
Therapy-Acquired Clonal Mutations in Thiopurine Drug-Response Genes Drive Majority of Early Relapses in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia
 , , and
, , and
Abstract
:1. Introduction
2. Materials & Methods
2.1. Patients and Samples
2.2. Primary Genetic Subtype Analysis
2.3. Deep Sequencing for Secondary Genetic Abnormalities
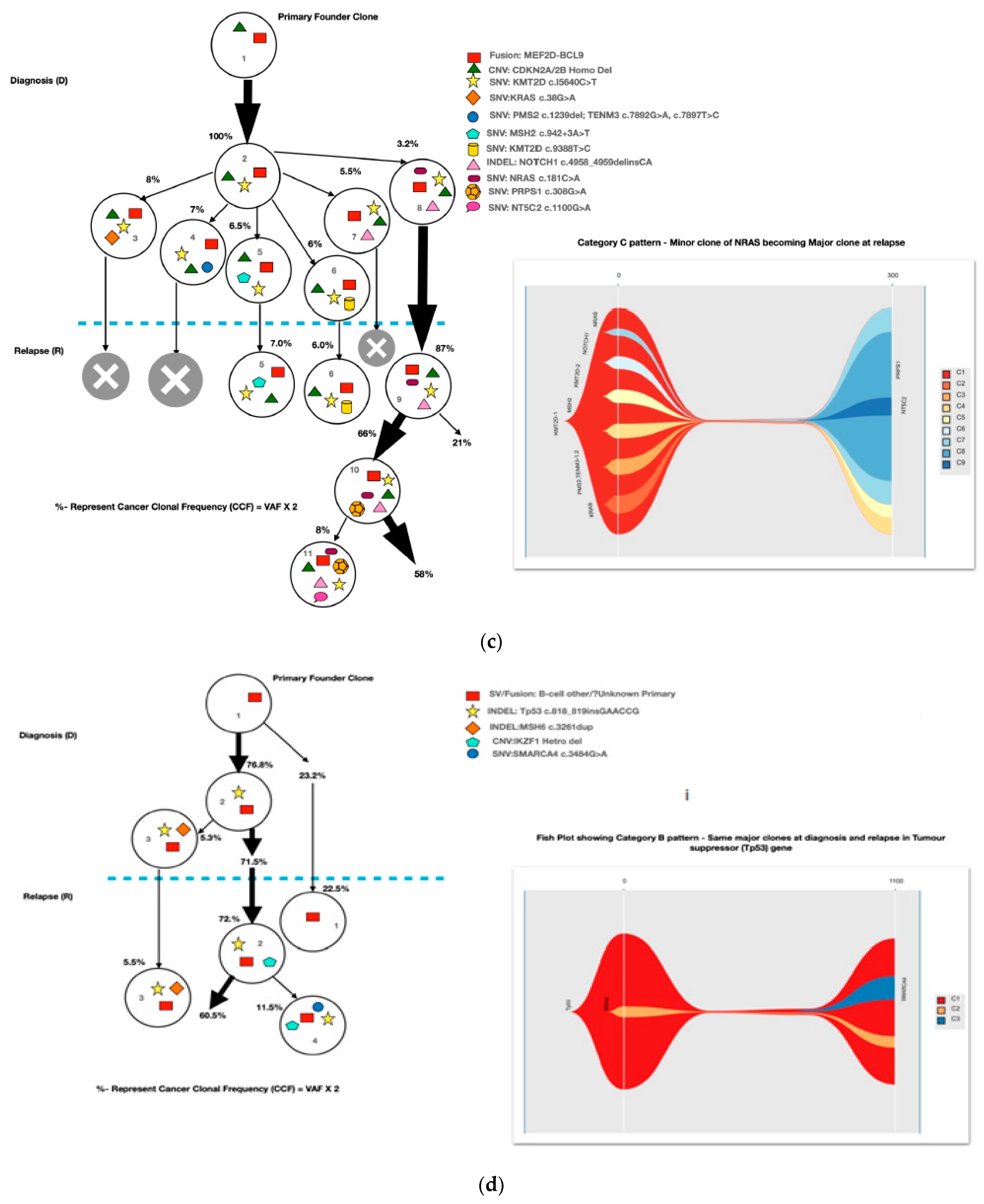
2.4. Bioinformatic Definitions and Clonal Evolution Patterns and Plots
3. Results
3.1. Primary Genetic Abnormality Data
3.2. Deep Sequencing Data for Secondary Genetic Abnormalities of SNV/INDELs and CNVs in Paired Cases and Non-Relapsed Control Arm Samples
3.3. Clonal Evolution Pattern
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pui, C.H.; Evans, W.E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med 2006, 354, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Lu, X.; Devidas, M.; Camitta, B.M.; Gaynon, P.S.; Winick, N.J.; Reaman, G.H.; Carroll, W.L. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: A report from the children’s oncology group. J. Clin. Oncol 2012, 30, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, M.; Ohara, A.; Manabe, A.; Kumagai, M.; Shimada, H.; Kikuchi, A.; Mori, T.; Saito, M.; Akiyama, M.; Fukushima, T.; et al. Long-term results of Tokyo children’s cancer study group trials for childhood acute lymphoblastic leukemia, 1984-1999. Leukemia 2010, 24, 383–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, K.P.; Marwaha, R.K.; Trehan, A.; Bansal, D. Survival outcome in childhood ALL: Experience from a tertiary care centre in North India. Pediatr. Blood Cancer 2009, 53, 168–173. [Google Scholar] [CrossRef]
- Lins, M.M.; Santos, M.O.; de Albuquerque, M.; de Castro, C.C.L.; Mello, M.J.G.; de Camargo, B. Incidence and survival of childhood leukemia in Recife, Brazil: A population-based analysis. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef]
- Wiangnon, S.; Jetsrisuparb, A.; Komvilaisak, P.; Suwanrungruang, K. Childhood cancer incidence and survival 1985-2009, Khon Kaen, Thailand. Asian Pacific J. Cancer Prev. 2014, 15, 7989–7993. [Google Scholar] [CrossRef] [Green Version]
- Inaba, H.; Greaves, M.; Mullighan, C.G. Acute lymphoblastic leukaemia. Lancet 2013, 381, 1943–1955. [Google Scholar] [CrossRef] [Green Version]
- Hefazi, M.; Litzow, M.R. Recent advances in the biology and treatment of B-cell acute lymphoblastic leukemia. Blood Lymphat. Cancer Targets Ther. 2018, 8, 47–61. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Manabe, A. Treatment and biology of pediatric acute lymphoblastic leukemia. Pediatr. Int. 2018, 60, 4–12. [Google Scholar] [CrossRef]
- Huang, F.L.; Liao, E.C.; Li, C.L.; Yen, C.Y.; Yu, S.J. Pathogenesis of pediatric B-cell acute lymphoblastic leukemia: Molecular pathways and disease treatments (Review). Oncol. Lett. 2020, 20, 448–454. [Google Scholar] [CrossRef]
- Martin, A.; Morgan, E.; Hijiya, N. Relapsed or refractory pediatric acute lymphoblastic leukemia: Current and emerging treatments. Pediatr. Drugs 2012, 14, 377–387. [Google Scholar] [CrossRef]
- Kaspers, G.J.L.; Veerman, A.J.P.; Pieters, R.; Van Zantwijk, C.H.; Smets, L.A.; Van Wering, E.R.; Van Der Does-Van Den Berg, A. In vitro cellular drug resistance and prognosis in newly diagnosed childhood acute lymphoblastic leukemia. Blood 1997, 90, 2723–2729. [Google Scholar] [CrossRef]
- Bhojwani, D.; Kang, H.; Moskowitz, N.P.; Min, D.-J.; Lee, H.; Potter, J.W.; Davidson, G.; Willman, C.L.; Borowitz, M.J.; Belitskaya-Levy, I.; et al. Biologic pathways associated with relapse in childhood acute lymphoblastic leukemia: A Children’s Oncology Group study. Blood 2006, 108, 711–717. [Google Scholar] [CrossRef] [Green Version]
- Hogan, L.E.; Meyer, J.A.; Yang, J.; Wang, J.; Wong, N.; Yang, W.; Condos, G.; Hunger, S.P.; Raetz, E.; Saffery, R.; et al. Integrated genomic analysis of relapsed childhood acute lymphoblastic leukemia reveals therapeutic strategies. Blood 2011, 118, 5218–5226. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Bhojwani, D.; Yang, W.; Cai, X.; Stocco, G.; Crews, K.; Wang, J.; Morrison, D.; Devidas, M.; Hunger, S.P.; et al. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood 2008, 112, 4178–4183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullighan, C.G.; Phillips, L.A.; Su, X.; Ma, J.; Miller, C.B.; Shurtleff, S.A.; Downing, J.R. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 2008, 322, 1377–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Brady, S.W.; Ma, X.; Shen, S.; Zhang, Y.; Li, Y.; Szlachta, K.; Dong, L.; Liu, Y.; Yang, F.; et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2020, 135, 41–55. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Zhang, J.; Kasper, L.H.; Lerach, S.; Payne-Turner, D.; Phillips, L.A.; Heatley, S.L.; Holmfeldt, L.; Collins-Underwood, J.R.; Ma, J.; et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011, 471, 235–239. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Li, H.; Bai, Y.; Kirschner-Schwabe, R.; Yang, J.J.; Chen, Y.; Lu, G.; Tzoneva, G.; Ma, X.; Wu, T.; et al. Negative feedback- defective PRPS1 mutants drive thiopurine resistance in relapsed childhood. Nat. Med. 2015, 21, 563–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, J.A.; Wang, J.; Hogan, L.E.; Yang, J.J.; Dandekar, S.; Patel, J.P.; Tang, Z.; Zumbo, P.; Li, S.; Zavadil, J.; et al. Relapse- specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 290–294. [Google Scholar] [CrossRef] [Green Version]
- Tzoneva, G.; Perez-Garcia, A.; Carpenter, Z.; Khiabanian, H.; Tosello, V.; Allegretta, M.; Paietta, E.; Racevskis, J.; Rowe, J.M.; Tallman, M.S.; et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed. Nat. Med. 2013, 19, 368–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Edmonson, M.; Yergeau, D.; Muzny, D.M.; Hampton, O.A.; Rusch, M.; Song, G.; Easton, J.; Harvey, R.C.; Wheeler, D.A.; et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat. Commun. 2015, 6, 6604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshima, K.; Khiabanian, H.; da Silva-Almeida, A.C.; Tzoneva, G.; Abate, F.; Ambesi-Impiombato, A.; Sanchez-Martin, M.; Carpenter, Z.; Penson, A.; Perez-Garcia, A.; et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2016, 113, 11306–11311. [Google Scholar] [CrossRef] [Green Version]
- Das, N.; Banavali, S.; Bakhshi, S.; Trehan, A.; Radhakrishnan, V.; Seth, R.; Arora, B.; Narula, G.; Sinha, S.; Roy, P.; et al. Protocol for ICiCLe-ALL-14 (InPOG-ALL-15-01): A prospective, risk stratified, randomised, multicentre, open label, controlled therapeutic trial for newly diagnosed childhood acute lymphoblastic leukaemia in India. Trials 2022, 23, 102. [Google Scholar] [CrossRef]
- Roberts, K.G.; Gu, Z.; Payne-Turner, D.; McCastlain, K.; Harvey, R.C.; Chen, I.-M.; Pei, D.; Iacobucci, I.; Valentine, M.; Pounds, S.B.; et al. High Frequency and Poor Outcome of Philadelphia Chromosome-Like Acute Lymphoblastic Leukemia in Adults. J. Clin. Oncol. 2017, 35, 394–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.T.; Abelson, S.; Zou, J.; Li, T.; Zhao, Z.; Dick, J.; Shlush, L.I.; Pugh, T.J.; Bratman, S.V. High efficiency error suppression for accurate detection of low-frequency variants. Nucleic Acids Res. 2019, 47, e87. [Google Scholar] [CrossRef]
- Waanders, E.; Gu, Z.; Dobson, S.M.; Antić, Ž.; Crawford, J.C.; Ma, X.; Edmonson, M.N.; Payne-Turner, D.; van de Vorst, M.; Jongmans, M.C.J.; et al. Mutational landscape and patterns of clonal evolution in relapsed pediatric acute lymphoblastic leukemia. Blood Cancer Discov. 2020, 1, 96–111. [Google Scholar] [CrossRef]
- Spinella, J.F.; Richer, C.; Cassart, P.; Ouimet, M.; Healy, J.; Sinnett, D. Mutational dynamics of early and late relapsed childhood ALL: Rapid clonal expansion and long-term dormancy. Blood Adv. 2018, 2, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Malinowska-Ozdowy, K.; Frech, C.; Schönegger, A.; Eckert, C.; Cazzaniga, G.; Stanulla, M. KRAS and CREBBP mutations: A relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia 2015, 29, 1656–1667. [Google Scholar] [CrossRef]
- Yu, J.; Waanders, E.; van Reijmersdal, S.V.; Antic, Z.; van Bosbeek, C.M.; Sonneveld, E.; Yu, J.; Waanders, E.; van Reijmersdal, S.V.; Antić van Bosbeek, C.M.; et al. Upfront treatment influences the composition of genetic alterations in relapsed pediatric B-cell precursor acute lymphoblastic leukemia. Hemasphere 2020, 4, e318. [Google Scholar] [CrossRef]
- Antić, Ž.; Yu, J.; Bornhauser, B.C.; Lelieveld, S.H.; Ham, C.G.; Reijmersdal, S.V.; Morgado, L.; Elitzur, S.; Bourquin, J.; Cazzaniga, G.; et al. Clonal dynamics in pediatric B-cell precursor acute lymphoblastic leukemia with very early relapse. Pediatr. Blood Cancer 2022, 69, e29361. [Google Scholar] [CrossRef] [PubMed]
- Ishida, H.; Iguchi, A.; Aoe, M.; Takahashi, T.; Tamefusa, K.; Kanamitsu, K.; Fujiwara, K.; Washio, K.; Matsubara, T.; Tsukahara, H.; et al. Panel-based next-generation sequencing identifies prognostic and actionable genes in childhood acute lymphoblastic leukemia and is suitable for clinical sequencing. Ann. Hematol. 2019, 98, 657–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Age/Gender | Primary Genetic Event | TLC (109/L) | Day 35 MRD | Final Risk (ICiCLe) | Site of Relapse | Relapse Time (Months) | Relapse Type | Clonal Pattern | Pathways of Relapse (Major Gene Clone) | Hypermutation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| C12 | 2 Y/M | P2Y8-CRLF2 | 56 | Negative ≤ 0.01% | IR | BM | 8 | Very early | Minor–major | RAS pathway (KRAS) | No |
| C15 | 7 Y/M | MEF2D-BCL9 | 31.3 | Negative ≤ 0.01% | SR | BM | 8.5 | Very early | Minor–major and therapy-acquired clone | RAS (NRAS) + nucleotide metabolism (PRPS1) | No |
| C18 | 7 Y/M | BCR-ABL1 | 51.2 | Negative ≤ 0.01% | HR | BM + CNS | 8 | Very early | Unclassified | No major clones noted at diagnosis and relapse | No |
| C2 | 8 Y/M | Hypodiploidy | 121 | Positive 1.56% | HR | BM | 12 | Early | Therapy-acquired | Mismatch repair—thiopurine-dose response (PMS2) | No |
| C3 | 4 Y/F | B-cell other | 182 | Negative ≤ 0.01% | IR | CNS | 15 | Early | Therapy-acquired | Epigenetic (KMT2D) and nucleotide metabolism (NT5C2) | No |
| C4 | 5 Y/M | BCR-ABL1 | 96 | Positive 0.76% | HR | BM + CNS | 31 | Early | Major–major | Epigenetic (UHRF1) | Yes |
| C5 | 3 Y/M | B-cell other | 45.2 | Negative ≤ 0.01% | SR | BM + CNS | 31 | Early | Major–major | B-cell development (PAX-5) | Yes |
| C6 | 4 Y/M | B-cell other | 180 | Negative ≤ 0.01% | IR | BM + CNS | 31 | Early | Minor–major and therapy acquired clone | RAS (KRAS) + nucleotide metabolism (NT5C2) | No |
| C8 | 8 Y/M | BCR-ABL1 | 29.6 | Positive 3.5% | HR | BM | 31 | Early | Therapy-acquired | Mismatch repair—thiopurine-dose response (PMS2) | Yes |
| C9 | 10 Y/F | KMT2A-MLLT1 | 224 | Negative ≤ 0.01% | HR | BM | 14 | Early | Therapy-acquired | Epigenetic (KMT2D) and mismatch repair—thiopurine-dose response (PMS2) | Yes |
| C10 | 5 Y/F | EBF1-PDGFRB | 165 | Positive 0.76% | HR | CNS | 25 | Early | Therapy-acquired | B-cell development (ETV6) | No |
| C13 | 10 Y/M | BCR-ABL1 | 67 | Negative ≤ 0.01% | HR | BM + Testicular | 30 | Early | Therapy-acquired | Epigenetic (KMT2D) and nucleotide metabolism (NT5C2) and RAS pathways (NRAS) | Yes |
| C16 | 6 Y/M | ETV6-RUNX1 | 6.4 | Positive 0.02% | HR | BM | 20 | Early | Major–major | Epigenetic (UHRF1) and RAS pathway (KRAS) | No |
| C17 | 3 Y/M | ETV6-RUNX1 | 8.1 | Negative ≤ 0.01% | IR | BM | 33 | Early | Therapy-acquired | Epigenetic (UHRF1) | Yes |
| C19 | 11 Y/M | TAF15-ZNF384 | 20.5 | Positive (0.1%) | HR | BM | 32 | Early | Minor–major | RAS Pathway (Flt3) | No |
| C1 | 5 Y/M | ETV6-RUNX1 | 41 | Negative ≤ 0.01% | SR | BM | 39 | Late | Therapy-acquired | Epigenetic (UHRF1) | No |
| C7 | 6 Y/M | ETV6-RUNX1 | 5.5 | Negative ≤ 0.01% | SR | BM | 40 | Late | Unclassified | - | Yes |
| C11 | 11 Y/M | B-cell other | 30.1 | Positive 0.02% | HR | BM | 37 | Late | Major–major | Epigenetic (KMT2D), RAS (NRAS) and Cell cycle (Tp53) clones increased in size at relapse | No |
| C14 | 4 Y/M | TCF3-PBX1 | 27 | Negative ≤ 0.01% | SR | Testicular | 38 | Late | Therapy-acquired | RAS pathway (KRAS) | No |
| C20 | 5 y/M | B-cell other | 150.6 | Negative ≤ 0.01% | IR | BM + Testicular | 37 | Late | Major–major | Cell cycle (Tp53) | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thakur, R.; Bhatia, P.; Singh, M.; Sreedharanunni, S.; Sharma, P.; Singh, A.; Trehan, A. Therapy-Acquired Clonal Mutations in Thiopurine Drug-Response Genes Drive Majority of Early Relapses in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. Diagnostics 2023, 13, 884. https://doi.org/10.3390/diagnostics13050884
Thakur R, Bhatia P, Singh M, Sreedharanunni S, Sharma P, Singh A, Trehan A. Therapy-Acquired Clonal Mutations in Thiopurine Drug-Response Genes Drive Majority of Early Relapses in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. Diagnostics. 2023; 13(5):884. https://doi.org/10.3390/diagnostics13050884
Chicago/Turabian StyleThakur, Rozy, Prateek Bhatia, Minu Singh, Sreejesh Sreedharanunni, Pankaj Sharma, Aditya Singh, and Amita Trehan. 2023. "Therapy-Acquired Clonal Mutations in Thiopurine Drug-Response Genes Drive Majority of Early Relapses in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia" Diagnostics 13, no. 5: 884. https://doi.org/10.3390/diagnostics13050884
APA StyleThakur, R., Bhatia, P., Singh, M., Sreedharanunni, S., Sharma, P., Singh, A., & Trehan, A. (2023). Therapy-Acquired Clonal Mutations in Thiopurine Drug-Response Genes Drive Majority of Early Relapses in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. Diagnostics, 13(5), 884. https://doi.org/10.3390/diagnostics13050884