; Senger, D.L.; et al. Targeting human medulloblastoma: Oncolytic virotherapy with myxoma virus is enhanced by rapamycin. Cancer Res. 2007, 67, 8818–8827. [Google Scholar] [CrossRef]
- Baryawno, N.; Rahbar, A.; Wolmer-Solberg, N.; Taher, C.; Odeberg, J.; Darabi, A.; Khan, Z.; Sveinbjörnsson, B.; FuskevÅg, O.M.; Segerström, L.; et al. Detection of human cytomegalovirus in medulloblastomas reveals a potential therapeutic target. J. Clin. Investig. 2011, 121, 4043–4055. [Google Scholar] [CrossRef]
- Lacroix, J.; Schlund, F.; Leuchs, B.; Adolph, K.; Sturm, D.; Bender, S.; Hielscher, T.; Pfister, S.M.; Witt, O.; Rommelaere, J.; et al. Oncolytic effects of parvovirus H-1 in medulloblastoma are associated with repression of master regulators of early neurogenesis. Int. J. Cancer 2014, 134, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.M.; Brown, M.; Dobrikova, E.; Ramaswamy, V.; Taylor, M.D.; McLendon, R.; Sanks, J.; Chandramohan, V.; Bigner, D.; Gromeier, M. Poliovirus Receptor (CD155) Expression in Pediatric Brain Tumors Mediates Oncolysis of Medulloblastoma and Pleomorphic Xanthoastrocytoma. J. Neuropathol. Exp. Neurol. 2018, 77, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Carrera, D.; Phillips, J.J.; Weiss, W.A.; Raffel, C. An oncolytic measles virus-sensitive Group 3 medulloblastoma model in immune-competent mice. Neuro Oncol. 2018, 20, 1606–1615. [Google Scholar] [CrossRef]
- Studebaker, A.W.; Hutzen, B.; Pierson, C.R.; Russell, S.J.; Galanis, E.; Raffel, C. Oncolytic measles virus prolongs survival in a murine model of cerebral spinal fluid-disseminated medulloblastoma. Neuro Oncol. 2012, 14, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Studebaker, A.W.; Hutzen, B.J.; Pierson, C.R.; Haworth, K.B.; Cripe, T.P.; Jackson, E.M.; Leonard, J.R. Oncolytic Herpes Virus rRp450 Shows Efficacy in Orthotopic Xenograft Group 3/4 Medulloblastomas and Atypical Teratoid/Rhabdoid Tumors. Mol. Ther. Oncolytics 2017, 6, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Baxter, P.A.; Zhao, X.; Liu, Z.; Wadhwa, L.; Zhang, Y.; Su, J.M.; Tan, X.; Yang, J.; Adesina, A.; et al. A single intravenous injection of oncolytic picornavirus SVV-001 eliminates medulloblastomas in primary tumor-based orthotopic xenograft mouse models. Neuro Oncol. 2011, 13, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Friedman, G.K.; Moore, B.P.; Nan, L.; Kelly, V.M.; Etminan, T.; Langford, C.P.; Xu, H.; Han, X.; Markert, J.M.; Beierle, E.A.; et al. Pediatric medulloblastoma xenografts including molecular subgroup 3 and CD133+ and CD15+ cells are sensitive to killing by oncolytic herpes simplex viruses. Neuro Oncol. 2016, 18, 227–235. [Google Scholar] [CrossRef]
- Menyhárt, O.; Győrffy, B. Molecular stratifications, biomarker candidates and new therapeutic options in current medulloblastoma treatment approaches. Cancer Metastasis Rev. 2020, 39, 211–233. [Google Scholar] [CrossRef]
- Schuelke, M.R.; Gundelach, J.H.; Coffey, M.; West, E.; Scott, K.; Johnson, D.R.; Samson, A.; Melcher, A.; Vile, R.G.; Bram, R.J. Phase I trial of sargramostim/pelareorep therapy in pediatric patients with recurrent or refractory high-grade brain tumors. Neurooncol. Adv. 2022, 4, vdac085. [Google Scholar] [CrossRef]
- Hill, R.M.; Richardson, S.; Schwalbe, E.C.; Hicks, D.; Lindsey, J.C.; Crosier, S.; Rafiee, G.; Grabovska, Y.; Wharton, S.B.; Jacques, T.S.; et al. Time, pattern, and outcome of medulloblastoma relapse and their association with tumour biology at diagnosis and therapy: A multicentre cohort study. Lancet Child Adolesc. Health 2020, 4, 865–874. [Google Scholar] [CrossRef]
- Menyhárt, O.; Giangaspero, F.; Győrffy, B. Molecular markers and potential therapeutic targets in non-WNT/non-SHH (group 3 and group 4) medulloblastomas. J. Hematol. Oncol. 2019, 12, 29. [Google Scholar] [CrossRef]
- Taylor, L.; Wade, P.K.; Johnson, J.E.C.; Aldighieri, M.; Morlando, S.; Di Leva, G.; Kerr, I.D.; Coyle, B. Drug Resistance in Medulloblastoma Is Driven by YB-1, ABCB1 and a Seven-Gene Drug Signature. Cancers 2023, 15, 1086. [Google Scholar] [CrossRef]
- Gabriel, N.; Balaji, K.; Jayachandran, K.; Inkman, M.; Zhang, J.; Dahiya, S.; Goldstein, M. Loss of H3K27 Trimethylation Promotes Radiotherapy Resistance in Medulloblastoma and Induces an Actionable Vulnerability to BET Inhibition. Cancer Res. 2022, 82, 2019–2030. [Google Scholar] [CrossRef]
- Gareev, I.; Beylerli, O.; Liang, Y.; Xiang, H.; Liu, C.; Xu, X.; Yuan, C.; Ahmad, A.; Yang, G. The Role of MicroRNAs in Therapeutic Resistance of Malignant Primary Brain Tumors. Front. Cell Dev. Biol. 2021, 9, 740303. [Google Scholar] [CrossRef]
- Ge, J.; Wang, B.; Zhao, S.; Xu, J. Inhibition of lncRNA NEAT1 sensitizes medulloblastoma cells to cisplatin through modulating the miR-23a-3p-glutaminase (GLS) axis. Bioengineered 2022, 13, 7670–7682. [Google Scholar] [CrossRef]
- Marabitti, V.; Giansanti, M.; De Mitri, F.; Gatto, F.; Mastronuzzi, A.; Nazio, F. Pathological implications of metabolic reprogramming and its therapeutic potential in medulloblastoma. Front. Cell Dev. Biol. 2022, 10, 1007641. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Faria, C.C.; Perreault, S.; Cho, Y.-J.; Shih, D.J.; Luu, B.; Dubuc, A.M.; Northcott, P.A.; et al. Recurrence patterns across medulloblastoma subgroups: An integrated clinical and molecular analysis. Lancet Oncol. 2013, 14, 1200–1207. [Google Scholar] [CrossRef]
- Wang, X.; Dubuc, A.M.; Ramaswamy, V.; Mack, S.; Gendoo, D.M.A.; Remke, M.; Wu, X.; Garzia, L.; Luu, B.; Cavalli, F.; et al. Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol. 2015, 129, 449–457. [Google Scholar] [CrossRef]
- Morrissy, A.S.; Garzia, L.; Shih, D.J.H.; Zuyderduyn, S.; Huang, X.; Skowron, P.; Remke, M.; Cavalli, F.M.G.; Ramaswamy, V.; Lindsay, P.E.; et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature 2016, 529, 351–357. [Google Scholar] [CrossRef]
- Treisman, D.; Li, Y.; Zhu, Y. Stem-Like Cell Populations, p53-Pathway Activation and Mechanisms of Recurrence in Sonic Hedgehog Medulloblastoma. Neuromol. Med. 2022, 24, 13–17. [Google Scholar] [CrossRef]
- Raso, A.; Mascelli, S.; Biassoni, R.; Nozza, P.; Kool, M.; Pistorio, A.; Ugolotti, E.; Milanaccio, C.; Pignatelli, S.; Ferraro, M.; et al. High levels of PROM1 (CD133) transcript are a potential predictor of poor prognosis in medulloblastoma. Neuro-Oncology 2011, 13, 500–508. [Google Scholar] [CrossRef]
- Blazek, E.R.; Foutch, J.L.; Maki, G. Daoy medulloblastoma cells that express CD133 are radioresistant relative to CD133- cells, and the CD133+ sector is enlarged by hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Glumac, P.M.; Lebeau, A.M. The role of CD133 in cancer: A concise review. Clin. Transl. Med. 2018, 7, e18. [Google Scholar] [CrossRef]
- Zhang, L.; He, X.; Liu, X.; Zhang, F.; Huang, L.F.; Potter, A.S.; Xu, L.; Zhou, W.; Zheng, T.; Luo, Z.; et al. Single-Cell Transcriptomics in Medulloblastoma Reveals Tumor-Initiating Progenitors and Oncogenic Cascades during Tumorigenesis and Relapse. Cancer Cell 2019, 36, 302–318.e307. [Google Scholar] [CrossRef]
- Treisman, D.M.; Li, Y.; Pierce, B.R.; Li, C.; Chervenak, A.P.; Tomasek, G.J.; Lozano, G.; Zheng, X.; Kool, M.; Zhu, Y. Sox2(+) cells in Sonic Hedgehog-subtype medulloblastoma resist p53-mediated cell-cycle arrest response and drive therapy-induced recurrence. Neurooncol. Adv. 2019, 1, vdz027. [Google Scholar] [CrossRef] [PubMed]
- Hendrikse, L.D.; Haldipur, P.; Saulnier, O.; Millman, J.; Sjoboen, A.H.; Erickson, A.W.; Ong, W.; Gordon, V.; Coudière-Morrison, L.; Mercier, A.L.; et al. Failure of human rhombic lip differentiation underlies medulloblastoma formation. Nature 2022, 609, 1021–1028. [Google Scholar] [CrossRef]
- Gibson, P.; Tong, Y.; Robinson, G.; Thompson, M.C.; Currle, D.S.; Eden, C.; Kranenburg, T.A.; Hogg, T.; Poppleton, H.; Martin, J.; et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 2010, 468, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Wallace, V.A. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr. Biol. 1999, 9, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-J.; Ellis, T.; Markant, S.L.; Read, T.-A.; Kessler, J.D.; Bourboulas, M.; Schüller, U.; Machold, R.; Fishell, G.; Rowitch, D.H.; et al. Medulloblastoma Can Be Initiated by Deletion of Patched in Lineage-Restricted Progenitors or Stem Cells. Cancer Cell 2008, 14, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Hovestadt, V.; Smith, K.S.; Bihannic, L.; Filbin, M.G.; Shaw, M.L.; Baumgartner, A.; Dewitt, J.C.; Groves, A.; Mayr, L.; Weisman, H.R.; et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature 2019, 572, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Vladoiu, M.C.; El-Hamamy, I.; Donovan, L.K.; Farooq, H.; Holgado, B.L.; Sundaravadanam, Y.; Ramaswamy, V.; Hendrikse, L.D.; Kumar, S.; Mack, S.C.; et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 2019, 572, 67–73. [Google Scholar] [CrossRef]
- Folgiero, V.; Miele, E.; Carai, A.; Ferretti, E.; Alfano, V.; Po, A.; Bertaina, V.; Goffredo, B.M.; Benedetti, M.C.; Camassei, F.D.; et al. IDO1 involvement in mTOR pathway: A molecular mechanism of resistance to mTOR targeting in medulloblastoma. Oncotarget 2016, 7, 52900–52911. [Google Scholar] [CrossRef]
- Eckerdt, F.; Beauchamp, E.; Bell, J.; Iqbal, A.; Su, B.; Fukunaga, R.; Lulla, R.R.; Goldman, S.; Platanias, L.C. Regulatory effects of a Mnk2-eIF4E feedback loop during mTORC1 targeting of human medulloblastoma cells. Oncotarget 2014, 5, 8442–8451. [Google Scholar] [CrossRef] [PubMed]
- Raaf, J. Relation of abnormal collections of cells in posterior medullary velum of cerebellum to origin of medulloblastoma. Arch. Neurol. Psychiatry 1944, 52, 163. [Google Scholar] [CrossRef]
- Rorke, L.B.; Fogelson, M.H.; Riggs, H.E. Cerebellar heterotopia in infancy. Dev. Med. Child Neurol. 1968, 10, 644–650. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| WNT | SHH | Non-WNT/Non-SHH | |||
|---|---|---|---|---|---|
| Group 3 | Group 4 | ||||
| % of medulloblastoma | 10 | 30 | 25 | 35 | |
| Gender (M:F) | 1:1 | 1:1 | 2:1 | 3:1 | |
| Age group | Child > adult | Infant, adult > child | Infant, child | Child > infant, adult | |
| Histology | Classic, rarely LCA | Desmoplastic/nodular> classic, MBEN, LCA | Classic, LCA | Classic, LCA | |
| Immunoprofile | B-catenin nuc (+) YAP1/filamin A (+) GAB1 (−) | B-catenin nuc (−) YAP1/filamin A (+) GAB1 (+) | B-catenin nuc (−) YAP1/filamin A (+) GAB1 (−) | ||
| Proposed cell of origin | Lower rhombic lip progenitor cells | CGNPs of the EGL | Undifferentiated cerebellar stem cells | Unipolar brush cells | |
| Tumor location | Fourth ventricle; infiltrating brainstem | Cerebellar hemispheres; rarely midline | Fourth ventricle; midline | Fourth ventricle; midline | |
| Metastasis at diagnosis | 5–10% | 15–20% | 40–45% | 35–40% | |
| Prognosis (5-year overall survival %) | >95% | TP53-wild type: 80% | TP53 mutated (SHH-α): 40% | 50% | 75% |
| WNT | SHH | Non-WNT/non-SHH | ||
|---|---|---|---|---|
| Group 3 | Group 4 | |||
| Proposed number of subtypes | 2 (WNT-α and WNT-ß) | 4 (SHH-α, SHH-ß, SHH-γ, and SHH-δ) | 8 (Group 3/Group 4 subtypes I-VIII) | |
| Cytogenetics | Monosomy 6 | Loss of 9q, 10q, 14q, and 17p Gain of 3q and 9p | Loss of 8q, 10q, 11q, 15q, 16q, and 17p Gain of 1q, 7, and 18 Isochromosome: 17q | Loss of 8p, 10p, 11, and 17p Gain of 4, 7q, 17, and 18q Isochromosome: 17q |
| Genomic abnormalities (most prevalent) | CTNNB1, DDX3X, SMARCA4, KMT2D, CREBBP, CDH1, MYC, APC, ARD1A, ARID2, and TP53 | PTCH1, PALB2, BRCA2, TP53, MYCN, KMT2D, SUFU, SMO, GLI2, YAP1, IDH1, and TERT | MYC, GLI1B, GFI1, OTX2, DDX31, SMARCA4, PALB2, and BRCA2 | MYCN, CDK6, SNCAIP, KDM6A, PALB2, and BRCA2 |
| Expression signature | WNT signaling | SHH signaling | MYC signature; photoreceptor/GABAergic signature | Neuronal/glutamatergic signature |
| Genetic targets | PARP, EGFR, WEE-1, and ALK | PARP, EGFR, WEE-1, and ALK | PARP, EGFR, WEE-1, and ALK | PARP, EGFR, WEE-1, and ALK |
| Epigenetic targets | HDAC and BET/BRD | SMO, HDAC, and BET/BRD | HDAC, BET/BRD, and EZH2 | HDAC, BET/BRD, EZH2, and CDK4/6 |
| Therapy Type | Title | Intervention | Patient Age | Enrollment | Phase | Status | Trial ID |
|---|---|---|---|---|---|---|---|
| Adoptive cellular therapy | HER2-specific CAR T Cell Locoregional Immunotherapy for HER2-positive Recurrent/Refractory Pediatric CNS Tumors | Biological: HER2-specific chimeric antigen receptor (CAR) T cell | 1 year to 26 years | 48 | 1 | Recruiting | NCT03500991 |
| EGFR806-specific CAR T Cell Locoregional Immunotherapy for EGFR-positive Recurrent or Refractory Pediatric CNS Tumors | Biological: EGFR806-specific chimeric antigen receptor (CAR) T cell | 1 year to 26 years | 11 | 1 | Active but not recruiting | NCT03638167 | |
| NKG2D-based CAR T-cells Immunotherapy for Patient With r/r NKG2DL+ Solid Tumors | Biological: NKG2D-based CAR T-cells | 18 years to 75 years | 3 | 1 | Recruiting | NCT05131763 | |
| Study of B7-H3-Specific CAR T Cell Locoregional Immunotherapy for Diffuse Intrinsic Pontine Glioma/Diffuse Midline Glioma and Recurrent or Refractory Pediatric Central Nervous System Tumors | Biological: SCRI-CARB7H3(s); B7H3-specific chimeric antigen receptor (CAR) T cells | 1 year to 26 years | 90 | 1 | Recruiting | NCT04185038 | |
| GD2-CAR T Cells for Pediatric Brain Tumors | Biological: GD2-CART01 (iC9-GD2-CAR T-cells) | 6 months to 30 years | 54 | 1 | Not yet recruiting | NCT05298995 | |
| Brain Tumor-Specific Immune Cells (IL13Ralpha2-CAR T Cells) for the Treatment of Leptomeningeal Glioblastoma, Ependymoma, or Medulloblastoma | Biological: IL13Ralpha2-specific hinge-optimized 41BB-co-stimulatory CAR truncated CD19-expressing autologous T lymphocytes | 18 years and older | 30 | 1 | Recruiting | NCT04661384 | |
| Expanded Natural Killer Cell Infusion in Treating Younger Patients with Recurrent/Refractory Brain Tumors | Biological: natural killer cell therapy | 0 years to 21 years | 12 | 1 | Completed | NCT02271711 | |
| Immune checkpoint inhibition | Immune Checkpoint Inhibitor Nivolumab in People with Recurrent Select Rare CNS Cancers | Drug: nivolumab | 18 years to 99 years | 180 | 2 | Recruiting | NCT03173950 |
| Pembrolizumab in Treating Younger Patients with Recurrent, Progressive, or Refractory High-Grade Gliomas, Diffuse Intrinsic Pontine Gliomas, Hypermutated Brain Tumors, Ependymoma or Medulloblastoma | Biological: pembrolizumab | 1 year to 29 years | 110 | 1 | Recruiting | NCT02359565 | |
| Durvalumab in Pediatric and Adolescent Patients | Drug: durvalumab (MEDI4736) | 1 year to 17 years | 36 | 1 | Unknown | NCT02793466 | |
| A Study to Evaluate the Safety and Efficacy of Nivolumab Monotherapy and Nivolumab in Combination with Ipilimumab in Pediatric Participants with High Grade Primary Central Nervous System (CNS) Malignancies | Biological: nivolumab Biological: ipilimumab | 6 months to 21 years | 166 | 2 | Completed | NCT03130959 | |
| Chemo-immunotherapy Using Ibrutinib Plus Indoximod for Patients with Pediatric Brain Cancer | Drug: ibrutinib Drug: indoximod Drug: cyclophosphamide Drug: etoposide | 12 years to 25 years | 37 | 1 | Recruiting | NCT05106296 | |
| Pediatric Trial of Indoximod with Chemotherapy and Radiation for Relapsed Brain Tumors or Newly Diagnosed DIPG | Drug: indoximod Radiation: partial radiation Radiation: full-dose radiation Drug: temozolomide Drug: cyclophosphamide Drug: etoposide Drug: lomustine | 3 years to 21 years | 140 | 2 | Recruiting | NCT04049669 | |
| 131I-Omburtamab, in Recurrent Medulloblastoma and Ependymoma | Drug: irinotecan Drug: temozolomide Drug: bevacizumab Drug: omburtamab I-131 Drug: liothyronine Drug: SSKI Drug: dexamethasone | Up to 21 years | 62 | 2 | Active but not recruiting | NCT04743661 | |
| 131I-omburtamab for the Treatment of Central Nervous System/Leptomeningeal Neoplasms in Children and Young Adults | Drug: 131I-omburtamab | Child, adult, and older adult | 52 | 2/3 | Available | NCT05064306 | |
| Cancer vaccination | Vaccine Immunotherapy for Recurrent Medulloblastoma and Primitive Neuroectodermal Tumor | Biological: TTRNA-xALT Biological: TTRNA-DCs | Up to 30 years | 26 | 2 | Active but not recruiting | NCT01326104 |
| Decitabine/Vaccine Therapy in Relapsed/Refractory Pediatric High Grade Gliomas/Medulloblastomas/CNS PNETs | Biological: vaccine (autologous dendritic cells) Drugs: decitabine and hiltonol | 2 years to 25 years | 1 | 1/2 | Terminated | NCT02332889 | |
| Vaccination With Dendritic Cells Loaded with Brain Tumor Stem Cells for Progressive Malignant Brain Tumor | Biological: dendritic cells Drug: imiquimod | Child, adult, and older adult | 8 | 1 | Completed | NCT01171469 | |
| Chemotherapy and Vaccine Therapy Followed by Bone Marrow or Peripheral Stem Cell Transplantation and Interleukin-2 in Treating Patients with Recurrent or Refractory Brain Cancer | Biological: aldesleukin Biological: autologous tumor cell vaccine Biological: filgrastim Biological: sargramostim Biological: therapeutic autologous lymphocytes Drug: carmustine Drug: cisplatin Drug: cyclophosphamide Drug: paclitaxel | Up to 65 years | N/A | 2 | Completed | NCT00014573 | |
| Oncolytic virotherapy | PEP-CMV in Recurrent Medulloblastoma/Malignant Glioma | Drug: PEP-CMV | 3 years to 35 years | 30 | 1 | Active but not recruiting | NCT03299309 |
| Cytomegalovirus (CMV) RNA-Pulsed Dendritic Cells for Pediatric Patients and Young Adults with WHO Grade IV Glioma, Recurrent Malignant Glioma, or Recurrent Medulloblastoma | Biological: CMV-DCs with GM-CSF Biological: Td (tetanus toxoid) | 0 years to 35 years | 11 | 1 | Completed | NCT03615404 | |
| Phase 1b Study PVSRIPO for Recurrent Malignant Glioma in Children | Biological: polio/rhinovirus recombinant (PVSRIPO) | 12 years to 21 years | 12 | 1 | Active but not recruiting | NCT03043391 | |
| Modified Measles Virus (MV-NIS) for Children and Young Adults with Recurrent Medulloblastoma or Recurrent ATRT | Biological: modified measles virus | 12 months to 39 years | 46 | 1 | Recruiting | NCT02962167 | |
| Wild-Type Reovirus in Combination with Sargramostim in Treating Younger Patients with High-Grade Relapsed or Refractory Brain Tumors | Biological: sargramostim Biological: Wild-type reovirus | 10 years to 21 years | 6 | 1 | Active but not recruiting | NCT02444546 | |
| HSV G207 in Children with Recurrent or Refractory Cerebellar Brain Tumors | Biological: G207 | 3 years to 18 years | 15 | 1 | Recruiting | NCT03911388 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rechberger, J.S.; Toll, S.A.; Vanbilloen, W.J.F.; Daniels, D.J.; Khatua, S. Exploring the Molecular Complexity of Medulloblastoma: Implications for Diagnosis and Treatment. Diagnostics 2023, 13, 2398. https://doi.org/10.3390/diagnostics13142398
Rechberger JS, Toll SA, Vanbilloen WJF, Daniels DJ, Khatua S. Exploring the Molecular Complexity of Medulloblastoma: Implications for Diagnosis and Treatment. Diagnostics. 2023; 13(14):2398. https://doi.org/10.3390/diagnostics13142398
Chicago/Turabian StyleRechberger, Julian S., Stephanie A. Toll, Wouter J. F. Vanbilloen, David J. Daniels, and Soumen Khatua. 2023. "Exploring the Molecular Complexity of Medulloblastoma: Implications for Diagnosis and Treatment" Diagnostics 13, no. 14: 2398. https://doi.org/10.3390/diagnostics13142398
APA StyleRechberger, J. S., Toll, S. A., Vanbilloen, W. J. F., Daniels, D. J., & Khatua, S. (2023). Exploring the Molecular Complexity of Medulloblastoma: Implications for Diagnosis and Treatment. Diagnostics, 13(14), 2398. https://doi.org/10.3390/diagnostics13142398