
Airway Epithelium: A Neglected but Crucial Cell Type in Asthma Pathobiology

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Need to Change the Existing Concepts of Asthma Pathogenesis (Figure 1)
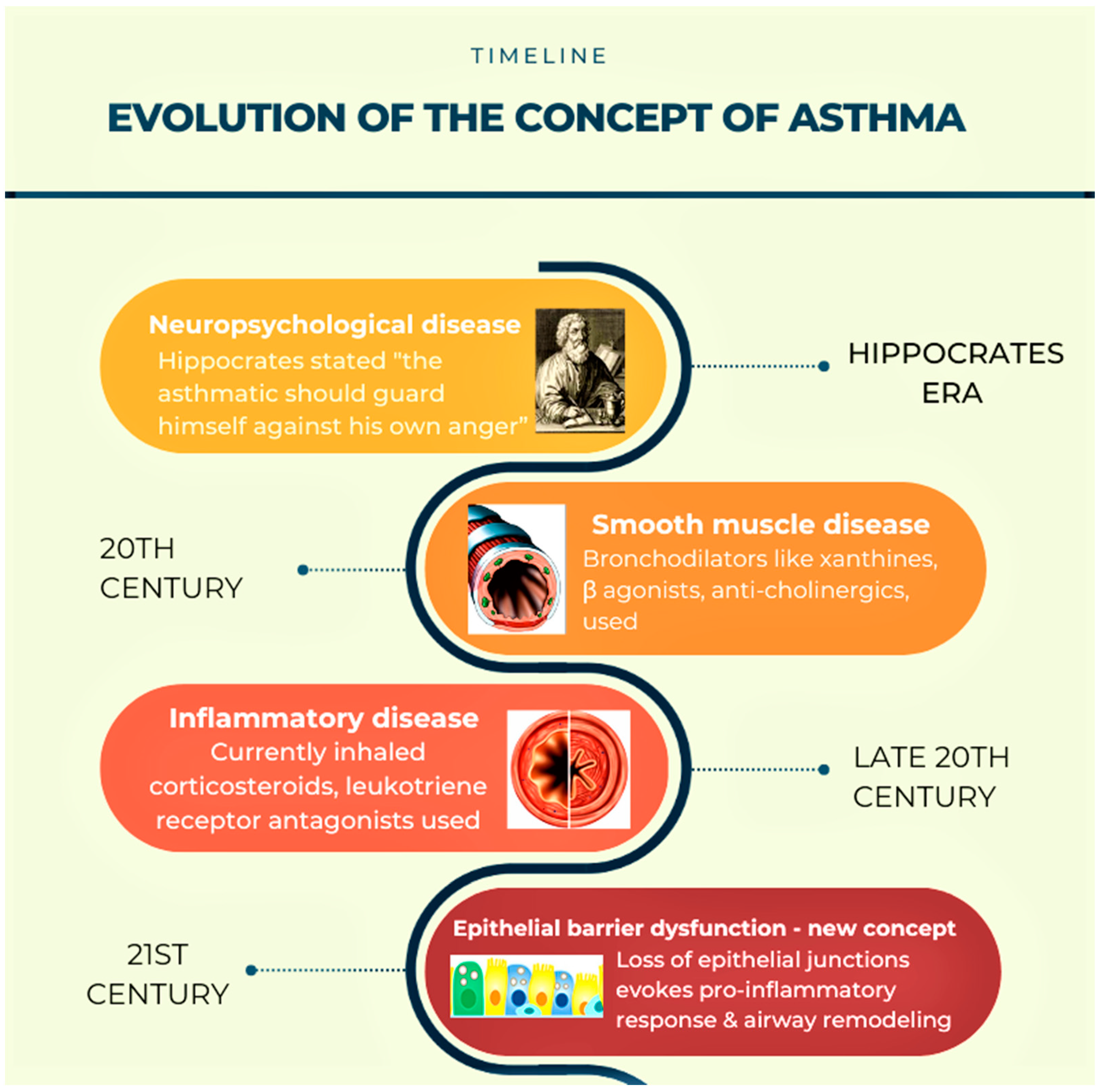
3. Barrier Function of Airway Epithelium
3.1. Importance of Epithelial Barrier in Maintaining Homeostasis
3.2. Anatomical Barrier Role of Airway Epithelium (Figure 2)

3.3. Chemical Barrier Role of Airway Epithelium
3.4. Physiological Barrier in Airway
3.5. Special Cellular Machinery in Airway Epithelial Barrier
3.6. Barrier Function of Airway Epithelium against Air Pollutants and Pathogens
4. The Victim Role of Airway Epithelium in Asthma Pathogenesis
4.1. Role of Th2 Cytokines in Airway Epithelial Barrier Dysfunction
4.2. Mitochondrial Dysfunction in Asthmatic Airway Epithelium
5. Governing/Immune Role of Airway Epithelium in Allergic Airway Inflammation
5.1. Less Dominant Role of Inflammation in Causing Epithelial Barrier Dysfunction
5.2. Less Dominant Role of Inflammation in Causing Mitochondrial Dysfunction in Airway Epithelia
5.3. Airway Epithelium Induces ILC2-Mediated Type 2 Immune Response through Alarmins (Figure 3)

5.3.1. TSLP
5.3.2. IL-33
5.3.3. IL-25
5.3.4. Discovery of ILC2 and Its Role in Asthma
5.3.5. Determinant Role of Airway Epithelium in Mounting the Type of Immune Response
5.3.6. Airway Epithelium as a Target for Innovative Treatments in Asthma
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention. 2022. Available online: www.ginasthma.org (accessed on 18 January 2023).
- Vos, T.; Lim, S.S.; Abbafati, C.; Abbas, K.M.; Abbasi, M.; Abbasifard, M.; Bhutta, Z.A. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- van Ree, R.; Hummelshøj, L.; Plantinga, M.; Poulsen, L.K.; Swindle, E. Allergic sensitization: Host-immune factors. Clin. Transl. Allergy 2014, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, D.; Lehman, H. Asthma Phenotypes as a Guide for Current and Future Biologic Therapies. Clin. Rev. Allergy Immunol. 2020, 59, 160–174. [Google Scholar] [CrossRef]
- Sze, E.; Bhalla, A.; Nair, P. Mechanisms and therapeutic strategies for non-T2 asthma. Allergy 2020, 75, 311–325. [Google Scholar] [CrossRef] [Green Version]
- Lambrecht, B.N.; Hammad, H.; Fahy, J.V. The Cytokines of Asthma. Immunity 2019, 50, 975–991. [Google Scholar] [CrossRef]
- Calvén, J.; Ax, E.; Rådinger, M. The Airway Epithelium-A Central Player in Asthma Pathogenesis. Int. J. Mol. Sci. 2020, 21, 8907. [Google Scholar] [CrossRef]
- Diamant, Z.; Boot, J.D.; Virchow, J.C. Summing up 100 years of asthma. Respir. Med. 2007, 101, 378–388. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, M.; Ray, A.; Wenzel, S.E. Evolving Concepts of Asthma. Am. J. Respir. Crit. Care Med. 2015, 192, 660–668. [Google Scholar] [CrossRef] [Green Version]
- Holgate, S.T. A brief history of asthma and its mechanisms to modern concepts of disease pathogenesis. Allergy Asthma Immunol. Res. 2010, 2, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Banno, A.; Reddy, A.T.; Lakshmi, S.P.; Reddy, R.C. Bidirectional interaction of airway epithelial remodeling and inflammation in asthma. Clin. Sci. 2020, 134, 1063–1079. [Google Scholar] [CrossRef]
- Heijink, I.H.; Kuchibhotla, V.N.S.; Roffel, M.P.; Maes, T.; Knight, D.A.; Sayers, I.; Nawijn, M.C. Epithelial cell dysfunction, a major driver of asthma development. Allergy 2020, 75, 1902–1917. [Google Scholar] [CrossRef]
- Davies, D.E. The role of the epithelium in airway remodeling in asthma. Proc. Am. Thorac. Soc. 2009, 6, 678–682. [Google Scholar] [CrossRef] [Green Version]
- Loxham, M.; Davies, D.E.; Blume, C. Epithelial function and dysfunction in asthma. Clin. Exp. Allergy 2014, 44, 1299–1313. [Google Scholar] [CrossRef]
- Sparr, E.; Millecamps, D.; Isoir, M.; Burnier, V.; Larsson, Å.; Cabane, B. Controlling the hydration of the skin though the application of occluding barrier creams. J. R. Soc. Interface 2012, 10, 20120788. [Google Scholar] [CrossRef]
- Guillot, L.; Nathan, N.; Tabary, O.; Thouvenin, G.; Le Rouzic, P.; Corvol, H.; Amselem, S.; Clement, A. Alveolar epithelial cells: Master regulators of lung homeostasis. Int. J. Biochem. Cell Biol. 2013, 45, 2568–2573. [Google Scholar] [CrossRef]
- Georas, S.N.; Rezaee, F. Epithelial barrier function: At the front line of asthma immunology and allergic airway inflammation. J. Allergy Clin. Immunol. 2014, 134, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef] [Green Version]
- Overgaard, C.E.; Mitchell, L.A.; Koval, M. Roles for claudins in alveolar epithelial barrier function. Ann. N. Y. Acad. Sci. 2012, 1257, 167–174. [Google Scholar] [CrossRef]
- Soini, Y. Claudins in lung diseases. Respir. Res. 2011, 12, 70. [Google Scholar] [CrossRef] [Green Version]
- Wittekindt, O.H. Tight junctions in pulmonary epithelia during lung inflammation. Pflug. Arch 2017, 469, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Sweerus, K.; Lachowicz-Scroggins, M.; Gordon, E.; LaFemina, M.; Huang, X.; Parikh, M.; Kanegai, C.; Fahy, J.V.; Frank, J.A. Claudin-18 deficiency is associated with airway epithelial barrier dysfunction and asthma. J. Allergy Clin. Immunol. 2017, 139, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Niessen, C.M.; Gottardi, C.J. Molecular components of the adherens junction. Biochim. Biophys. Acta 2008, 1778, 562–571. [Google Scholar] [CrossRef] [Green Version]
- Fu, R.; Jiang, X.; Li, G.; Zhu, Y.; Zhang, H. Junctional complexes in epithelial cells: Sentinels for extracellular insults and intracellular homeostasis. FEBS J. 2022, 289, 7314–7333. [Google Scholar] [CrossRef]
- Mitamura, Y.; Ogulur, I.; Pat, Y.; Rinaldi, A.O.; Ardicli, O.; Cevhertas, L.; Brüggen, M.C.; Traidl-Hoffmann, C.; Akdis, M.; Akdis, C.A. Dysregulation of the epithelial barrier by environmental and other exogenous factors. Contact Dermat. 2021, 85, 615–626. [Google Scholar] [CrossRef]
- Luissint, A.C.; Parkos, C.A.; Nusrat, A. Inflammation and the Intestinal Barrier: Leukocyte-Epithelial Cell Interactions, Cell Junction Remodeling, and Mucosal Repair. Gastroenterology 2016, 151, 616–632. [Google Scholar] [CrossRef] [Green Version]
- Perez-Moreno, M.; Davis, M.A.; Wong, E.; Pasolli, H.A.; Reynolds, A.B.; Fuchs, E. p120-catenin mediates inflammatory responses in the skin. Cell 2006, 124, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Qin, S.; Zhang, Y.; Zhang, C.; Ma, H.; Li, N.; Liu, L.; Wang, X.; Wu, R. p120 modulates LPS-induced NF-κB activation partially through RhoA in bronchial epithelial cells. BioMed Res. Int. 2014, 2014, 932340. [Google Scholar] [CrossRef] [Green Version]
- Hart, L.A.; Krishnan, V.L.; Adcock, I.M.; Barnes, P.J.; Chung, K.F. Activation and localization of transcription factor, nuclear factor-kappaB, in asthma. Am. J. Respir. Crit. Care Med. 1998, 158 Pt 1, 1585–1592. [Google Scholar] [CrossRef] [Green Version]
- Yuksel, H.; Turkeli, A. Airway epithelial barrier dysfunction in the pathogenesis and prognosis of respiratory tract diseases in childhood and adulthood. Tissue Barriers 2017, 5, e1367458. [Google Scholar] [CrossRef]
- Whitsett, J.A.; Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 2015, 16, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, M.S.; Chakraborty, S.; Veleri, S.; Kateriya, S. Mucociliary Respiratory Epithelium Integrity in Molecular Defense and Susceptibility to Pulmonary Viral Infections. Biology 2021, 10, 95. [Google Scholar] [CrossRef]
- Zhang, N.; Van Crombruggen, K.; Gevaert, E.; Bachert, C. Barrier function of the nasal mucosa in health and type-2 biased airway diseases. Allergy 2016, 71, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Leiva-Juárez, M.M.; Kolls, J.K.; Evans, S.E. Lung epithelial cells: Therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol. 2018, 11, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Karp, P.H.; Brody, S.L.; Pierce, R.A.; Welsh, M.J.; Holtzman, M.J.; Ben-Shahar, Y. Chemosensory functions for pulmonary neuroendocrine cells. Am. J. Respir. Cell Mol. Biol. 2014, 50, 637–646. [Google Scholar] [CrossRef] [Green Version]
- Gu, Q.; Lee, L.Y. Neurophysiology: Neural Control of Airway Smooth Muscle. In Encyclopedia of Respiratory Medicine, Four-Volume Set 2006; Laurent, G.J., Shapiro, S.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; pp. 138–145. [Google Scholar] [CrossRef]
- Cutz, E.; Yeger, H.; Pan, J. Pulmonary neuroendocrine cell system in pediatric lung disease-recent advances. Pediatr. Dev. Pathol. Off. J. Soc. Pediatr. Pathol. Paediatr. Pathol. Soc. 2007, 10, 419–435. [Google Scholar] [CrossRef]
- Song, H.; Yao, E.; Lin, C.; Gacayan, R.; Chen, M.H.; Chuang, P.T. Functional characterization of pulmonary neuroendocrine cells in lung development, injury, and tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 17531–17536. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, M.; Furukawa, K.T.; Morimoto, M. Pulmonary neuroendocrine cells: Physiology, tissue homeostasis and disease. Dis. Model Mech. 2020, 13, dmm046920. [Google Scholar] [CrossRef]
- Gu, Q.; Lee, l.-y. Neural Control of Airway Smooth Muscle. World J. Anesthesiol. 2020. [Google Scholar] [CrossRef]
- Sui, P.; Wiesner, D.L.; Xu, J.; Zhang, Y.; Lee, J.; Van Dyken, S.; Lashua, A.; Yu, C.; Klein, B.S.; Locksley, R.M.; et al. Pulmonary neuroendocrine cells amplify allergic asthma responses. Science 2018, 360, eaan8546. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Xu, J.; Jiang, C.; Lu, S. Neuro-Immune Regulation in Inflammation and Airway Remodeling of Allergic Asthma. Front. Immunol. 2022, 13, 894047. [Google Scholar] [CrossRef]
- Schuller, H.M.; Plummer, H.K., 3rd; Jull, B.A. Receptor-mediated effects of nicotine and its nitrosated derivative NNK on pulmonary neuroendocrine cells. Anat. Record. Part A Discov. Mol. Cell. Evol. Biol. 2003, 270, 51–58. [Google Scholar] [CrossRef]
- Laucho-Contreras, M.E.; Polverino, F.; Gupta, K.; Taylor, K.L.; Kelly, E.; Pinto-Plata, V.; Divo, M.; Ashfaq, N.; Petersen, H.; Stripp, B.; et al. Protective role for club cell secretory protein-16 (CC16) in the development of COPD. Eur. Respir. J. 2015, 45, 1544–1556. [Google Scholar] [CrossRef] [Green Version]
- Hellings, P.W.; Steelant, B. Epithelial barriers in allergy and asthma. J. Allergy Clin. Immunol. 2020, 145, 1499–1509. [Google Scholar] [CrossRef]
- Gohy, S.; Carlier, F.M.; Fregimilicka, C.; Detry, B.; Lecocq, M.; Ladjemi, M.Z.; Verleden, S.; Hoton, D.; Weynand, B.; Bouzin, C.; et al. Altered generation of ciliated cells in chronic obstructive pulmonary disease. Sci. Rep. 2019, 9, 17963. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.Y.; Lee, Y.S.; Min, K.H.; Hur, G.Y.; Lee, S.Y.; Kang, K.H.; Rhee, C.K.; Park, S.J.; Shim, J.J. Decreased serum club cell secretory protein in asthma and chronic obstructive pulmonary disease overlap: A pilot study. Int. J. Chronic Obstr. Pulm. Dis. 2018, 13, 3411–3417. [Google Scholar] [CrossRef] [Green Version]
- Verckist, L.; Pintelon, I.; Timmermans, J.P.; Brouns, I.; Adriaensen, D. Selective activation and proliferation of a quiescent stem cell population in the neuroepithelial body microenvironment. Respir. Res. 2018, 19, 207. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Driskell, R.R.; Engelhardt, J.F. Stem cells in the lung. Methods Enzymol. 2006, 419, 285–321. [Google Scholar] [CrossRef]
- Guarnieri, M.; Balmes, J.R. Outdoor air pollution and asthma. Lancet 2014, 383, 1581–1592. [Google Scholar] [CrossRef] [Green Version]
- Toskala, E.; Kennedy, D.W. Asthma risk factors. Int. Forum. Allergy Rhinol. 2015, 5 (Suppl. 1), S11–S16. [Google Scholar] [CrossRef]
- Tiotiu, A.I.; Novakova, P.; Nedeva, D.; Chong-Neto, H.J.; Novakova, S.; Steiropoulos, P.; Kowal, K. Impact of Air Pollution on Asthma Outcomes. Int. J. Environ. Res. Public Health 2020, 17, 6212. [Google Scholar] [CrossRef]
- Tatsuta, M.; Kan, O.K.; Ishii, Y.; Yamamoto, N.; Ogawa, T.; Fukuyama, S.; Ogawa, A.; Fujita, A.; Nakanishi, Y.; Matsumoto, K. Effects of cigarette smoke on barrier function and tight junction proteins in the bronchial epithelium: Protective role of cathelicidin LL-37. Respir. Res. 2019, 20, 251. [Google Scholar] [CrossRef] [Green Version]
- Aghapour, M.; Ubags, N.D.; Bruder, D.; Hiemstra, P.S.; Sidhaye, V.; Rezaee, F.; Heijink, I.H. Role of air pollutants in airway epithelial barrier dysfunction in asthma and COPD. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2022, 31, 210112. [Google Scholar] [CrossRef]
- Wang, F.; Liu, J.; Zeng, H. Interactions of particulate matter and pulmonary surfactant: Implications for human health. Adv. Colloid Interface Sci. 2020, 284, 102244. [Google Scholar] [CrossRef]
- Tsui, H.C.; Ronsmans, S.; Hoet, P.H.M.; Nemery, B.; Vanoirbeek, J.A.J. Occupational Asthma Caused by Low-Molecular-Weight Chemicals Associated With Contact Dermatitis: A Retrospective Study. J. Allergy Clin. Immunology Pract. 2022, 10, 2346–2354. [Google Scholar] [CrossRef]
- Douwes, J.; Pearce, N. Epidemiology of Respiratory Allergies and Asthma. In Handbook of Epidemiology; Springer: New York, NY, USA, 2014; pp. 2263–2319. [Google Scholar] [CrossRef]
- Meca, O.; Cruz, M.J.; Sánchez-Ortiz, M.; González-Barcala, F.J.; Ojanguren, I.; Munoz, X. Do Low Molecular Weight Agents Cause More Severe Asthma than High Molecular Weight Agents? PLoS ONE 2016, 11, e0156141. [Google Scholar] [CrossRef] [Green Version]
- Reid, M.J.; Moss, R.B.; Hsu, Y.P.; Kwasnicki, J.M.; Commerford, T.M.; Nelson, B.L. Seasonal asthma in northern California: Allergic causes and efficacy of immunotherapy. J. Allergy Clin. Immunol. 1986, 78 Pt 1, 590–600. [Google Scholar] [CrossRef]
- León, B.; Ballesteros-Tato, A. Modulating Th2 Cell Immunity for the Treatment of Asthma. Front. Immunol. 2021, 12, 637948. [Google Scholar] [CrossRef]
- Barnes, P.J. Th2 cytokines and asthma: An introduction. Respir. Res. 2001, 2, 64. [Google Scholar] [CrossRef]
- Saatian, B.; Rezaee, F.; Desando, S.; Emo, J.; Chapman, T.; Knowlden, S.; Georas, S.N. Interleukin-4 and interleukin-13 cause barrier dysfunction in human airway epithelial cells. Tissue Barriers 2013, 1, e24333. [Google Scholar] [CrossRef] [Green Version]
- Burgess, J.K.; Jonker, M.R.; Berg, M.; Ten Hacken, N.T.; Meyer, K.B.; van den Berge, M.; Nawijn, M.C.; Heijink, I.H. Periostin: Contributor to abnormal airway epithelial function in asthma? Eur. Respir. J. 2021, 57, 2001286. [Google Scholar] [CrossRef]
- Schmidt, H.; Braubach, P.; Schilpp, C.; Lochbaum, R.; Neuland, K.; Thompson, K.; Jonigk, D.; Frick, M.; Dietl, P.; Wittekindt, O.H. IL-13 Impairs Tight Junctions in Airway Epithelia. Int. J. Mol. Sci. 2019, 20, 3222. [Google Scholar] [CrossRef] [Green Version]
- Amin, K.; Janson, C.; Bystrom, J. Role of eosinophil granulocytes in allergic airway inflammation endotypes. Scand. J. Immunol. 2016, 84, 75–85. [Google Scholar] [CrossRef]
- Konrádová, V.; Čopová, C.; Suková, B.; Houštěk, J. Ultrastructure of the bronchial epithelium in three children with asthma. Pediatr. Pulmonol. 1985, 1, 182–187. [Google Scholar] [CrossRef]
- Mabalirajan, U.; Dinda, A.K.; Kumar, S.; Roshan, R.; Gupta, P.; Sharma, S.K.; Ghosh, B. Mitochondrial structural changes and dysfunction are associated with experimental allergic asthma. J. Immunol. 2008, 181, 3540–3548. [Google Scholar] [CrossRef] [Green Version]
- Heydeck, D.; Thomas, L.; Schnurr, K.; Trebus, F.; Thierfelder, W.E.; Ihle, J.N.; Kühn, H. Interleukin-4 and -13 induce upregulation of the murine macrophage 12/15-lipoxygenase activity: Evidence for the involvement of transcription factor STAT6. Blood 1998, 92, 2503–2510. [Google Scholar] [CrossRef]
- Rapoport, S.M.; Schewe, T.; Wiesner, R.; Halangk, W.; Ludwig, P.; JanickeHöhne, M.; Tannert, C.; Hiebsch, C.; Klatt, D. The lipoxygenase of reticulocytes. Purification, characterization and biological dynamics of the lipoxygenase; its identity with the 96 respiratory inhibitors of the reticulocyte. Eur. J. Biochem. 1979, 96, 545–561. [Google Scholar] [CrossRef]
- Watson, A.; Doherty, F.J. Calcium promotes membrane association of reticulocyte 15-lipoxygenase. Biochem. J. 1994, 298, 377–383. [Google Scholar] [CrossRef]
- van Leyen, K.; Duvoisin, R.M.; Engelhardt, H.; Wiedmann, M. A function for lipoxygenase in programmed organelle degradation. Nature 1998, 395, 392–395. [Google Scholar] [CrossRef]
- Chu, H.W.; Balzar, S.; Westcott, J.Y.; Trudeau, J.B.; Sun, Y.; Conrad, D.J.; Wenzel, S.E. Expression and activation of 15-lipoxygenase pathway in severe asthma: Relationship to eosinophilic phenotype and collagen deposition. Clin. Exp. Allergy 2002, 32, 1558–1565. [Google Scholar] [CrossRef]
- Chanez, P.; Bonnans, C.; Chavis, C.; Vachier, I. 15-lipoxygenase: A Janus enzyme? Am. J. Respir. Cell Mol. Biol. 2002, 27, 655–658. [Google Scholar] [CrossRef]
- Mabalirajan, U.; Rehman, R.; Ahmad, T.; Kumar, S.; Leishangthem, G.D.; Singh, S.; Dinda, A.K.; Biswal, S.; Agrawal, A.; Ghosh, B. 12/15-lipoxygenase expressed in non-epithelial cells causes airway epithelial injury in asthma. Sci. Rep. 2013, 3, 1540. [Google Scholar] [CrossRef] [Green Version]
- Mabalirajan, U.; Rehman, R.; Ahmad, T.; Kumar, S.; Singh, S.; Leishangthem, G.D.; Aich, J.; Kumar, M.; Khanna, K.; Singh, V.P.; et al. Linoleic acid metabolite drives severe asthma by causing airway epithelial injury. Sci. Rep. 2013, 3, 1349. [Google Scholar] [CrossRef] [Green Version]
- Mabalirajan, U.; Dinda, A.K.; Sharma, S.K.; Ghosh, B. Esculetin restores mitochondrial dysfunction and reduces allergic asthma features in experimental murine model. J. Immunol. 2009, 183, 2059–2067. [Google Scholar] [CrossRef] [Green Version]
- Mabalirajan, U.; Aich, J.; Leishangthem, G.D.; Sharma, S.K.; Dinda, A.K.; Ghosh, B. Effects of vitamin E on mitochondrial dysfunction and asthma features in an experimental allergic murine model. J. Appl. Physiol. 2009, 107, 1285–1292. [Google Scholar] [CrossRef]
- Mabalirajan, U.; Ahmad, T.; Rehman, R.; Leishangthem, G.D.; Dinda, A.K.; Agrawal, A.; Ghosh, B.; Sharma, S.K. Baicalein reduces airway injury in allergen and IL-13 induced airway inflammation. PLoS ONE 2013, 8, e62916. [Google Scholar] [CrossRef] [Green Version]
- Cocco, M.P.; White, E.; Xiao, S.; Hu, D.; Mak, A.; Sleiman, P.; Yang, M.; Bobbitt, K.R.; Gui, H.; Levin, A.M.; et al. Asthma and its relationship to mitochondrial copy number: Results from the Asthma Translational Genomics Collaborative (ATGC) of the Trans-Omics for Precision Medicine (TOPMed) program. PLoS ONE 2020, 15, e0242364. [Google Scholar] [CrossRef]
- Loxham, M.; Davies, D.E. Phenotypic and genetic aspects of epithelial barrier function in asthmatic patients. J. Allergy Clin. Immunol. 2017, 139, 1736–1751. [Google Scholar] [CrossRef] [Green Version]
- Koppelman, G.H.; Meyers, D.A.; Howard, T.D.; Zheng, S.L.; Hawkins, G.A.; Ampleford, E.J.; Xu, J.; Koning, H.; Bruinenberg, M.; Nolte, I.M.; et al. Identification of PCDH1 as a novel susceptibility gene for bronchial hyperresponsiveness. Am. J. Respir. Crit. Care Med. 2009, 180, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Bønnelykke, K.; Sleiman, P.; Nielsen, K.; Kreiner-Møller, E.; Mercader, J.M.; Belgrave, D.; den Dekker, H.T.; Husby, A.; Sevelsted, A.; Faura-Tellez, G.; et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat. Genet. 2014, 46, 51–55. [Google Scholar] [CrossRef]
- Basnet, S.; Bochkov, Y.A.; Brockman-Schneider, R.A.; Kuipers, I.; Aesif, S.W.; Jackson, D.J.; Lemanske RFJr Ober, C.; Palmenberg, A.C.; Gern, J.E. CDHR3 Asthma-Risk Genotype Affects Susceptibility of Airway Epithelium to Rhinovirus C Infections. Am. J. Respir. Cell Mol. Biol. 2019, 61, 450–458. [Google Scholar] [CrossRef]
- Runswick, S.; Mitchell, T.; Davies, P.; Robinson, C.; Garrod, D.R. Pollen proteolytic enzymes degrade tight junctions. Respirology 2007, 12, 834–842. [Google Scholar] [CrossRef]
- Guryanova, S.V.; Gigani, O.B.; Gudima, G.O.; Kataeva, A.M.; Kolesnikova, N.V. Dual Effect of Low-Molecular-Weight Bioregulators of Bacterial Origin in Experimental Model of Asthma. Life 2022, 12, 192. [Google Scholar] [CrossRef]
- Aguilera-Aguirre, L.; Bacsi, A.; Saavedra-Molina, A.; Kurosky, A.; Sur, S.; Boldogh, I. Mitochondrial dysfunction increases allergic airway inflammation. J. Immunol. 2009, 183, 5379–5387. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [CrossRef]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow–derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759. [Google Scholar] [CrossRef] [Green Version]
- Barton, J.L.; Berg, T.; Didon, L.; Nord, M. The pattern recognition receptor Nod1 activates CCAAT/enhancer binding protein beta signalling in lung epithelial cells. Eur. Respir. J. 2007, 30, 214–222. [Google Scholar] [CrossRef]
- Farkas, L.; Stoelcker, B.; Jentsch, N.; Heitzer, S.; Pfeifer, M.; Schulz, C. Muramyldipeptide modulates CXCL-8 release of BEAS-2B cells via NOD2. Scand. J. Immunol. 2008, 68, 315–322. [Google Scholar] [CrossRef]
- Kauffman, H.F. Innate immune responses to environmental allergens. Clin. Rev. Allergy Immunol. 2006, 30, 129–140. [Google Scholar] [CrossRef]
- Kato, A.; Favoreto, S.; Avila, P.C.; Schleimer, R.P. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J. Immunol. 2007, 179, 1080–1087. [Google Scholar] [CrossRef]
- Hammad, H.; Lambrecht, B.N. Dendritic cells and epithelial cells: Linking innate and adaptive immunity in asthma. Nat. Reviews Immunol. 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef]
- Licona-Limón, P.; Kim, L.K.; Palm, N.W.; Flavell, R.A. TH2, allergy and group 2 innate lymphoid cells. Nat. Immunol. 2013, 14, 536–542. [Google Scholar] [CrossRef]
- Smithgall, M.D.; Comeau, M.R.; Yoon, B.R.; Kaufman, D.; Armitage, R.; Smith, D.E. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int. Immunol. 2008, 20, 1019–1030. [Google Scholar] [CrossRef]
- Gauvreau, G.M.; Bergeron, C.; Boulet, L.P.; Cockcroft, D.W.; Côté, A.; Davis, B.E.; Leigh, R.; Myers, I.; O’Byrne, P.M.; Sehmi, R. Sounding the alarmins-The role of alarmin cytokines in asthma. Allergy 2022, 78, 402–417. [Google Scholar] [CrossRef]
- Whetstone, C.E.; Ranjbar, M.; Omer, H.; Cusack, R.P.; Gauvreau, G.M. The Role of Airway Epithelial Cell Alarmins in Asthma. Cells 2022, 11, 1105. [Google Scholar] [CrossRef]
- Lee, H.C.; Headley, M.B.; Loo, Y.M.; Berlin, A.; Gale, M.; Debley, J.S., Jr.; Lukacs, N.W.; Ziegler, S.F. Thymic stromal lymphopoietin is induced by respiratory syncytial virus-infected airway epithelial cells and promotes a type 2 response to infection. J. Allergy Clin. Immunol. 2012, 130, 1187–1196. [Google Scholar] [CrossRef] [Green Version]
- Allakhverdi, Z.; Comeau, M.R.; Jessup, H.K.; Yoon, B.R.; Brewer, A.; Chartier, S.; Paquette, N.; Ziegler, S.F.; Sarfati, M.; Delespesse, G. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J. Exp. Med. 2007, 204, 253–258. [Google Scholar] [CrossRef]
- Kamijo, S.; Takeda, H.; Tokura, T.; Suzuki, M.; Inui, K.; Hara, M.; Matsuda, H.; Matsuda, A.; Oboki, K.; Ohno, T.; et al. IL-33-mediated innate response and adaptive immune cells contribute to maximum responses of protease allergen-induced allergic airway inflammation. J. Immunol. (Baltim. Md. 1950) 2013, 190, 4489–4499. [Google Scholar] [CrossRef] [Green Version]
- Jackson, D.J.; Makrinioti, H.; Rana, B.M.; Shamji, B.W.; Trujillo-Torralbo, M.B.; Footitt, J.; Del-Rosario, J.; Telcian, A.G.; Nikonova, A.; Zhu, J.; et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am. J. Respir. Crit. Care Med. 2014, 190, 1373–1382. [Google Scholar] [CrossRef] [Green Version]
- Ballantyne, S.J.; Barlow, J.L.; Jolin, H.E.; Nath, P.; Williams, A.S.; Chung, K.F.; Sturton, G.; Wong, S.H.; McKenzie, A.N. Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J. Allergy Clin. Immunol. 2007, 120, 1324–1331. [Google Scholar] [CrossRef]
- Cheng, D.; Xue, Z.; Yi, L.; Shi, H.; Zhang, K.; Huo, X.; Bonser, L.R.; Zhao, J.; Xu, Y.; Erle, D.J.; et al. Epithelial interleukin-25 is a key mediator in Th2-high, corticosteroid-responsive asthma. Am. J. Respir. Crit. Care Med. 2014, 190, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Fort, M.M.; Cheung, J.; Yen, D.; Li, J.; Zurawski, S.M.; Lo, S.; Menon, S.; Clifford, T.; Hunte, B.; Lesley, R.; et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity 2001, 15, 985–995. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Yoshimoto, T.; Yasuda, K.; Futatsugi-Yumikura, S.; Morimoto, M.; Hayashi, N.; Hoshino, T.; Fujimoto, J.; Nakanishi, K. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int. Immunol. 2008, 20, 791–800. [Google Scholar] [CrossRef] [Green Version]
- Fallon, P.G.; Ballantyne, S.J.; Mangan, N.E.; Barlow, J.L.; Dasvarma, A.; Hewett, D.R.; McIlgorm, A.; Jolin, H.E.; McKenzie, A.N. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J. Exp. Med. 2006, 203, 1105–1116. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [Green Version]
- Halim, T.Y.; Krauss, R.H.; Sun, A.C.; Takei, F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity 2012, 36, 451–463. [Google Scholar] [CrossRef] [Green Version]
- Klein Wolterink, R.G.; Kleinjan, A.; van Nimwegen, M.; Bergen, I.; de Bruijn, M.; Levani, Y.; Hendriks, R.W. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur. J. Immunol. 2012, 42, 1106–1116. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.E.; Maizels, R.M. Diversity and dialogue in immunity to helminths. Nat. Rev. Immunol. 2011, 11, 375–388. [Google Scholar] [CrossRef]
- Rigas, D.; Lewis, G.; Aron, J.L.; Wang, B.; Banie, H.; Sankaranarayanan, I.; Galle-Treger, L.; Maazi, H.; Lo, R.; Freeman, G.J.; et al. Type 2 innate lymphoid cell suppression by regulatory T cells attenuates airway hyperreactivity and requires inducible T-cell costimulator-inducible T-cell costimulator ligand interaction. J. Allergy Clin. Immunol. 2017, 139, 1468–1477. [Google Scholar] [CrossRef] [Green Version]
- Thorey, I.S.; Roth, J.; Regenbogen, J.; Halle, J.P.; Bittner, M.; Vogl, T.; Kaesler, S.; Bugnon, P.; Reitmaier, B.; Durka, S.; et al. The Ca2+-binding proteins S100A8 and S100A9 are encoded by novel injury-regulated genes. J. Biol. Chem. 2001, 276, 35818–35825. [Google Scholar] [CrossRef] [Green Version]
- Ryckman, C.; Vandal, K.; Rouleau, P.; Talbot, M.; Tessier, P.A. Proinflammatory activities of S100: Proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J. Immunol. 2003, 170, 3233–3242. [Google Scholar] [CrossRef] [Green Version]
- Nenci, A.; Becker, C.; Wullaert, A.; Gareus, R.; van Loo, G.; Danese, S.; Huth, M.; Nikolaev, A.; Neufert, C.; Madison, B.; et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 2007, 446, 557–561. [Google Scholar] [CrossRef]
- Swamy, M.; Jamora, C.; Havran, W.; Hayday, A. Epithelial decision makers: In search of the ‘epimmunome’. Nat. Immunol. 2010, 11, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Kabata, H.; Moro, K.; Fukunaga, K.; Suzuki, Y.; Miyata, J.; Masaki, K.; Betsuyaku, T.; Koyasu, S.; Asano, K. Thymic stromal lymphopoietin induces corticosteroid resistance in natural helper cells during airway inflammation. Nat. Commun. 2013, 4, 2675. [Google Scholar] [CrossRef] [Green Version]
- Holgate, S.T.; Polosa, R. The mechanisms, diagnosis, and management of severe asthma in adults. Lancet 2006, 368, 780–793. [Google Scholar] [CrossRef]
- Marshall, C.L.; Hasani, K.; Mookherjee, N. Immunobiology of Steroid-Unresponsive Severe Asthma. Front. Allergy 2021, 2, 718267. [Google Scholar] [CrossRef]
- Jonckheere, A.-C.; Seys, S.F.; Steelant, B.; Decaesteker, T.; Dekoster, K.; Cremer, J.; Dilissen, E.; Schols, D.; Iwakura, Y.; Vande Velde, G.; et al. Innate Lymphoid Cells Are Required to Induce Airway Hyperreactivity in a Murine Neutrophilic Asthma Model. Front. Immunol. 2022, 13, 849155. [Google Scholar] [CrossRef]
- Gauvreau, G.M.; O’Byrne, P.M.; Boulet, L.P.; Wang, Y.; Cockcroft, D.; Bigler, J.; FitzGerald, J.M.; Boedigheimer, M.; Davis, B.E.; Dias, C.; et al. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. N. Engl. J. Med. 2014, 370, 2102–2110. [Google Scholar] [CrossRef]
- Menzies-Gow, A.; Corren, J.; Bourdin, A.; Chupp, G.; Israel, E.; Wechsler, M.E.; Brightling, C.E.; Griffiths, J.M.; Hellqvist, Å.; Bowen, K.; et al. Tezepelumab in Adults and Adolescents with Severe, Uncontrolled Asthma. N. Engl. J. Med. 2021, 384, 1800–1809. [Google Scholar] [CrossRef]
- Porsbjerg, C.M.; Sverrild, A.; Lloyd, C.M.; Menzies-Gow, A.N.; Bel, E.H. Anti-alarmins in asthma: Targeting the airway epithelium with next-generation biologics. Eur. Respir. J. 2020, 56, 2000260. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.; Dutta, J.; Ray, A.; Karmakar, A.; Mabalirajan, U. Airway Epithelium: A Neglected but Crucial Cell Type in Asthma Pathobiology. Diagnostics 2023, 13, 808. https://doi.org/10.3390/diagnostics13040808
Singh S, Dutta J, Ray A, Karmakar A, Mabalirajan U. Airway Epithelium: A Neglected but Crucial Cell Type in Asthma Pathobiology. Diagnostics. 2023; 13(4):808. https://doi.org/10.3390/diagnostics13040808
Chicago/Turabian StyleSingh, Sabita, Joytri Dutta, Archita Ray, Atmaja Karmakar, and Ulaganathan Mabalirajan. 2023. "Airway Epithelium: A Neglected but Crucial Cell Type in Asthma Pathobiology" Diagnostics 13, no. 4: 808. https://doi.org/10.3390/diagnostics13040808
APA StyleSingh, S., Dutta, J., Ray, A., Karmakar, A., & Mabalirajan, U. (2023). Airway Epithelium: A Neglected but Crucial Cell Type in Asthma Pathobiology. Diagnostics, 13(4), 808. https://doi.org/10.3390/diagnostics13040808